


Abstract
Methamphetamine (MA) impairs vesicular monoamine transporter 2 (VMAT2) and dopamine transporter (DAT) function and expression, increasing intracellular DA levels that lead to neurotoxicity. The trace amine–associated receptor 1 (TAAR1) is activated by MA, but when the receptor is not activated, MA-induced neurotoxicity is increased. To investigate interactions among TAAR1, VMAT2, and DAT, transporter function and expression were measured in transgenic Taar1 knockout (KO) and wild-type (WT) mice 24 hours following a binge-like regimen (four intraperitoneal injections, 2 hours apart) of MA (5 mg/kg) or the same schedule of saline treatment. Striatal synaptosomes were separated by fractionation to examine the function and expression of VMAT2 localized to cytosolic and membrane-associated vesicles. DAT was measured in whole synaptosomes. VMAT2-mediated [3H]DA uptake inhibition was increased in Taar1 KO mice in synaptosomal and vesicular fractions, but not the membrane-associated fraction, compared with Taar1 WT mice. There was no difference in [3H]dihydrotetrabenazine binding to the VMAT2 or [125I]RTI-55 binding to the DAT between genotypes, indicating activation of TAAR1 does not affect VMAT2 or DAT expression. There was also no difference between Taar1 WT and KO mice in DAT-mediated [3H]DA uptake inhibition following in vitro treatment with MA. These findings provide the first evidence of a TAAR1-VMAT2 interaction, as activation of TAAR1 mitigated MA-induced impairment of VMAT2 function, independently of change in VMAT2 expression. Additionally, the interaction is localized to intracellular VMAT2 on cytosolic vesicles and did not affect expression or function of DAT in synaptosomes or VMAT2 at the plasmalemmal surface, i.e., on membrane-associated vesicles.
SIGNIFICANCE STATEMENT Methamphetamine stimulates the G protein-coupled receptor TAAR1 to affect dopaminergic function and neurotoxicity. Here we demonstrate that a functional TAAR1 protects a specific subcellular fraction of VMAT2, but not the dopamine transporter, from methamphetamine-induced effects, suggesting new directions in pharmacotherapeutic development for neurodegenerative disorders.
Introduction
The neurotoxic effects of methamphetamine (MA) are attributed to dysregulation of the dopamine (DA) system through impairment of the dopamine transporter (DAT) and the vesicular monoamine transporter 2 (VMAT2) (Fleckenstein et al., 2007). Taken into DA axon terminals via DAT, MA impairs VMAT2 function, inhibiting vesicular DA uptake, and inducing DA release, leading to excessive levels of cytosolic DA (Sulzer et al., 2005). This elevation of intracellular DA leads to the formation of reactive oxygen species, increasing oxidative stress and ultimately DA terminal degeneration (Cadet and Brannock, 1998; McDonnell-Dowling and Kelly, 2017).
MA is also a full agonist at the trace amine–associated receptor 1 (TAAR1). A Gαs protein-coupled receptor, TAAR1 is activated by endogenous trace amines, such as β-phenethylamine and tyramine, as well as many exogenous amines, including the psychostimulants amphetamine (AMPH) and 3-4 methylenedioxymethamphetamine (MDMA) (Bunzow et al., 2001; Simmler et al., 2016). TAAR1 is found in the periphery and many areas of the central nervous system, notably monoaminergic nuclei and projections: the ventral tegmental area, striatum, substantia nigra, nucleus accumbens, dorsal raphe nucleus, locus coeruleus, and the frontal cortex (Borowsky et al., 2001; Lindemann et al., 2008; Espinoza et al., 2015). Uncharacteristically of most G protein-coupled receptors, TAAR1 is not located on the cell membrane but is predominantly localized to the cytoplasm (Bunzow et al., 2001; Xie et al., 2008a; Harmeier et al., 2015). Although there is currently no published research on an interaction between TAAR1 and VMAT2, their intracellular localization suggests a link and there is evidence that TAAR1 and DAT are coexpressed in the substantia nigra (Xie et al., 2007).
A modulator of the DA system, TAAR1’s activation has an overall inhibitory effect on the firing of dopaminergic neurons (Geracitano et al., 2004; Lindemann et al., 2008; Revel et al., 2011). The advent of the transgenic Taar1 knockout (KO) mouse led to the discovery that activation of TAAR1 alters sensitivity to the effects of amphetamines (MA, AMPH, and MDMA). Amphetamines elicit increased monoamine release, locomotor activity, and voluntary MA drinking when TAAR1 is not activated (Wolinsky et al., 2007; Lindemann et al., 2008; Di Cara et al., 2011; Harkness et al., 2015; Reed et al., 2018). Recently, we demonstrated that activation of TAAR1 also diminishes sustained MA-induced neurotoxicity (Miner et al., 2017a). Repeated administration of a non-neurotoxic dose of MA (2.5 mg/kg, four injections, 2 hours apart) decreases DA levels 7 days later in Taar1 KO but not wild-type (WT) mice, indicating that activation of the receptor is neuroprotective. Following a neurotoxic regimen of MA (5 mg/kg, four injections, 2 hours apart), striatal DA levels were further decreased in Taar1 KO compared with WT mice 7 days later.
This binge-like dosing regimen not only induces neurotoxicity but also rapidly impairs transporter function and reduces expression. MA inhibits VMAT2-mediated [3H]DA uptake in an enriched vesicular fraction isolated from striatal synaptosomes at both 1 and 24 hours later (Brown et al., 2000; Ugarte et al., 2003). MA diminishes VMAT2 expression in the vesicular fraction, although expression is unaltered in the synaptosomal and membrane-associated fractions, indicating rapid vesicular trafficking (Hogan et al., 2000; Riddle et al., 2002; Chu et al., 2008). DAT function is also rapidly impaired, but the alteration is transient. MA inhibits [3H]DA uptake within 30 minutes, but uptake returns to baseline after 24 hours (Fleckenstein et al., 1997; Sandoval et al., 2000). DAT expression does not correlate with function, as striatal DAT levels remain diminished 7 days following MA administration and serve as a marker of neurotoxicity (Zhu et al., 2005; McConnell et al., 2015).
On the basis of the key roles of VMAT2 and DAT in the modulation of MA-induced neurotoxicity and our previous findings that activation of TAAR1 mitigates neurotoxicity, we hypothesized interactions between these transporters and TAAR1. We examined the impact of TAAR1 activation on MA-induced dysregulation of striatal VMAT2 and DAT function and expression. Using subcellular fractionation, membrane-associated and cytosolic vesicles were isolated from whole synaptosomes to investigate and localize interactions between TAAR1 and VMAT2. In vivo treatment with MA further impaired VMAT2 function when TAAR1 was not activated compared, with TAAR1 activation, but only in whole synaptosomes. In cytosolic vesicles, VMAT2 function was impaired in mice lacking TAAR1, regardless of treatment. TAAR1 activation did not alter VMAT2 function in membrane-associated vesicles, nor did it alter synaptosomal DAT function or expression, indicating the receptor does not interact with these transporters at the plasma membrane. These findings reveal a novel TAAR1-VMAT2 interaction, a potential mechanism underlying the ability of TAAR1 activation to mitigate MA-induced neurotoxicity.
Materials and Methods
Drugs and Chemicals.
Racemic MA hydrochloride was generously provided by the National Institute on Drug Abuse (NIDA) Research Resources Drug Supply Program (Bethesda, MD). [3H]DA and [125I]RTI-55 (radioligand for neurotransmitter transporters) were purchased from Perkin Elmer (Boston, MA). [3H]Dihydrotetrabenazine (DHTB), which binds to the VMAT2, was purchased from American Radiolabeled Chemicals (St. Louis, MO). All other reagents were obtained from standard commercial sources, unless otherwise noted.
Taar1 KO Mouse Breeding and Genotyping.
The Taar1 KO mice were obtained from the U.C. Davis Knockout Mouse Project (KOMP; www.komp.org) as previously described (Harkness et al., 2015). Briefly, chimeric mice were created using C57BL/6N ES cells in which the entire Taar1 coding region was deleted by homologous recombination, using the Veloci-Gene Null Allele Bac vector, and then injected into BALB/c blastocysts. The chimeras were bred with wild-type C57BL/6N mice and their offspring genotyped according to the strategy recommended by KOMP using the following primers: ACTCTTCACCAAGAATGTGG (forward); CCAACAGCGCTCAACAGTTC (reverse, wild-type allele); GTCGTCCTAGCTTCCTCACTG (reverse, null allele). Male and female siblings, identified as heterozygous for the targeted locus, were subsequently bred to produce Taar1 WT, Taar1 KO, and Taar1 HET (heterozygous) littermates on a C57BL/6N background.
Animal Maintenance and Housing.
Mice of both sexes were used in all experiments and tested at 10–20 weeks of age. Before experiment initiation, mice were group-housed (three to five mice) in filtered acrylic plastic shoebox cages (28 × 18 × 13 cm; l × w × h), fitted with wire tops. Cages were lined with either ECO-Fresh bedding (Absorption Corporation, Ferndale, WA) or Bed-O-Cob (The Andersons, Maumee, OH). Mice had free access to rodent chow (5LOD, 5.0% fat content; Purina Mills, St. Louis, MO) and water. The colony room was maintained at an environmental temperature of 21 ± 1°C with lights on a 12:12-hour light/dark schedule, beginning at 0600 hours. All procedures were approved by the VA Portland Health Care System’s Institutional Animal Care and Use Committee and followed the requirements of the Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to use alternatives to in vivo techniques when available.
Temperature Recording.
Two days prior to drug administration, mice were implanted with IPTT-300 temperature transponders from BioMedic Data Systems (Seaford, DE) to assess body temperature via telemetry. Animals were anesthetized with isoflurane (5% induction, 2.5% maintenance) and transponders were subcutaneously injected dorsally between the shoulders. On the day of MA administration, animals were weighed and transferred from group to individual cages to avoid temperature changes associated with interaction (e.g., huddling) and remained singly housed until euthanized 24 hours after MA treatment. Individual cages were the same type as used for group housing. After a 1-hour acclimation period, temperature recording began and was measured every 15 minutes for 8 hours. All experiments were conducted between 0700 and 1700 hours. Temperatures were noninvasively recorded using the DAS-8001 reader console and smart probe from BioMedic Data Systems. Animals were removed from the cage and the smart probe placed within 5 cm of the embedded transponder to acquire temperature readings. The environmental temperature of the testing environment was 23 ± 1°C. This temperature was selected to differentiate neurotoxic effects of the drugs from those exacerbated by elevated environmental temperatures.
Drug Treatment.
Following the first temperature recording (baseline), each animal received four intraperitoneal injections of saline or MA (5 mg/kg, 2 hours apart). This regimen and dose were selected on the basis of the ability to elicit differences between Taar1 WT and KO mice on neurotoxicity measures (Miner et al., 2017a). A higher dose of MA (10 mg/kg) was not used as it induced nearly the same high levels of neurotoxicity in both genotypes. MA was dissolved in 0.9% saline and injected in a final volume of 10 ml/kg. Mice were euthanized by cervical dislocation 24 hours after the final treatment, followed by decapitation. The brain was immediately transferred to ice and the striatum was dissected and removed using blunt curved microforceps. The striatal tissue was either kept on ice to be immediately used for the [3H]DA uptake assay or flash-frozen and stored at −80°C until time of use for [3H]DHTB or [125I]RTI-55 binding.
Subcellular Fractionation of Striatal Tissue for VMAT2-Mediated [3H]DA Uptake and [3H]DHTB Binding.
Three separate subcellular fractions (synaptosomal, membrane-associated, and enriched vesicular) were isolated as previously described (Teng et al., 1998; Hogan et al., 2000), with minor modifications. All homogenization was performed using a glass/Teflon homogenizer. Striata were homogenized (12 strokes) in ice-cold sucrose (0.32 M, with protease inhibitors) before being centrifuged (800g, 12 minutes, 4°C). The supernatant (S1) was removed and centrifuged (22,000g, 15 minutes, 4°C). The resulting pellet (P2) was osmotically shocked with ice-cold water to disrupt the synaptosomes and release membrane-associated structures, followed by homogenization (six strokes). Osmolarity was restored by addition of Tris (25 mM, pH 7.4) and potassium tartrate (100 mM). A sample was removed to be used for the crude synaptosomal fraction and the remaining P2 fraction was centrifuged (20,000g, 20 minutes, 4°C). The resulting supernatant (S3) was removed and saved. The pellet (P3) was resuspended in VMAT2 assay buffer (25 mM Tris, 100 mM potassium tartrate, 2 mM MgSO4, 4 mM KCl, 0.5 mM EGTA, 0.1 mM EDTA, 1.7 mM ascorbic acid, 10 μM pargyline, and 100 μM tropolone; pH 7.4) and homogenized (six strokes) to generate the membrane-associated fraction. MgSO4 (0.9 mM) was added to the supernatant (S3) and centrifuged (100,000g, 45 minutes, 4°C). The supernatant was discarded and the pellet (P4) resuspended in VMAT2 assay buffer and homogenized (six strokes) to generate the enriched vesicular fraction. Protein concentrations from all three fractions were determined using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA).
Synaptosome Preparation for DAT-Mediated [3H]DA Uptake and [125I]RTI-55 Binding.
Synaptosomes were isolated as previously described (Janowsky et al., 2001). Striatal tissue from untreated mice was homogenized (12 strokes) in ice-cold sucrose (0.32 M, with protease inhibitors) before being centrifuged (800g, 12 minutes, 4°C). The supernatant (S1) was then removed and centrifuged (22,000g, 15 minutes, 4°C). The resulting pellet (P2) was resuspended in DAT assay buffer (Krebs-HEPES buffer: 25 mM HEPES, 122 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 0.2% glucose, 0.02% ascorbic acid, 10 μM pargyline, 100 μM tropolone; pH 7.4) and homogenized (six strokes). Protein density was determined using the bicinchoninic acid protein assay (Thermo Fisher Scientific, Waltham, MA).
Radioligand Binding Assays.
Radioligand binding assays were terminated by filtration using a Wallac 96-well harvester through Perkin Elmer filtermat A filters presoaked in 0.05% polyethyleneimine, unless otherwise noted. Scintillation fluid was added and radioactivity was determined using a Perkin Elmer microbeta plate counter. Assays were conducted in duplicate or triplicate, and data from three or more independent experiments were analyzed. All results were normalized to the amount of total protein loaded.
VMAT2: [3H]DA Uptake Assay.
The [3H]DA uptake assay was adapted from a previously described protocol (Hogan et al., 2000). [3H]DA uptake was performed using pooled striatal tissue from four to five Taar1 WT or KO mice/treatment group. Uptake was separately measured in each fraction preparation: synaptosomal (approx. 5 μg protein), membrane-associated (approx. 5 μg protein), and vesicular (approx. 2.5 μg protein). Preparations were supplemented with MgATP (2 mM) and the assay initiated with the addition of [3H]DA (50 nM). Specific uptake was defined as the difference in uptake observed in the presence or absence of the VMAT2 uptake blocker reserpine (1 μM), and the final assay volume was 250 μl. Incubation (6 minutes, 30°C) was terminated using ice-cold saline (0.9% NaCl) and radioactivity was measured in triplicate as described above. As reserpine does not bind to DAT, the serotonin transporter, or the norepinephrine transporter (Pristupa et al., 1994; Rudd et al., 2005; Mandela et al., 2010), all specific uptake in tissue preparations was considered to be VMAT2-mediated, whereas nonspecific uptake may have been mediated via other transporters.
VMAT2: [3H]DHTB Saturation Binding.
The [3H]DHTB binding assay was adapted from a previously described protocol (Teng et al., 1998). [3H]DHTB binding was performed using pooled striatal tissue from 12 to 15 Taar1 WT or KO mice/treatment group. Binding was separately measured in each subcellular fraction: synaptosomal (approx.15 μg protein), membrane-associated (approx. 15 μg protein), and vesicular (approx. 10 μg protein). The assay was initiated with the addition of [3H]DHTB (1–16 nM). Specific binding was defined as the difference in binding observed in the presence or absence of the VMAT2 inhibitor tetrabenazine (10 μM). The final assay volume was 1 ml. Incubation (60 minutes, 25°C) was terminated using ice-cold Tris buffer (50 mM) and radioactivity was measured in duplicate as described above.
DAT: Effects of MA In Vitro in the [3H]DA Uptake Assay Across Genotypes.
To examine the role of TAAR1 on MA-induced effects in vitro, the [3H]DA uptake assay was adapted from a previously described protocol (Janowsky et al., 2001). Striatal synaptosomes (approx. 10 μg of protein) from untreated Taar1 WT and KO mice were preincubated with MA (1 nM–10 μM) for 10 minutes at 37°C in DAT assay buffer supplemented with a serotonin uptake blocker, fluoxetine (1 μM), and the norepinephrine uptake blocker desipramine (500 nM). The uptake assay was initiated with the addition of [3H]DA (8 nM). Specific uptake was defined as the difference in binding observed in the presence or absence of the DA uptake blocker mazindol (5 μM), and the final assay volume was 500 μl. Incubation (10 minutes, 37°C) was terminated using ice-cold saline (0.9% NaCl) and radioactivity was measured in triplicate as described above.
DAT: [125I]RTI-55 Saturation Binding.
The [125I]RTI-55 binding assay was adapted from a previously described protocol (Eshleman et al., 1999). [125I]RTI-55 binding was performed using striatal tissue from Taar1 WT and KO mice (one per treatment group per assay). Binding was measured in synaptosomes only (approx., 15 μg protein). DAT assay buffer was supplemented with fluoxetine (50 nM) and the assay initiated with the addition of [125I]RTI-55 (40 pM). The specific activity of [125I]RTI-55 was diluted with unlabeled RTI-55 ranging in concentrations from 0.02 to 20 nM, and specific binding was defined as the difference in binding observed in the presence or absence of mazindol (5 μM). The final assay volume was 500 μl. Incubation (90 minutes, 25°C) was terminated by filtration using ice-cold saline (0.9% NaCl) and radioactivity was measured in triplicate as described above, with the exception that filters were not soaked in polyethyleneimine owing to the increase in nonspecific binding of RTI-55. A preliminary experiment was also conducted to compare DAT expression among the three fractions using pooled striatal tissue from six to eight untreated Taar1 HET mice and [125I]RTI-55 (40 pM) diluted with a single concentration of unlabeled RTI-55 (4 nM), under the same conditions outlined above.
Data Analysis.
The IC50 values for the DA uptake assays were calculated by analyzing the sigmoidal dose-response curves using a nonlinear curve-fitting program and further analyzed using unpaired t tests of the logarithm of the IC50 values. Saturation binding data were analyzed by nonlinear regression to generate KD and Bmax values. Radioligand binding data were analyzed by two-way analysis of variance (ANOVA) with treatment and genotype as between-group factors. Sex was not included as a variable in radioligand binding data because tissue from both sexes was pooled. The basis of the decision to pool across sex was the absence of sex effects on temperature response to MA in the current study and on previous measures of MA-induced neurotoxicity (Miner et al., 2017a). Temperature data were analyzed using a repeated measures four-way ANOVA with time as a within-subject factor and sex, genotype, and treatment as between-group factors. Group size was 11–12 (half of each sex), consistent with other experiments detecting differential MA effects on body temperature in these mice (Harkness et al., 2015; Miner et al., 2017a). As there were no significant interactions involving sex in the initial analysis, this factor was excluded from further analyses. Significant two-way interactions were further investigated using post-hoc mean comparisons with the Newman-Keuls test, when appropriate. For temperature data, subsequent analyses were conducted at 30 minutes after each injection, because under the described conditions the maximum hypothermic drop would occur 30 minutes following administration of MA (Harkness et al., 2015; Miner et al., 2017a). Data were analyzed for outliers using Dixon’s Q test at 90% confidence. Statistical analyses were performed using Statistica version 13 software (StatSoft Inc., Tulsa, OK) and Prism version 7 (GraphPad Software, La Jolla, CA). Differences were considered significant at P < 0.05.
Results
VMAT2: [3H]DA Uptake.
In the synaptosomal fraction, MA treatment in vivo reduced [3H]DA uptake compared with saline treatment, and reduction of uptake was significantly greater in Taar1 KO compared with – mice. A two-way ANOVA revealed a significant genotype x treatment interaction (F1,23 = 6.26, P < 0.05) (Fig. 1A). Simple main effect analysis of genotype for each treatment revealed no difference between genotypes for saline-treated animals, but the MA-induced decrease in [3H]DA uptake was significantly greater in Taar1 KO (–57%) compared with Taar1 WT (–32%) mice (P < 0.01). Simple main effect analysis of treatment of each genotype indicated that MA treatment significantly inhibited [3H]DA uptake in both Taar1 WT and Taar1 KO mice compared with levels in saline-treated animals (P < 0.0001). In the membrane-associated fraction, a two-way ANOVA for [3H]DA uptake revealed only a main effect of treatment (F1,19 = 164.5, P < 0.0001) (Fig. 1B). MA treatment inhibited [3H]DA uptake (−51%) compared with saline-treated controls, collapsed across genotype. In the vesicular fraction, a two-way ANOVA revealed a main effect of genotype (F1,19 = 9.14, P < 0.01) and treatment (F1,19 = 81.56, P < 0.0001), but no significant interaction (Fig. 1C). MA treatment inhibited [3H]DA uptake compared with saline-treated controls, collapsed across genotype, and [3H]DA uptake was decreased in Taar1 KO compared with Taar1 WT mice, collapsed across treatment.

VMAT2: [3H]DA uptake in subcellular fractions: synaptosomal (A), membrane-associated (B), and vesicular (C). Taar1 WT and KO mice received four intraperitoneal injections of saline or MA (5 mg/kg), 2 hours apart, and were euthanized 24 hours following the final injection. Striatal tissue from four to five mice per genotype per treatment group was pooled and values normalized to the amount of protein in each sample. Experiments were conducted as described in the text. Data represent means ± S.E.M. of five to seven independent experiments, expressed as percentage of the specific uptake in Taar1 WT control (saline) pooled tissue in each experiment. Mean ± S.E.M. of Taar1 WT control group: synaptosomal, 13.4 ± 3.1 pmol/mg protein; membrane-associated, 13.9 ± 3.5 pmol/mg protein; vesicular, 72.9 ± 21.6 pmol/mg. *P < 0.0001 compared with saline-treated controls; +P < 0.05 between genotypes; #P < 0.0001 for the main effect of treatment; †P < 0.01 for the main effect of genotype.
VMAT2: [3H]DHTB Binding.
Saturation analysis was conducted in each tissue fraction to determine the Bmax and KD values for [3H]DHTB binding (Fig. 2). There were no significant interactions or main effects of genotype for Bmax or KD within any of the tissue fractions (Table 1). A two-way ANOVA of Bmax revealed a main effect of treatment only in the vesicular fraction (F1,8 = 6.97 P < 0.05). MA decreased Bmax in the vesicular fraction compared with saline-treated animals, regardless of genotype. A two-way ANOVA of KD also revealed a main effect of treatment only in the vesicular fraction (F1,8 = 9.82 P < 0.05), where the KD value was decreased in MA-treated animals compared with saline-treated controls. Baseline [3H]DHTB binding to the VMAT2 was compared among fractions in saline-treated animals, collapsed across genotype. A one-way ANOVA revealed a significant difference in Bmax among the three fractions (F2,15 = 5.29 P < 0.05). Bmax was highest in the membrane-associated fraction (1.92 ± 0.25 pmol/mg protein), followed by the synaptosomal fraction (1.43 ± 0.13 pmol/mg protein) and vesicular fraction (1.10 ± 0.12 pmol/mg protein). There was no significant difference in baseline KD value among the three fractions: synaptosomal (2.31 ± 0.27 nM), membrane-associated (2.31 ± 0.38 nM), and vesicular (2.79 ± 0.53 nM) fractions.

[3H]DHTB saturation binding in subcellular fractions: synaptosomal (A), membrane-associated (B), and vesicular (C). Taar1 WT and KO mice received four intraperitoneal injections of saline or MA (5 mg/kg), 2 hours apart, and were euthanized 24 hours following the final injection. Striatal tissue from 12 to 15 mice per genotype per treatment group was pooled and values normalized to the amount of protein in each sample. Experiments were conducted as described in the text. Shown are representative saturation curves from each fraction. Inset: Scatchard transformation of [3H]DHTB saturation binding data.
[3H]DHTB binding in subcellular fractions
Data represent mean ± S.E.M. for each treatment group from three independent experiments, using pooled tissue from 12–15 mice per genotype per treatment group.
DAT: In Vitro MA-Induced Changes in [3H]DA Uptake.
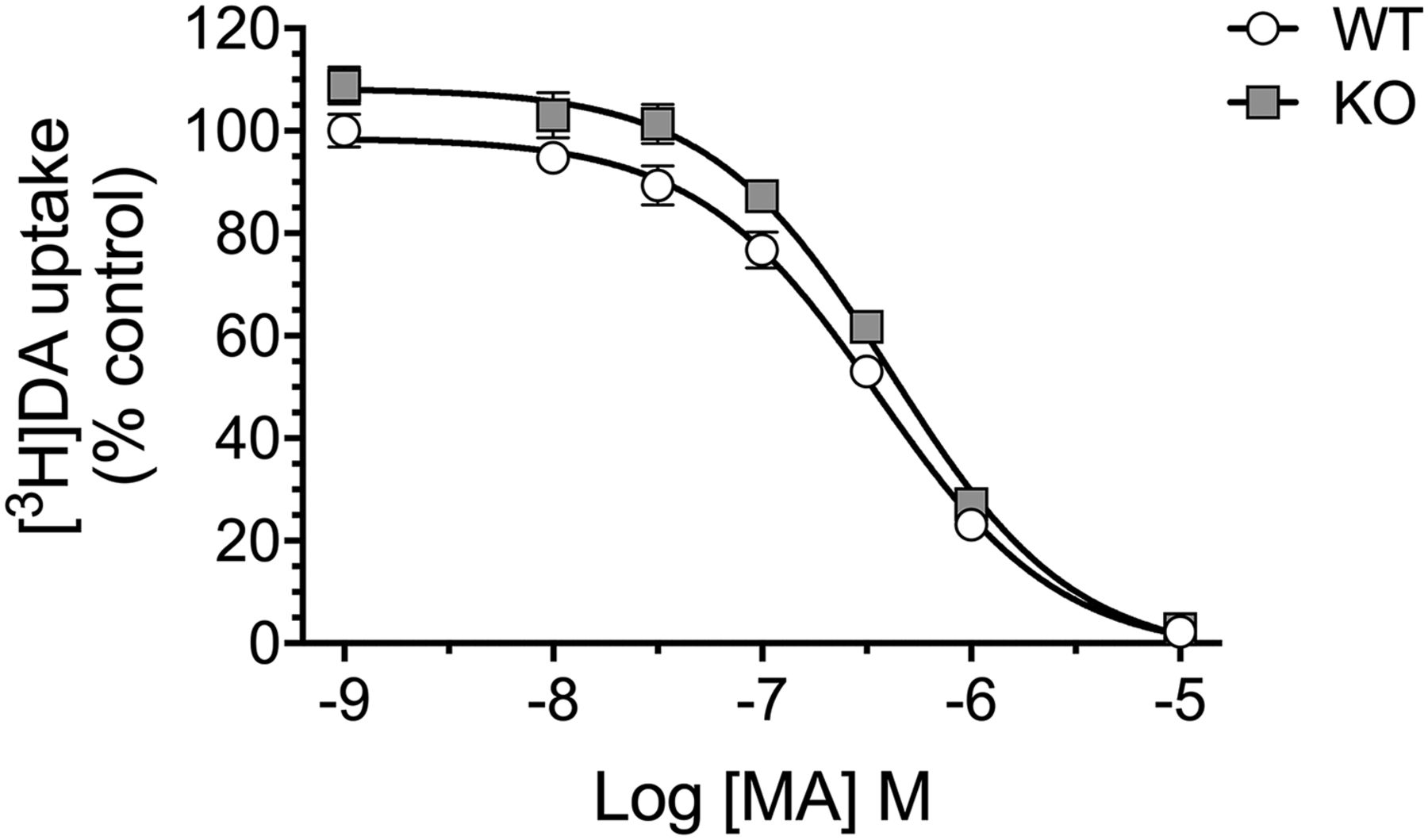
There was no significant difference in IC50 values for DAT-mediated [3H]DA uptake between striatal synaptosomes from Taar1 WT (371 ± 55 nM) and Taar1 KO (426 ± 39 nM) mice treated with MA, P > 0.05 (Fig. 3).

DAT: MA-induced changes in [3H]DA uptake in synaptosomes. Striatal synaptosomes from untreated Taar1 WT and KO mice were preincubated with MA (1 nM–10 μM) for 10 minutes before [3H]DA (8 nM) was added and incubated for 10 minutes. Values were normalized to the amount of protein in each sample. Data represent mean ± S.E.M. for each genotype from three independent experiments with two mice of each genotype per experiment, expressed as percentage of the Taar1 WT control (saline) group-specific uptake in each experiment. Mean ± S.E.M. of Taar1 WT control group: 39.9 ± 5.2 pmol/mg protein.
DAT: [125I]RTI-55 Binding.
Subcellular fractionation analysis of DAT expression was conducted using a single concentration of RTI-55 (4 nM) in untreated Taar1 HET mice to investigate DAT distribution, particularly the purity of the vesicular fraction. A one-way ANOVA revealed a significant difference in binding among the three fractions (F2,15 = 23.53 P < 0.0001). [125I]RTI-55 binding was highest in the isolated membrane-associated fraction, followed by the synaptosomal fraction, with negligible binding in the vesicular fraction. Post-hoc mean comparisons revealed binding in all fractions to be significantly different from each other (Table 2).
[125I]RTI-55 specific binding in subcellular fractions
Data represent mean ± S.E.M. for each subcellular fraction from three independent experiments, using pooled tissue from six to eight untreated Taar1 HET mice with one to two independently pooled preparations of tissue per experiment.
Subsequent saturation binding analysis was conducted in the synaptosomal fraction of treated Taar1 WT and KO mice to determine the Bmax and KD values for [125I]RTI-55 binding (Fig. 4; Table 3). A two-way ANOVA revealed a main effect of MA treatment on Bmax (F1,12 = 10.83 P < 0.01), but no significant interaction or main effect of genotype. MA treatment resulted in a decreased Bmax in comparison with saline-treated animals, regardless of genotype. There were no significant interaction or main effects on KD value.

MA-induced [125I]RTI-55 saturation binding in synaptosomes. Taar1 WT and KO mice received four intraperitoneal injections of saline or MA (5 mg/kg), 2 hours apart, and were euthanized 24 hours following the final injection. Values were normalized to the amount of protein in each sample. Experiments were conducted as described in text. Shown is a representative saturation curve. Inset: Scatchard transformation of [125I]RTI-55 binding data.
[125I]RTI-55 binding in synaptosomes
Data represent mean ± S.E.M. for each genotype and treatment group from four independent experiments, n = 4 mice per genotype per treatment group.
Thermal Response.
Prior to the first MA injection, the mean baseline temperature of all mice was 38.0°C (S.D. = 0.4°C), with no significant between-genotype or -treatment differences. Profound genotype-dependent MA-induced hypothermia was observed. MA did not elicit hyperthermia in any mice, defined as a 0.5°C increase in body temperature above the initial baseline temperature. Analysis of the temperature data, using a three-way repeated-measures ANOVA, revealed a significant genotype × treatment × time interaction (F32,1376 = 5.15, P < 0.0001). There was no difference in temperature between Taar1 WT and KO mice receiving saline (Fig. 5A), although there was a main effect of time (F32,704 = 30.20, P < 0.0001), as temperatures in the control groups decreased over the 8-hour period, probably attributable to single housing. We have previously reported this effect, as have other studies (Fantegrossi et al., 2008; Miner et al., 2017a,b). Examination of the effects of MA revealed a significant genotype x time interaction (F32.672 = 7.60, P < 0.0001) (Fig. 5B). Data were examined for genotype differences using simple main effect analysis at each 30-minute post-injection time point to investigate the hypothermic drop in body temperature. The temperatures of Taar1 WT mice receiving MA were significantly lower 30 minutes after the first and second MA injections compared with their Taar1 KO counterparts.

Effects of repeated saline or MA injections on core body temperature. Taar1 WT and KO mice received four intraperitoneal injections (indicated by arrows) of saline (A) or MA (B; 5 mg/kg), 2 hours apart. Body temperature was measured every 15 minutes via telemetry over 8 hours in an ambient temperature environment of 23 ± 1°C. Data represent temperature for each genotype and treatment group (mean ± S.E.M.) at specified time points, n = 11–12 mice per genotype per treatment group. Time points selected for detailed analysis were 30 minutes after each injection. *P < 0.01; **P < 0.0001 between genotypes.
Next, the data were analyzed by genotype, revealing a significant treatment × time interaction for both genotypes: Taar1 WT (F32,704 = 5.63, P < 0.0001) and Taar1 KO (F32,672 = 6.82, P < 0.0001). Simple main effects analysis revealed that MA significantly decreased body temperature 30 minutes following the first and second injections in Taar1 WT mice, whereas MA significantly increased temperatures in Taar1 KO mice following the second injection, compared with saline-treated control mice (Table 4).
MA-induced change in body temperature relative to saline-treated mice
Data represent change (ΔT) in temperature (mean ± S.E.M.) for each treatment group from saline-treated mice at the specified time point; n = 11–12 mice per genotype per treatment group. Taar1 WT saline-treated group body temperature at each timepoint (mean ± S.E.M.): 1st inj, 37.9 ± 0.1°C; 2nd inj, 37.7 ± 0.1°C; 3rd inj, 37.3 ± 0.1°C; 4th inj, 37.1 ± 0.1°C. Taar1 KO saline-treated group body temperature at each timepoint: 1st inj, 37.7 ± 0.2°C; 2nd inj, 37.6 ± 0.1°C; 3rd inj, 37.2 ± 0.1°C; 4th inj, 36.7 ± 0.1°C.
Discussion
We investigated potential mechanisms underlying TAAR1 regulation of MA-induced neurotoxicity, specifically VMAT2 and DAT function and expression. Our findings reveal a novel localized interaction between TAAR1 and VMAT2 functionality: DA uptake by VMAT2 on cytosolic vesicles was further inhibited in the absence of TAAR1 compared with its presence. This effect was absent on VMAT2 located on membrane-associated vesicles. Activation of TAAR1 did not alter VMAT2 expression (assessed via radioligand binding), indicating that the interaction impairs function rather than decreasing DA uptake owing to decreased VMAT2 expression. Activation of TAAR1 also did not alter DAT function or expression, indicating a lack of interaction between the receptor and transporter at the plasmalemmal surface. These findings provide the first evidence of a relationship between TAAR1 and VMAT2 and indicate TAAR1 activation has an overall protective effect against MA-induced impairment of VMAT2 function.
The effects of TAAR1 activation on VMAT2 were quantified in whole striatal synaptosomes but also differentiated between VMAT2 localized to membrane-associated vesicles and transporters found on cytosolic vesicles. Whereas [3H]DHTB binding to VMAT2 was robust in all fractions, [125I]RTI-55 binding to the DAT was robust only in the synaptosomal and membrane-associated fractions, reflecting both the purity of the enriched vesicular fraction and the restricted localization of DAT to plasmalemmal membranes. Similar results have been reported in subcellular fractions using immunoreactivity assays of VMAT2 and DAT expression (Volz et al., 2007). MA decreased VMAT2-mediated [3H]DA uptake, but with different effects between genotypes across the three fractions (Fig. 1). In the synaptosomal fraction, MA-induced [3H]DA uptake inhibition was increased in Taar1 KO compared with WT mice. In the membrane-associated fraction, MA inhibited [3H]DA uptake equally across genotypes. Finally, in the vesicular fraction, [3H]DA uptake was diminished in Taar1 KO compared with WT mice, regardless of treatment. The absence of this effect in the plasma membrane-associated fraction is presumably the result of intracellular localization of TAAR1. Although TAAR1 is associated with the total cellular membrane fraction, including cytosol, nuclear, and cytoskeletal compartments, the receptor is notably lacking in expression on the cell surface membrane (Bunzow et al., 2001; Xie et al., 2008a; Harmeier et al., 2015). As impairment of VMAT2 function is both a biomarker and potential cause of increased MA-induced neurotoxicity (Fleckenstein et al., 2009), this interaction may be responsible for the increased MA-induced neurotoxicity in Taar1 KO mice using the same dosing regimen (Miner et al., 2017a).
Using [3H]DHTB saturation binding analysis, we were able to quantitatively determine VMAT2 expression levels in each subcellular fraction. Because diminished basal VMAT2 expression increases MA-induced neurotoxicity (Fumagalli et al., 1999; Guillot et al., 2008), we hypothesized Taar1 KO mice may express less VMAT2 than Taar1 WT mice, a potential contributing factor to their increased sensitivity to the neurotoxic effects of MA (Miner et al., 2017a). However, there was no difference in VMAT2 expression between Taar1 WT and KO mice in any of the fractions (Table 1). MA diminished [3H]DHTB binding in the purified vesicular fraction but did not alter binding in the synaptosomal and membrane-associated fractions. This differential pattern of VMAT2 expression among subcellular compartments has been previously reported (Hogan et al., 2000; Ugarte et al., 2003; Chu et al., 2008). Although decreased VMAT2 function has been attributed to trafficking of VMAT2 out of the vesicles (Riddle et al., 2002; Sandoval et al., 2003), it is difficult to separate impairment in VMAT2 function from decreased function caused by diminished expression when changes occur concurrently. Here, genotype differences in vesicular [3H]DA uptake, but not [3H]DHTB binding, suggest VMAT2 function is impaired when TAAR1 is not activated, as opposed to being decreased as a result of trafficking. Independent of the effects of TAAR1 activation in the synaptosomal and vesicular fractions, VMAT2 function and expression were also separated in the membrane-associated fraction, where MA inhibited [3H]DA uptake but did not alter [3H]DHTB binding. Although previously unreported in mice, MA administration to rats also diminishes VMAT2-mediated DA uptake in membrane-associated vesicles (Chu et al., 2010). These results further indicate that MA can impair VMAT2 function independently of decreasing VMAT2 expression. More research is warranted on potential contributing factors.
Previous research on TAAR1 modulation of DAT function has produced equivocal findings. In vitro, MA inhibits [3H]DA uptake, and [3H]DA release is increased in striatal tissue from Taar1 WT compared with KO mice (Xie and Miller, 2009). Similar findings were described in cells cotransfected with TAAR1 and DAT, compared with cells transfected only with DAT, in which MA-induced [3H]DA uptake inhibition and release were increased (Xie and Miller, 2007, 2009). However, these findings indicate MA-induced impairment of DAT function is increased when TAAR1 is activated, as opposed to in vivo treatment with AMPH or MDMA, by which striatal extracellular DA levels are increased when TAAR1 is not activated (Wolinsky et al., 2007; Lindemann et al., 2008; Di Cara et al., 2011). We were unable to replicate the results of Xie and Miller (2009) under similar in vitro conditions (Fig. 3). There was no difference in IC50 values for [3H]DA uptake inhibition by MA between synaptosomes from Taar1 WT and KO mice. As DAT expression, unlike function, remains diminished 24 hours following in vivo MA treatment (Sandoval et al., 2000), [125I]RTI-55 binding was measured in synaptosomes at this time point to more closely simulate a physiologic environment. Like the results for function, activation of TAAR1 did not alter DAT expression (Table 3). Baseline striatal DAT levels did not differ between saline-treated control Taar1 WT and KO mice, confirming previous research using [3H]GBR 12935 binding and quantitative reverse-transcription–polymerase chain reaction (Lindemann et al., 2008; Di Cara et al., 2011). There was also no difference in Bmax between genotypes 24 hours following MA administration. However, MA diminished [125I]RTI-55 binding in synaptosomes overall, indicating this was not owing to a lack of an effect of MA.
Taken together, our results do not support an earlier hypothesis that TAAR1 modulates DAT (Xie and Miller, 2007, 2009; Xie et al., 2008b), as there was no evidence of an interaction under conditions described above. Recent reports support our findings that the DAT is unaffected by TAAR1. Coadministration of MA and the TAAR1 partial agonist RO523648 did not alter [3H]DA uptake and release in striatal synaptosomes in rats (Cotter et al., 2015). Fast-scan cyclic voltammetry showed no difference in DA clearance, as mediated by DAT, in striatal tissue from Taar1 WT compared with KO mice (Leo et al., 2014). Finally, selective TAAR1 agonists diminished hyperlocomotion in DAT KO mice and DAT KO rats, providing behavioral evidence that TAAR1 signals independently of DAT (Revel et al., 2011; Leo et al., 2018). Given the lack of interaction, DAT is an improbable mediator of TAAR1 regulation of MA-induced neurotoxicity.
Hyperthermia is known to exacerbate MA-induced inhibition of both VMAT- and DAT-mediated DA uptake (Sandoval et al., 2000; Ugarte et al., 2003). As the thermal response to MA never exceeded the initial baseline temperature for either genotype, all effects can be deemed hyperthermia-independent. MA induced a biphasic temperature response in Taar1 WT mice, characterized by acute hypothermia within 30 minutes of the first injection, followed by a rise in body temperature toward baseline levels, a pattern that was absent in Taar1 KO mice (Fig. 5). It is improbable that the MA-induced acute hypothermia in Taar1 WT mice contributed to the mitigation of MA-induced impairment of VMAT2 function, as there were no differences in DAT function or expression of either transporter between Taar1 WT and KO mice. The thermal data also replicated previous findings that activation of TAAR1 is necessary for MA-induced hypothermia (Harkness et al., 2015; Miner et al., 2017a).
In conclusion, these findings reveal a novel interaction between TAAR1 and VMAT2 that is localized to cytosolic vesicles. Activation of TAAR1 mitigated MA-induced impairment of overall synaptosomal VMAT2 function in the striatum independently of changes in expression, and VMAT2 function on intracellular vesicles was impaired, regardless of treatment. However, activation of TAAR1 did not modulate in vitro MA-impairment of DAT function or DAT expression. As TAAR1 activation did not alter the function or expression of DAT in whole synaptosomes or VMAT2 located on membrane-associated vesicles, these results indicate TAAR1 does not interact with these transporters on the plasma membrane but does affect intracellular VMAT2 function. This is also the first report of TAAR1 activation altering the function of a monoamine transporter following an in vivo MA treatment. Although this interaction may be a potential underlying mechanism by which TAAR1 regulates MA-induced neurotoxicity, more research is necessary to further understand and characterize this relationship.
Acknowledgments
The authors thank Dr. David Grandy for providing the Taar1 KO and WT mice. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Authorship Contributions
Participated in research design: Miner, Phillips, Janowsky.
Conducted experiments: Miner.
Performed data analysis: Miner, Phillips, Janowsky.
Wrote or contributed to the writing of the manuscript: Miner, Phillips, Janowsky.
Footnotes
- Received April 22, 2019.
- Accepted July 10, 2019.
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grants R01DA046081 (T.J.P.), P50DA018165 (A.J., T.J.P.), and U01DA041579 (T.J.P.)], the Department of Veterans Affairs Merit Review Program [I01BX002106 (T.J.P.), 5I01BX002758 (A.J.)], and the VA Research Career Scientist program (T.J.P., A.J.).
Abbreviations
- AMPH
- amphetamine
- ANOVA
- analysis of variance
- DA
- dopamine
- DAT
- dopamine transporter
- DHTB
- dihydrotetrabenazine
- HET
- heterozygous
- KO
- knockout
- MA
- methamphetamine
- MDMA
- 3,4-methylenedioxymethamphetamine
- RTI-55
- methyl (1R,2S,3S)-3-(4-iodophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate
- TAAR1
- trace amine–associated receptor 1
- VMAT2
- vesicular monoamine transporter 2
- WT
- wild type
- U.S. Government work not protected by U.S. copyright




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}