


Abstract
In brain monoaminergic systems, common biogenic amines, including dopamine, norepinephrine, and serotonin, serve as neurotransmitters. Monoamine autoreceptors provide feedback regulation in neurotransmitter release, and monoamine transporters clear the released neurotransmitters to control synaptic signaling. Recently, trace amine-associated receptor 1 (TAAR1) has been found to be expressed in brain monoaminergic nuclei and activated by common biogenic amines in vitro. This study used transfected cells and brain synaptosomes to evaluate the interaction of common biogenic amines with TAAR1 and monoamine autoreceptors and explore their modulatory effects on monoamine transporters. We confirmed that TAAR1 was activated by dopamine, norepinephrine, and serotonin and demonstrated that TAAR1 signaling was attenuated by monoamine autoreceptors at exposure to dopamine, norepinephrine, and serotonin. In transfected cells, TAAR1 in response to dopamine, norepinephrine, and serotonin significantly inhibited uptake and promoted efflux of [3H]dopamine, [3H]norepinephrine, and [3H]serotonin, respectively, whereas the monoamine autoreceptors, D2s, α2A, and 5-HT1B enhanced the uptake function under the same condition. In brain synaptosomes, dopamine, norepinephrine, and serotonin significantly altered the uptake and efflux of [3H]dopamine, [3H]norepinephrine, and [3H]serotonin, respectively, when the monoamine autoreceptors were blocked. By comparing the effects of dopamine, norepinephrine, and serotonin in monkey and wild-type mouse synaptosomes to their effects in TAAR1 knockout mouse synaptosomes, we deduced that TAAR1 activity inhibited uptake and promoted efflux by monoamine transporters and that monoamine autoreceptors exerted opposite effects. These data provide the first evidence that common biogenic amines modulate monoamine transporter function via both TAAR1 and monoamine autoreceptors, which may balance monoaminergic activity.
Common biogenic amines are neurotransmitters in brain that are synthesized and packaged in monoaminergic neurons and released into synaptic clefts to interact with presynaptic and postsynaptic receptors. Monoamine transporters, including dopamine transporter (DAT), norepinephrine transporter (NET), and serotonin transporter (SERT), clear the released neurotransmitters to terminate neural signaling (Uhl and Johnson, 1994; Masson et al., 1999) and are functionally or dynamically regulated by various chemicals, including psychostimulants and antidepressants (Sulzer et al., 1995; Gutman and Owens, 2006; Fleckenstein et al., 2007). Aberrant transport of common biogenic amines by monoamine transporters in brain is associated with a variety of neuropsychiatric disorders/diseases, such as anxiety, depression, Parkinson's disease, schizophrenia, suicide, and drug abuse/addiction (Klimek et al., 1997; Arango et al., 2002; Schmitt et al., 2006; Serretti et al., 2006). Monoamine autoreceptors, residing on the presynaptic membranes of monoaminergic neurons, monitor and respond to the released common biogenic amines to provide feedback regulation of neurotransmitter release (Hjorth et al., 2000; Schmitz et al., 2002; Garcia et al., 2004). The reported D2 receptor modulation of DAT activity in vitro and in vivo (Meiergerd et al., 1993; Cass and Gerhardt, 1994; Dickinson et al., 1999) suggests that common biogenic amines may modulate monoamine transporter function via interacting with monoamine autoreceptors in the brain.
TAAR1 is a G protein-coupled receptor that is widely expressed in brain monoaminergic nuclei (Borowsky et al., 2001; Miller et al., 2005; Xie et al., 2007b) and is activated by a wide spectrum of compounds, including common biogenic amines, trace amines, and amphetamine-like psychostimulant drugs, such as methamphetamine (Bunzow et al., 2001; Miller et al., 2005; Xie et al., 2007b). We previously demonstrated that TAAR1 could modulate DAT function and that the dopamine D2 autoreceptor (D2s) could attenuate TAAR1 signaling in vitro (Xie and Miller, 2007; Xie et al., 2007b). We have also reported TAAR1 and DAT colocalization in a subset of dopamine neurons in rhesus monkey and mouse substantia nigra (Xie et al., 2007b). More recently, we observed TAAR1 expression in NET-positive neurons in the locus coeruleus of rhesus monkey (unpublished data), and Lindemann et al. (2008) has provided evidence that TAAR1 is expressed in mouse dorsal raphe nucleus. These findings have led us to hypothesize that TAAR1 serves as a neuromodulator of monoamine transporter function in brain.
In this study, we used transfected cells to evaluate the interaction of the common biogenic amines with TAAR1 and monoamine autoreceptors and clarify whether TAAR1 function is influenced by monoamine autoreceptors at exposure to the common biogenic amines. We also used transfected cells and brain striatal and thalamic synaptosomes derived from nonhuman primates, wild-type mice, and TAAR1 knockout mice to investigate the effects of TAAR1 and monoamine autoreceptors on the uptake and efflux functions of DAT, NET, and SERT following treatment with the common biogenic amines. We confirmed that TAAR1 is activated by the common biogenic amines in vitro and showed that its signaling is related with monoamine autoreceptors. We demonstrated that TAAR1 and monoamine autoreceptors in response to the common biogenic amines oppositely modulate the uptake and efflux function of the monoamine transporters. The data reveal a novel mechanism by which dopamine, norepinephrine, and serotonin alter DAT, NET, and SERT function, respectively, via concurrent interaction with TAAR1 and the respective monoamine autoreceptors, which are oppositional in signaling.
Materials and Methods
Materials. Dopamine, norepinephrine, serotonin, indatraline, methylphenidate, desipramine, citalopram, raclopride, SKF86466, and methiothepin were purchased from Sigma-Aldrich (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM), 100× nonessential amino acids, fetal bovine serum, 100× penicillin/streptomycin, Geneticin (G418), and trypsin/EDTA were purchased from Invitro-gen (Carlsbad, CA). Dual-Luciferase Reporter Assay System kit, pGL4.73, and Passive Lysis Buffer (5×) were obtained from Promega (Madison, WI). [3H]Dopamine (60 Ci/mmol), [3H]norepinephrine (49.5 Ci/mmol), and [3H]serotonin (20.3 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). ReadySafe cocktail was obtained from Beckman Coulter (Fullerton, CA). The expression constructs of human α2A, α2B, 5-HT1A, and 5-HT1B were purchased from UMR cDNA Resource Center (Rolla, MO). Rhesus monkey dopamine D2 receptor short isoform (D2s) was constructed in pcDNA3.1(+) in our laboratory.
Cell Culture and Transfection. Cells were grown in DMEM supplemented with fetal bovine serum (10%), penicillin (100 U/ml), streptomycin (100 μg/ml), and nonessential amino acids (0.1 mM) at 5% CO2 and 37°C, and G418 was used for selection or maintenance of selection of stable cell lines. Calcium phosphate transfection was performed as described elsewhere (Xie et al., 2005, 2007a) to introduce the necessary receptor and transporter into different cells for assays. For cotransfection, the ratio of constructs and the amount of total DNA was held constant with pcDNA3.1.
Generation of Cell Lines for Assays. All cell lines in this study are generated originally from HEK293 cells (American Type Culture Collection, Manassas, VA). TAAR1 are HEK293 cells transiently transfected with rhesus monkey TAAR1, CRE-Luc, and pGL4.73; HEK are HEK293 cells transiently transfected with pcDNA 3.1, CRE-Luc, and pGL4.73; D2s-TAAR1 are stable D2s cells transiently transfected with rhesus monkey TAAR1, CRE-Luc, and pGL4.73; α2A-TAAR1 are stable α2A cells transiently transfected with rhesus monkey TAAR1, CRE-Luc, and pGL4.73; α2B-TAAR1 are stable α2B cells transiently transfected with rhesus monkey TAAR1, CRE-Luc, and pGL4.73; 5-HT1A-TAAR1 are stable 5-HT1A cells transiently transfected with rhesus monkey TAAR1, CRE-Luc, and pGL4.73; 5-HT1B-TAAR1 are stable 5-HT1B cells transiently transfected with rhesus monkey TAAR1, CRE-Luc, and pGL4.73; DAT are HEK293 cells transiently transfected with human DAT; NET are HEK293 cells transiently transfected with human NET; SERT are HEK293 cells transiently transfected with human SERT; TAAR1-DAT are stable TAAR1 cells transiently transfected with human DAT; TAAR1-NET are stable TAAR1 cells transiently transfected with human NET; TAAR1-SERT are stable TAAR1 cells transiently transfected with human SERT; D2s-DAT are stable D2s cells transiently transfected with human DAT; α2A-NET are stable α2A cells transiently transfected with human NET; and 5-HT1B-SERT are stable 5-HT1B cells transiently transfected with human SERT.
Animals and Synaptosome Preparation. Monkey brains were obtained from juvenile rhesus (Macaca mulatta) and tamarin (Sanguinus Oedipus) monkeys that were sacrificed for other purposes, and the striatum (only putamen) and thalamus were collected. TAAR1 knockout and wild-type mouse colonies were established at the New England Primate Research Center (Southborough, MA) from six pairs of heterozygous mice given to us as a gift by Lundbeck Research USA, Inc. (Paramus, NJ). The mice were sacrificed at 8 to 10 weeks old to collect the striatum and thalamus. All procedures were conducted in accordance with the Animal Experimentation Protocol approved by the Harvard Medical Area Standing Committee on Animals. Fresh tissues were homogenized in 1.5-ml Eppendorf centrifuge tubes with 10× volume of ice-cold unbuffered 0.32 M sucrose solution (pH 7.0), using a motor-driven pellet pestle purchased from Sigma-Aldrich for 30 up-and-down strokes. The homogenate was centrifuged (1000g, 10 min at 4°C) to yield crude nuclear pellet and low-speed supernatant. The low-speed supernatant fraction was carefully transferred into another fresh tube and centrifuged at 10,000g and 4°C for 20 min to yield synaptosome-containing pellet. The resulting pellet was resuspended in an appropriate volume of ice-cold uptake buffer (a modified Krebs buffer: 25 mM HEPES, 120 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1 μM pargyline, 2 mg/ml glucose, and 0.2 mg/ml ascorbic acid, pH 7.5) for further assays.
Dual-Luciferase Reporter Assay. Cells were placed in wells of 48-well plates (75,000 cells/well in 0.5 ml of medium). At 60 to 70% cell confluence, a luciferase reporter construct CRE-Luc (cAMP-sensitive) (Miller et al., 2005; Xie et al., 2007b) and a reporter control construct pGL4.73 (cAMP-irresponsive) were introduced into the cells along with the expression construct of rhesus monkey TAAR1. After 12 h of incubation under transfection condition, the cells were exposed to vehicle or 1 μM dopamine, norepinephrine, or serotonin in serum-free DMEM for 18 h. To block the autoreceptors, 10 μM raclopride, SKF86466, or methiothepin was added to cells 5 min before and remained during drug treatment. Drugs were dissolved in distilled water or dimethyl sulfoxide to make stock solutions, diluted with DMEM, and then added to the cells within 0.5 h after the replacement of the transfection medium. The culture plates were gently swirled and placed back into the incubator. Passive lysis buffer (PLB) and luciferase assay substrate reagents were prepared according to manufacturer's protocol. Cell lysates were prepared by adding 100 μl of 1× PLB into each well to break the cells followed by incubation on a shaking platform at 25°C for 30 min. The lysate (20 μl) from each well was transferred into wells of opaque 96-well microplates (PerkinElmer, Shelton, CT). Luciferase substrate reagents (50 μl) were injected into each well, and after a 2-s delay, luciferase levels were measured as RLUs for 12 s on a Wallac 1420 multilabel counter, Victor 3V (PerkinElmer).
[3H]Monoamine Uptake Assays. Cells were placed in 145-mm cell culture dishes (1.5 × 105 cells/ml) and grown to 60 to 70% confluence for transfection. After transfection (incubation under transfection condition for 24 h and then in fresh growth medium for 12 h), the cells were harvested and prepared into suspension at a density of 5 × 106 cells/ml. Brain synaptosomes were prepared from fresh striatal and thalamic tissues of monkey and mouse brains. Fifty microliters of the cell suspension or synaptosome preparation was added into 1.5-ml Eppendorf centrifuge tubes and pretreated with vehicle or 1 μM dopamine, norepinephrine, or serotonin in serum-free DMEM (for cells) or in uptake buffer (for synaptosomes) at 25°C for 10 min. After the drug pretreatment, the samples were centrifuged at 1000g (for cells) or at 10,000g (for synaptosomes) at 4°C for 3 min and washed twice with 1 ml of ice-cold serum-free DMEM (for cells) or ice-cold uptake buffer (for synaptosomes). [3H]Dopamine (10 nM), [3H]norepinephrine (20 nM), or [3H]serotonin (20 nM) diluted with serum-free DMEM or uptake buffer was then used to load the cells or synaptosomes at 25°C for 5 min. The samples pretreated with serum-free DMEM (for cells) or uptake buffer (for synaptosomes) only were taken as baseline (100% uptake). For time course uptake assays, synaptosome samples were exposed to 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin alone or the mixture of 10 nM [3H]dopamine plus 100 nM dopamine, 20 nM [3H]norepinephrine plus 100 nM norepinephrine, or 20 nM [3H]serotonin plus 100 nM serotonin at 25°C in uptake buffer for 1, 2, 3, 4, 5, 10, 20, and 30 min. The uptake of the synaptosomes at 30 min in [3H]dopamine, [3H]norepinephrine, or [3H]serotonin alone was taken as maximal uptake (100%). Nonspecific uptake was defined in the presence of 10 μM indatraline for cells or 10 μM methylphenidate, desipramine, or citalopram for synaptosomes. To block the autoreceptors, 10 μM raclopride, SKF86466, or methiothepin was added during drug pretreatment or during uploading (time course assays). Uptake reactions were terminated by the addition of 1 ml of ice-cold serum-free DMEM (for cells) or uptake buffer (for synaptosomes) into the tubes and immediate centrifugation at 1000g (for cells) or 10,000g (for synaptosomes) at 4°C for 3 min. The resulting pellets were rinsed twice with 1 ml of ice-cold serum-free DMEM (for cells) or uptake buffer (for synaptosomes) through centrifugation as described above. The washed pellets were incubated in 1× PLB buffer for 30 min on a shaking platform at 200 rpm and then transferred into scintillation vials containing 4 ml of Beckman ReadySafe scintillation cocktail and counted on Beckman LS6000IC scintillation spectrophotometer for 1 min/sample.
[3H]Monoamine Efflux Assays. Cells were placed in wells of 48-well plates (75,000 cells/well in 0.5 ml of medium) and grown to 60 to 70% confluence for transfection (incubation under transfection condition for 24 h and then in fresh growth medium for 12 h). Brain synaptosomes were prepared from fresh striatal and thalamic tissues of monkey and mouse brains. The cells and synaptosomes were preloaded with [3H]dopamine (10 nM), [3H]norepinephrine (20 nM), or [3H]serotonin (20 nM) at 25°C for 20 min in serum-free DMEM (for cells) or uptake buffer (for synaptosomes), and the assays were conducted in plate wells for cells and in 1.5-ml Eppendorf tubes for synaptosomes. Loading of [3H]monoamine was terminated by the addition of 1 ml of serum-free DMEM into the wells or uptake buffer into the tubes and immediate aspiration of the DMEM or centrifugation at 10,000g at 4°C for 3 min (for synaptosomes). Nonspecific loading was defined in the presence of 10 μM indatraline for cells or 10 μM methylphenidate, desipramine, or citalopram for synaptosomes. After loading, the samples were washed once and exposed to vehicle or 1 μM dopamine, norepinephrine, or serotonin in serum-free DMEM (for cells) or efflux buffer (25 mM HEPES, 120 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1 μM pargyline, 2 mg/ml glucose, and 0.2 mg/ml ascorbic acid, pH 7.5) (for synaptosomes) at 25°C for 30 min. Raclopride (10 μM), SKF86466, or methiothepin was added to block the autoreceptors, and 10 μM indatraline was added to block the transporters during drug treatment following loading of [3H]monoamine. Efflux process was terminated by the addition of 1 ml of ice-cold serum-free DMEM into the wells or efflux buffer into the tubes. After aspiration of the DMEM (for cells) or centrifugation at 10,000g at 4°C for 3 min (for synaptosome), the samples were then washed twice and incubated in 1× PLB buffer for 30 min on a shaking platform at 200 rpm. The lysates were transferred into scintillation vials containing 4 ml of Beckman ReadySafe scintillation cocktail and counted on Beckman LS6000IC scintillation spectrophotometer for 1 min/sample. The samples treated with efflux buffer following loading of [3H]monoamine were taken as baseline (100% retention).
Data Analysis. RLUs represent the luciferase level (firefly and Renilla). The average ratio of firefly luciferase RLU to Renilla luciferase RLU was calculated for each triplicate and then divided by the value at baseline (vehicle treatment), and the results were converted into a percentage value. The RLU increase, which is the percentage value above baseline, represents cAMP accumulation in response to the drug challenge. Specific [3H]monoamine uptake and retention were compared to the baseline or the maximal level and expressed as percentage values in each set of assays. Results were finalized as mean ± S.E.M. of the indicated number of observations. Student's t test was used to analyze the data with two treatments, and one-way ANOVA with Tukey honestly significant difference comparison was used to analyze the data with more than two treatments for judgement of significance of the differences between the groups; the results were considered significantly different at p < 0.05.
Results
Common Biogenic Amines Interact with TAAR1 and Monoamine Autoreceptors in Transfected Cells. Rhesus monkey TAAR1 along with CRE-Luc and pGL4.73 was transiently transfected into HEK293 and stable D2s, α2A, α2B, 5-HT1A, and 5-HT1B cells to generate different cell lines: TAAR1, D2s-TAAR1, α2A-TAAR1, α2B-TAAR1, 5-HT1A-TAAR1, and 5-HT1B-TAAR1. HEK293 cells transiently transfected with pcDNA3.1, CRE-Luc, and pGL4.73 were used as control (HEK). As the first step, we evaluated TAAR1 response to common biogenic amines. TAAR1 and HEK cells were exposed to vehicle or 1 μM dopamine, norepinephrine, or serotonin for 18 h, and CRE-Luc expression was determined as a measurement of cAMP accumulation. Relative to vehicle treatment, dopamine, norepinephrine, and serotonin increased CRE-Luc expression by 189.4 ± 28.5, 213.0 ± 27.6, and 111.5 ± 19.3% in TAAR1 cells and 19.7 ± 11.0, 35.1 ± 18.2, and 15.1 ± 8.7% in HEK cells, respectively (Fig. 1A). The levels of CRE-Luc expression in TAAR1 cells were significantly higher than those in HEK cells (p < 0.01). These data showed that TAAR1 is recognized and excited by dopamine, norepinephrine, and serotonin.
We next evaluated the influence of monoamine autoreceptor activation on TAAR1 signaling in response to common biogenic amines. The indicated cells were exposed to the common biogenic amines in the presence or absence of the autoreceptor inhibitors for 18 h, and CRE-Luc expression was determined to evaluate the receptor effect. After dopamine (1 μM) treatment, CRE-Luc expression in D2s-TAAR1 cells (RLU increase: 25.6 ± 10.6%) was significantly lower than that in TAAR1 cells (RLU increase: 189.4 ± 28.5%) (p < 0.01); however, in the presence of raclopride (10 μM), the CRE-Luc expression in D2s-TAAR1 cells (RLU increase: 174.2 ± 30.5%) was significantly increased (p < 0.01) (Fig. 1B). These data showed that the D2s receptor activation significantly attenuated the TAAR1 response to dopamine. Likewise, α2A or α2B receptor activation significantly attenuated the TAAR1 response to norepinephrine. After norepinephrine (1 μM) treatment, the levels of CRE-Luc expression in α2A-TAAR1 and α2B-TAAR1 cells (RLU increase: 63.0 ± 17.6 and 103.0 ± 20.6%, respectively) were significantly lower than those in TAAR1 cells (RLU increase: 213.0 ± 27.6%) (p < 0.01); however, in the presence of SKF86466 (10 μM), the CRE-Luc expression in either α2A-TAAR1 or α2B-TAAR1 cells (RLU increase: 203.0 ± 31.6 and 173.0 ± 29.6%, respectively) was significantly increased (p < 0.01) (Fig. 1C). Likewise, 5-HT1A or 5-HT1B receptor activation significantly attenuated the TAAR1 response to serotonin. After serotonin (1 μM) treatment, the levels of CRE-Luc expression in 5-HT1A-TAAR1 and 5-HT1B-TAAR1 cells (RLU increase: 61.5 ± 20.3 and 45.5 ± 12.3%, respectively) were significantly lower than those in TAAR1 cells (RLU increase: 111.5 ± 19.3%) (p < 0.01); however, in the presence of methiothepin (10 μM), the CRE-Luc expression in either 5-HT1A-TAAR1 or 5-HT1B-TAAR1 cells (RLU increase: 110.6 ± 21.5 and 115.6 ± 19.5%, respectively) was significantly increased (p < 0.01) (Fig. 1D). Raclopride, SKF86466, and methiothepin lacked direct excitatory effects on TAAR1 at the concentration of 10 μM (Fig. 1E). Our previous study showed that the expression of D2s, α2A, α2B, 5-HT1A, or 5-HT1B receptor did not alter TAAR1 expression in the cells (Xie and Miller, 2008). These results indicate that the common biogenic amines interact with both TAAR1 and monoamine autoreceptors that are oppositional in their signaling.

Interaction of the common biogenic amines with TAAR1 and monoamine autoreceptors in transfected cells. HEK293 and stable D2s, α2A, α2B, 5-HT1A, and 5-HT1B cells were transiently transfected with rhesus monkey TAAR1, CRE-Luc, and pGL4.73 to generate different cell lines (TAAR1, D2s-TAAR1, α2A-TAAR1, α2B-TAAR1, 5-HT1A-TAAR1, and 5-HT1B-TAAR1), and HEK293 cells transiently transfected with pcDNA 3.1 CRE-Luc and pGL4.73 were used as control (HEK). The cells were exposed to vehicle and 1 μM dopamine (DA), norepinephrine (NE), or serotonin (5-HT) for 18 h, and for blockade of the autoreceptors, 10 μM raclopride (RAC), SKF86466 (SKF), or methiothepin (MET) was added 5 min before and remained during the common biogenic amine challenge. The level of CRE-Luc expression was normalized to vehicle treatment and presented as RLU increase (percentage). Data are reported as mean ± S.E.M. for three independent experiments performed in triplicate. **, p < 0.01. A, induction of CRE-Luc expression in TAAR1 and HEK cells by dopamine, norepinephrine, and serotonin. B, influence of the D2s receptor on TAAR1 activation following dopamine treatment. C, influence of the α2A and α2B receptors on TAAR1 activation following norepinephrine treatment. D, influence of the 5-HT1A and 5-HT1B receptors on TAAR1 activation following serotonin treatment. E, direct influences of the autoreceptor inhibitors (10 μM) on TAAR1.
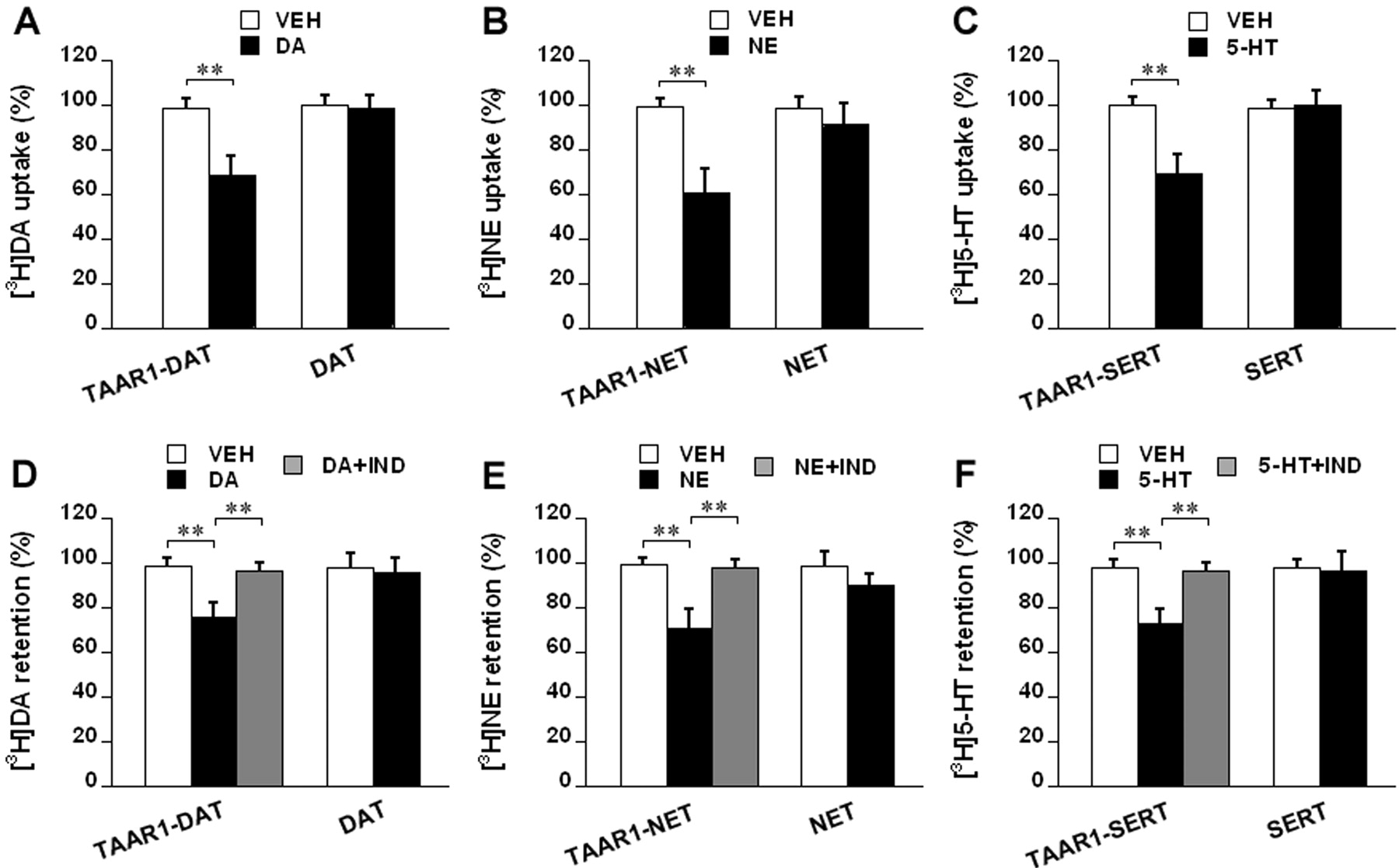
Common Biogenic Amines Modulate Monoamine Transporter Function via TAAR1 and Monoamine Autoreceptors in Transfected Cells. In the following studies, 10 to 20 nM [3H]monoamine is used because these lower concentrations do not activate TAAR1 (Zie et al., 2007). Human DAT, NET, or SERT was transiently transfected into HEK293, stable TAAR1, and stable D2s, α2A, or 5-HT1B cells to generate different cell lines: DAT, NET, SERT, TAAR1-DAT, TAAR1-NET, TAAR1-SERT, D2s-DAT, α2A-NET, and 5-HT1B-SERT. To evaluate [3H]monoamine uptake, the cells were pretreated with vehicle or 1 μM dopamine, norepinephrine, or serotonin for 10 min and then washed twice and loaded with 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin for 5 min. The uptake values were reported as percentage of baseline (uptake in cells pretreated with DMEM for 10 min). To evaluate [3H]monoamine efflux, the cells were loaded with 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin for 20 min and then washed twice and treated with vehicle or 1 μM dopamine, norepinephrine, or serotonin for 30 min. For blockade of the transporters in the efflux process, 10 μM indatraline was added to the cells during the exposure to dopamine, norepinephrine, or serotonin following loading of [3H]monoamine. The [3H]monoamine retention values were reported as percentage of baseline (retention in cells treated with DMEM for 30 min following loading of [3H]monoamine).
Dopamine pretreatment reduced [3H]dopamine uptake in TAAR1-DAT cells by 30.0 ± 5.4%, relative to vehicle treatment (p < 0.01), but had no such effect in DAT cells (Fig. 2A). Norepinephrine pretreatment reduced [3H]norepinephrine uptake in TAAR1-NET cells by 38.6 ± 6.2%, relative to vehicle treatment (p < 0.01), but only caused a reduction of 6.8 ± 2.4% in NET cells (Fig. 2B). Serotonin pretreatment reduced [3H]serotonin uptake in TAAR1-SERT cells by 30.2 ± 4.8%, relative to vehicle treatment (p < 0.01), but had no such effect in SERT cells (Fig. 2C). After loading of [3H]dopamine, dopamine reduced [3H]dopamine retention by 23.2 ± 3.8% in TAAR1-DAT cells, in comparison to vehicle treatment (p < 0.01), but had no such effect in DAT cells. Indatraline (10 μM) mixed with dopamine significantly attenuated the dopamine effect in TAAR1-DAT cells (p < 0.01) (Fig. 2D). After loading of [3H]norepinephrine, norepinephrine reduced [3H]norepinephrine retention by 28.4 ± 4.6% in TAAR1-NET cells, in comparison to vehicle treatment (p < 0.01), but only caused a reduction of 9.0 ± 3.2% in NET cells. Indatraline (10 μM) mixed with norepinephrine significantly attenuated the norepinephrine effect in TAAR1-NET cells (p < 0.01) (Fig. 2E). After loading of [3H]serotonin, serotonin reduced [3H]serotonin retention by 25.2 ± 4.2% in TAAR1-SERT cells, in comparison to vehicle treatment (p < 0.01), but had no such effect in SERT cells. Indatraline (10 μM) mixed with serotonin significantly attenuated the serotonin effect in TAAR1-SERT cells (p < 0.01) (Fig. 2F).

TAAR1 modulation of monoamine transporter function in response to common biogenic amines in transfected cells. HEK293 and stable TAAR1 cells were transiently transfected with human DAT, NET, or SERT to generate different cell lines: DAT, NET, SERT, TAAR1-DAT, TAAR1-NET, and TAAR1-SERT. For uptake assays (A, B, and C), the cells were pretreated with vehicle (VEH) or 1 μM dopamine (DA), norepinephrine (NE), or serotonin (5-HT) for 10 min and then washed twice and loaded with [3H]dopamine (10 nM), [3H]norepinephrine (20 nM), or [3H]serotonin (20 nM) for 5 min. The uptake values are percentage of baseline (uptake in cells pretreated with DMEM for 10 min). For efflux assays (D, E, and F), the cells were preloaded with [3H]dopamine (10 nM), [3H]norepinephrine (20 nM), or [3H]serotonin (20 nM) for 20 min and then washed twice and exposed to vehicle (VEH) or 1 μM dopamine (DA), norepinephrine (NE), or serotonin (5-HT) for 30 min. Indatraline (IND; 10 μM) was added to the cells during the exposure to common biogenic amines following preloading of [3H]monoamine, as indicated. The retention values are percentage of baseline (retention in cells treated with DMEM for 30 min following preloading of [3H]monoamine). Data are reported as mean ± S.E.M. Experiments were performed in triplicate for three times. **, p < 0.01. A, influence of dopamine pretreatment on [3H]dopamine uptake in TAAR1-DAT and DAT cells. B, influence of norepinephrine pretreatment on [3H]norepinephrine uptake in TAAR1-NET and NET cells. C, influence of serotonin pretreatment on [3H]serotonin uptake in TAAR1-SERT and SERT cells. D, effect of dopamine on [3H]dopamine efflux in TAAR1-DAT and DAT cells and influence of indatraline on the dopamine effect in TAAR1-DAT cells. E, effect of norepinephrine on [3H]norepinephrine efflux in TAAR1-NET and NET cells and influence of indatraline on the norepinephrine effect in TAAR1-NET cells. F, effect of serotonin on [3H]serotonin efflux in TAAR1-SERT and SERT cells and influence of indatraline on the serotonin effect in TAAR1-SERT cells.
In D2s-DAT cells, dopamine pretreatment enhanced [3H]dopamine uptake by 15.8 ± 3.5%, relative to vehicle treatment (p < 0.05), and raclopride (10 μM) blocked the dopamine effect (Fig. 3A). In α2A-NET cells, norepinephrine pretreatment increased [3H]norepinephrine uptake by 14.0 ± 3.2%, relative to vehicle treatment (p < 0.05), and SKF86466 blocked the norepinephrine effect (Fig. 3B). In 5-HT1B-SERT cells, serotonin pretreatment promoted [3H]serotonin uptake by 15.4 ± 3.7%, relative to vehicle treatment (p < 0.05), and methiothepin blocked the serotonin effect (Fig. 3C).

Monoamine autoreceptor modulation of monoamine transporter function in response to common biogenic amines in transfected cells. Stable D2s, α2A, and 5-HT1B cells were transiently transfected with human DAT, NET, or SERT to generate different cell lines: D2s-DAT, α2A-NET, and 5-HT1B-SERT. The cells were pretreated with vehicle (VEH) or 1 μM dopamine (DA), norepinephrine (NE), or serotonin (5-HT) for 10 min and then washed twice and loaded with [3H]dopamine (10 nM), [3H]norepinephrine (20 nM), or [3H]serotonin (20 nM) for 5 min. To block the autoreceptors, 10 μM raclopride (RAC), SKF86466 (SKF), or methiothepin (MET) was added during the exposure to the common biogenic amines. The uptake values are percentage of baseline (uptake in cells pretreated with DMEM for 10 min). Data are reported as mean ± S.E.M. for three independent experiments performed in triplicate. **, p < 0.01. A, effect of dopamine on [3H]dopamine uptake and influence of raclopride on dopamine effect in D2s-DAT cells. B, effect of norepinephrine on [3H]norepinephrine uptake and influence of SKF86466 on norepinephrine effect in α2A-NET cells. C, effect of serotonin on [3H]serotonin uptake and influence of methiothepin on serotonin effect in 5-HT1B-SERT cells.
Common Biogenic Amines Modulate [3H]Monoamine Uptake in Brain Synaptosomes. To investigate whether the common biogenic amines modulate monoamine transporter function via TAAR1 and monoamine autoreceptors in the brain, we collected monkey and mouse striatal and thalamic tissues to prepare synaptosomes to evaluate the influence of the receptors on monoamine transporter function at exposure to dopamine, norepinephrine, or serotonin. It has been reported that there is high density of DAT (Ciliax et al., 1995; Miller et al., 2001) and SERT (Sur et al., 1996; Kish et al., 2005) in striatum and NET in thalamus (Ding et al., 2003; Madras et al., 2006). Therefore, we used striatal synaptosomes for evaluation of DAT and SERT function and thalamic synaptosomes for evaluation of NET function. To improve specificity of the assays for DAT, NET, and SERT, we used the selective transporter inhibitors methylphenidate, desipramine, and citalopram, respectively, to block the transporters in determination of the nonspecific uptake. TAAR1-knockout mice (Wolinsky et al., 2007) were used as controls to deduce the role of TAAR1.
We first investigated the influence of dopamine, norepinephrine, or serotonin concurrent loading with [3H]monoamine on the [3H]monoamine uptake using time course studies. The synaptosomes were loaded with 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin alone (doses that do not activate TAAR1) or with a mixture of 10 nM [3H]dopamine plus 100 nM dopamine, 20 nM [3H]norepinephrine plus 100 nM norepinephrine, or 20 nM [3H]serotonin plus 100 nM serotonin for different periods. To block the monoamine autoreceptors, 10 μM raclopride, SKF86466, and methiothepin was added during loading. The uptake values were reported as percentage of the maximal uptake (uptake of 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin alone at 30 min). Dopamine concurrent loading with [3H]dopamine inhibited [3H]dopamine uptake—the uptake curve shifted right and down in rhesus monkey striatal synaptosomes (p < 0.05 compared to [3H]dopamine alone at each time point after 2 min) (Fig. 4A). Methiothepin did not alter the dopamine effect, but in the presence of raclopride, dopamine concurrent loading induced additional inhibition of [3H]dopamine uptake, including uptake halting that occurred after 3 min (p < 0.05 at 4, 5, and 10 min compared to the value in the absence of the autoreceptor inhibitor) (Fig. 4B). Norepinephrine concurrent loading with [3H]norepinephrine inhibited [3H]norepinephrine uptake—the uptake curve shifted right and down in rhesus monkey thalamic synaptosomes (p < 0.05 at 2, 3, 4, 5, and 10 min and p < 0.01 at 20 min compared to [3H]norepinephrine alone) (Fig. 4C). Raclopride did not alter the norepinephrine effect, but in the presence of SKF86466, norepinephrine concurrent loading induced additional inhibition of [3H]norepinephrine, including uptake halting that occurred after 3 min (p < 0.05 at 5 min and p < 0.01 at 10 min compared to the value in the absence of the autoreceptor inhibitor) (Fig. 4D). Serotonin concurrent loading with [3H]serotonin inhibited [3H]serotonin uptake—the uptake curve shifted right and down in rhesus monkey striatal synaptosomes (p < 0.05 at 2, 3, 4, 5, and 10 min and p < 0.01 at 20 min compared to [3H]serotonin alone) (Fig. 4E). Raclopride did not alter the serotonin effect, but in the presence of methiothepin, serotonin concurrent loading induced an additional inhibition of [3H]serotonin uptake, including uptake halting that occurred after 4 min (p < 0.05 at 5 min and p < 0.01 at 10 min compared to the value in the absence of the autoreceptor inhibitor) (Fig. 4F). Parallel experiments performed in tamarin striatal synaptosomes showed high similarity in the influence of dopamine on [3H]dopamine uptake (Fig. 4, G and H). Dopamine concurrent loading with [3H]dopamine inhibited [3H]dopamine uptake—the uptake curve shifted right and down in tamarin monkey striatal synaptosomes (p < 0.05 at 2, 3, 4, 5, and 10 min and p < 0.01 at 20 min compared to [3H]dopamine alone) (Fig. 4G). Methiothepin did not alter the dopamine effect, but in the presence of raclopride, dopamine concurrent loading induced additional inhibition of [3H]dopamine uptake, including uptake halting that occurred after 3 min (p < 0.05 at 4 and 5 min and p < 0.01 at 10 min compared to the value in the absence of the autoreceptor inhibitor) (Fig. 4H).

Influence of common biogenic amines on [3H]monoamine uptake in rhesus and tamarin monkey brain synaptosomes. The synaptosomes were loaded with 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin alone ([3H]DA, [3H]NE, [3H]5-HT), or with a mixture of 10 nM [3H]dopamine plus 100 nM dopamine ([3H]DA+DA), 20 nM [3H]norepinephrine plus 100 nM norepinephrine ([3H]NE+NE) or 20 nM [3H]serotonin plus 100 nM serotonin ([3H]5-HT + 5-HT) for the indicated times. To block the monoamine autoreceptors, 10 μM raclopride (RAC), SKF86466 (SKF), or methiothepin (MET) was added during loading. The uptake values are percentage of the maximal uptake (uptake of 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin at 30 min). Data are presented as mean ± S.E.M. for the experiments performed in triplicate for three rhesus monkeys and two tamarin monkeys. A, effect of dopamine on [3H]dopamine uptake in rhesus monkey striatal synaptosomes. *, p < 0.05 compared to [3H]DA. B, effect of dopamine on [3H]dopamine uptake in the presence of raclopride or methiothepin in rhesus monkey striatal synaptosomes. *, p < 0.05 compared to [3H]DA + DA. C, effect of norepinephrine on [3H]norepinephrine uptake in rhesus monkey thalamic synaptosomes. *, p < 0.05 and **, p < 0.01 compared to [3H]NE. D, effect of norepinephrine on [3H]norepinephrine uptake in the presence of SKF86466 or raclopride in rhesus monkey thalamic synaptosomes. *, p < 0.05 and **, p < 0.01 compared to [3H]NE + NE. E, effect of serotonin on [3H]serotonin uptake in rhesus monkey striatal synaptosomes. *, p < 0.05 and **, p < 0.01 compared to [3H]5-HT. F, effect of serotonin on [3H]serotonin uptake in rhesus monkey striatal synaptosomes in the presence of methiothepin or raclopride in striatal synaptosomes. *, p < 0.05 and **, p < 0.01 compared to [3H]5-HT + 5-HT. G, effect of dopamine on [3H]dopamine uptake in tamarin monkey striatal synaptosomes. *, p < 0.05 and **, p < 0.01 compared to [3H]DA. H, effect of dopamine on [3H]dopamine uptake in the presence of raclopride or methiothepin in tamarin monkey striatal synaptosomes. *, p < 0.05 and **, p < 0.01 compared to [3H]DA + DA.
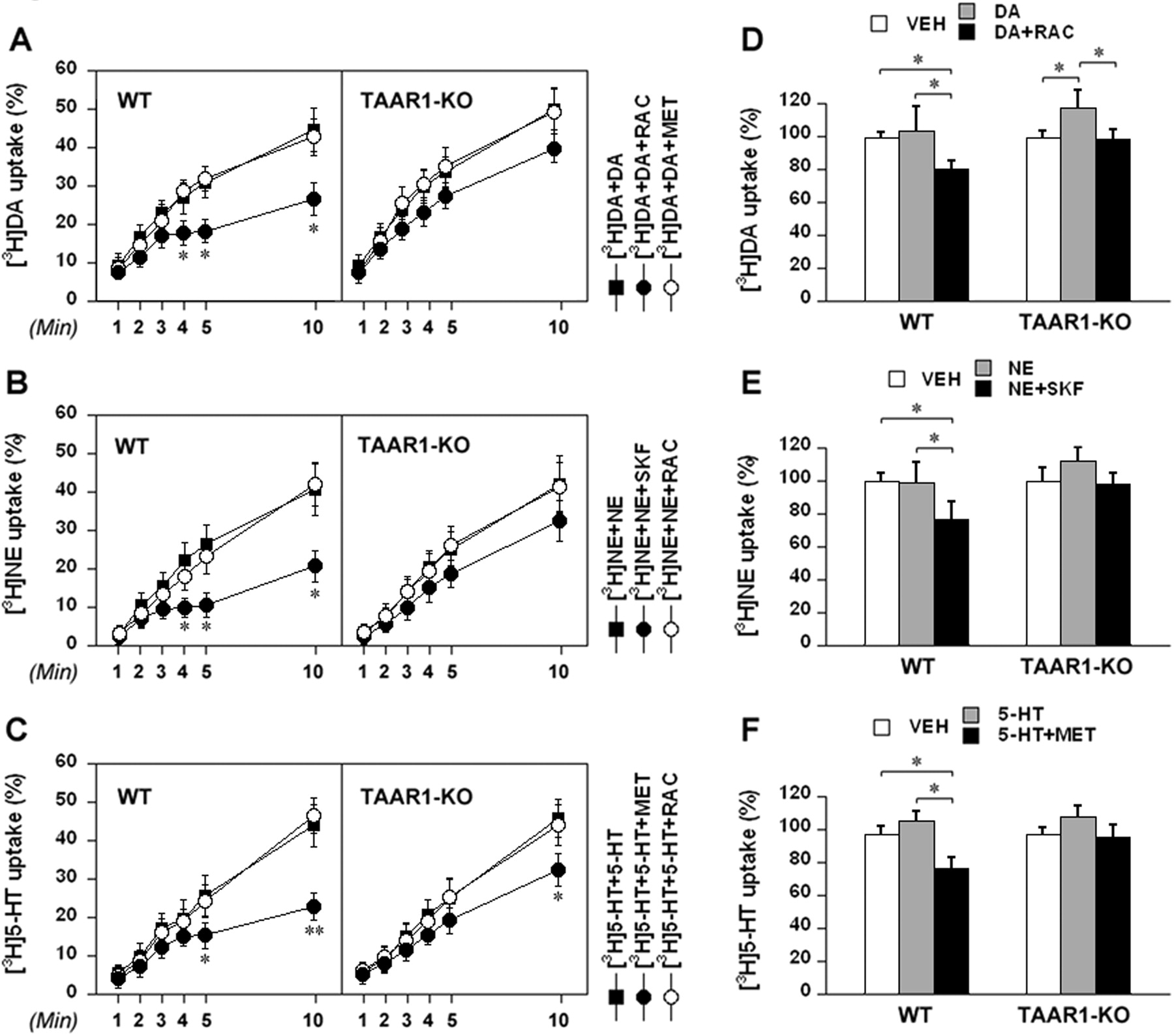
In mouse brain synaptosomes, methiothepin did not alter the dopamine effect; however, in the presence of raclopride, dopamine concurrent loading induced additional inhibition of [3H]dopamine uptake, including uptake halting that occurred after 3 min in wild-type mouse striatal synaptosomes (p < 0.05 at 4, 5, and 10 min compared to the value in the absence of the autoreceptor inhibitor), but such uptake halting was not observed in TAAR1 knockout mouse striatal synaptosomes (Fig. 5A). Because of the uptake halting, we can see that the reduction in the percentage value of [3H]dopamine uptake at 10 min in the wild-type mouse striatal synaptosomes (18.3 ± 3.2%) was significantly greater than that in TAAR1 knockout mouse striatal synaptosomes (10.4 ± 2.4%) (p < 0.05). With respect to [3H]norepinephrine uptake, raclopride did not alter the norepinephrine effect; however, in the presence of SKF86466, norepinephrine concurrent loading induced additional inhibition of [3H]norepinephrine uptake, including uptake halting that occurred after 3 min in wild-type mouse thalamic synaptosomes (p < 0.05 at 4, 5, and 10 min compared to the value in the absence of the autoreceptor inhibitor), but such uptake halting was not observed in TAAR1 knockout mouse thalamic synaptosomes (Fig. 5B). Because of the uptake halting, we can see that the reduction in the percentage value of [3H]norepinephrine uptake at 10 min in the wild-type mouse thalamic synaptosomes (20.2 ± 4.2%) was significantly greater than that in TAAR1 knockout mouse thalamic synaptosomes (9.8 ± 2.2%) (p < 0.05). For [3H]serotonin uptake, raclopride did not alter the serotonin effect, but in the presence of methiothepin, serotonin concurrent loading induced additional inhibition of [3H]serotonin uptake, including uptake halting that occurred after 4 min in wild-type mouse striatal synaptosomes (p < 0.05 at 5 min and p < 0.01 at 10 min compared to the value in the absence of the autoreceptor inhibitor). This pattern of uptake halting was not observed in TAAR1 knockout mouse striatal synaptosomes, although methiothepin did cause a significant inhibition of [3H]serotonin uptake at 10 min (p < 0.05) (Fig. 5C). Because of the uptake halting, the reduction in the percentage value of [3H]serotonin uptake at 10 min in the wild-type mouse striatal synaptosomes (21.4 ± 4.6%) was significantly greater than that in TAAR1 knockout mouse striatal synaptosomes (13.6 ± 3.3%) (p < 0.05).

Influence of common biogenic amines on [3H]monoamine uptake in wild-type (WT) and TAAR1 knockout (TAAR1-KO) mouse brain synaptosomes. For time course assays (A, B, and C), the synaptosomes were treated as described in Fig. 4 legend, and the uptake values are percentage of the maximal uptake (uptake of 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin at 30 min). For pretreatment assays (D, E, and F), the synaptosomes were treated as described in Fig. 4 legend, and the uptake values are percentage of baseline (uptake in synaptosomes pretreated with uptake buffer for 10 min). Data are reported as mean ± S.E.M. Experiments were performed in triplicate for three pairs of wild-type and TAAR1 knockout mice. A, uptake of 10 nM [3H]dopamine in the presence of 100 nM dopamine ([3H]DA + DA) and the influence of raclopride (RAC) or methiothepin (MET) on the uptake in striatal synaptosomes. *, p < 0.05 compared to [3H]DA + DA. B, uptake of 20 nM [3H]norepinephrine in the presence of 100 nM norepinephrine ([3H]NE + NE) and the influence of SKF86466 (SKF) or raclopride (RAC) on the uptake in thalamic synaptosomes. *, p < 0.05 compared to [3H]NE + NE. C, uptake of 20 nM [3H]serotonin in the presence of 100 nM serotonin ([3H]5-HT + 5-HT) and the influence of methiothepin (MET) or raclopride (RAC) on the uptake in striatal synaptosomes. *, p < 0.05 and **, p < 0.01 compared to [3H]5-HT + 5-HT. D, effect of dopamine (DA) pretreatment on [3H]dopamine uptake and influence of raclopride (RAC) on the dopamine effect in striatal synaptosomes. *, p < 0.05. E, effect of norepinephrine (NE) pretreatment on [3H]norepinephrine uptake and influence of SKF86466 (SKF) on the norepinephrine effect in thalamic synaptosomes. *, p < 0.05. F, effect of serotonin (5-HT) pretreatment on [3H]serotonin uptake and influence of methiothepin (MET) on the serotonin effect in striatal synaptosomes. *, p < 0.05. Vehicle (VEH) treatment was used as control.
We next evaluated the influence of common biogenic amine pretreatment on [3H]monoamine uptake in wild-type and TAAR1 knockout mouse striatal or thalamic synaptosomes. The synaptosomes were pretreated with vehicle or 1 μM dopamine, norepinephrine, or serotonin for 10 min and then washed twice and loaded with 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin for 5 min. Raclopride (10 μM), SKF86466 (10 μM), or Ro32-0432 (10 μM) was added to the synaptosomes during pretreatment to block monoamine autoreceptors. The uptake values were reported as percentage of baseline (uptake in synaptosomes pretreated with uptake buffer for 10 min). Dopamine pretreatment did not alter [3H]dopamine uptake in striatal synaptosomes of wild-type mice, but in the presence of raclopride, dopamine pretreatment inhibited [3H]dopamine uptake by 20.1 ± 3.3% (p < 0.05) to a level significantly lower than that of vehicle treatment (p < 0.05). In TAAR1 knockout mouse striatal synaptosomes, dopamine pretreatment increased [3H]dopamine uptake by 17.0 ± 3.1% compared to vehicle treatment (p < 0.05), and raclopride blocked the dopamine effect and returned the uptake back to the level of vehicle treatment (p < 0.05) (Fig. 5D). Likewise, norepinephrine pretreatment did not alter [3H]norepinephrine uptake in thalamic synaptosomes of wild-type mice, but in the presence of SKF86466, norepinephrine pretreatment inhibited [3H]norepinephrine uptake by 22.7 ± 4.1% (p < 0.05) to a level significantly lower than that of vehicle treatment (p < 0.05). In TAAR1 knockout mouse thalamic synaptosomes, norepinephrine pretreatment increased [3H]norepinephrine uptake by 11.7 ± 2.5% compared to vehicle treatment, and SKF86466 blocked the norepinephrine effect and returned the uptake back to the level of vehicle treatment (Fig. 5E). Likewise, serotonin pretreatment did not alter [3H]serotonin uptake in striatal synaptosomes of wild-type mice, but in the presence of methiothepin, serotonin inhibited [3H]serotonin uptake by 22.4 ± 3.9% (p < 0.05) to a level significantly lower than that of vehicle treatment (p < 0.05). In TAAR1 knockout mouse striatal synaptosomes, serotonin pretreatment increased [3H]serotonin uptake by 17.0 ± 3.1% compared to vehicle treatment, and methiothepin blocked the serotonin effect and returned the uptake to the level of vehicle treatment (Fig. 5F).
Common Biogenic Amines Modulate [3H]Monoamine Efflux in Brain Synaptosomes. To evaluate [3H]monoamine efflux, synaptosomes were preloaded with 10 nM [3H]dopamine, 20 nM [3H]norepinephrine, or 20 nM [3H]serotonin for 20 min and then washed twice and treated with vehicle or 1 μM dopamine, norepinephrine, or serotonin for 30 min. To block monoamine autoreceptors, 10 μM raclopride, SKF86466, or methiothepin was added to the synaptosomes during the exposure to dopamine, norepinephrine, or serotonin after preloading. The [3H]monoamine retention was reported as percentage of baseline (retention in cells treated with DMEM or synaptosomes treated with efflux buffer for 30 min following preloading of [3H]monoamine), and the loss of the [3H]monoamine retention was used as the efflux values.
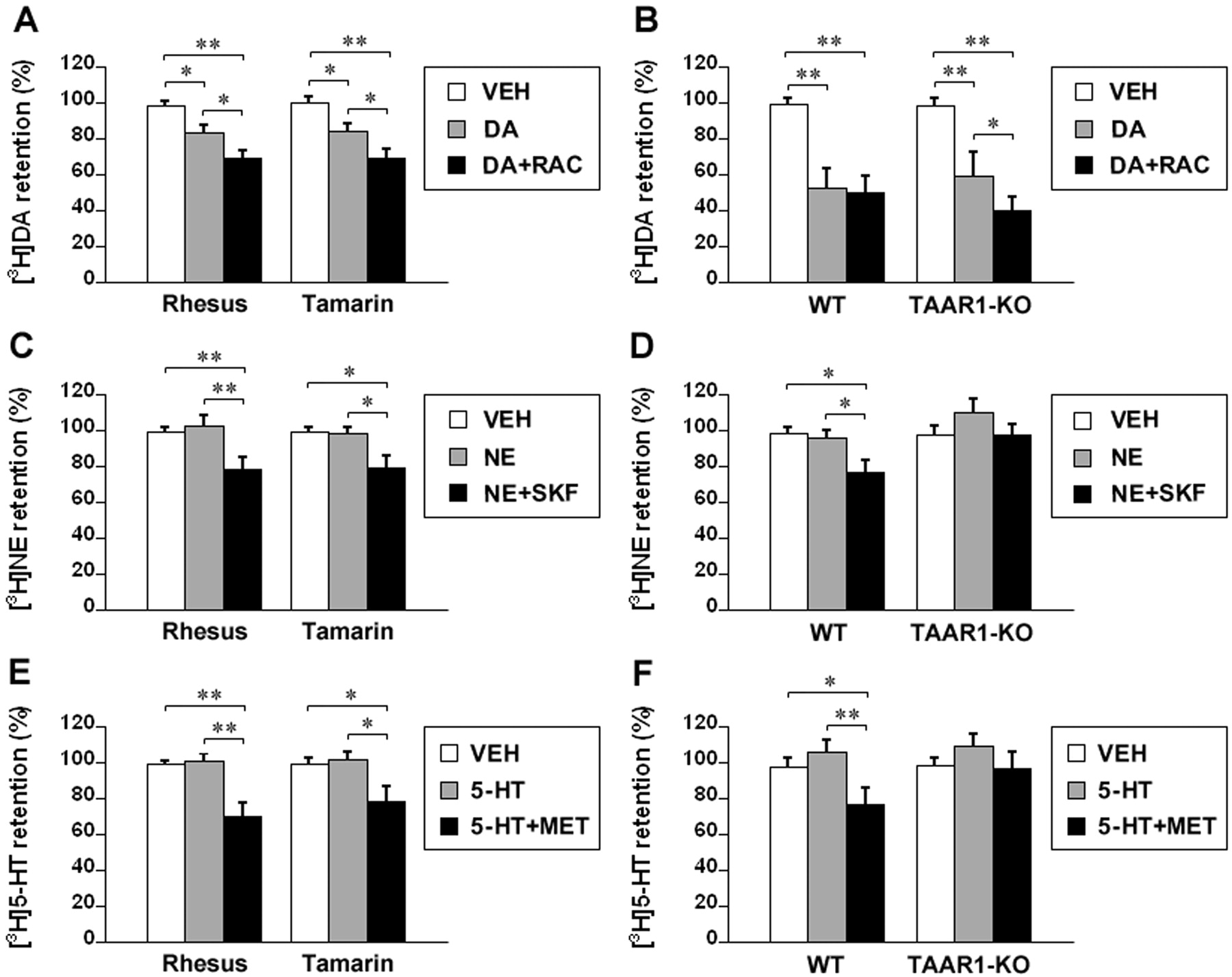
Dopamine induced efflux of [3H]dopamine by 16.5 ± 3.2 and 16.4 ± 2.9% in rhesus and tamarin monkey striatal synaptosomes, respectively, relative to vehicle treatment (p < 0.05). In the presence of raclopride, dopamine induced an additional efflux of 12.1 ± 2.6 and 11.1 ± 2.4%, respectively, in rhesus and tamarin monkey striatal synaptosomes (p < 0.05 compared to dopamine treatment and p < 0.01 compared to vehicle treatment) (Fig. 6A). In wild-type mouse striatal synaptosomes, dopamine induced efflux of [3H]dopamine by 49.2 ± 6.2%, relative to vehicle treatment (p < 0.01), and raclopride did not alter the dopamine effect. In TAAR1 knockout mouse striatal synaptosomes, dopamine induced efflux of [3H]dopamine by 39.1 ± 5.4%, relative to vehicle treatment (p < 0.01), and in the presence of raclopride, dopamine induced an additional efflux of 19.7 ± 3.1% (p < 0.05 compared to dopamine treatment and p < 0.01 compared to vehicle treatment) (Fig. 6B).
Norepinephrine did not significantly alter [3H]norepinephrine retention in either rhesus or tamarin monkey thalamic synaptosomes, but in the presence of SKF86466, norepinephrine induced efflux of [3H]norepinephrine by 20.6 ± 4.2 and 19.8 ± 3.6% in rhesus (p < 0.01) and tamarin (p < 0.05) monkey thalamic synaptosomes, respectively, relative to vehicle treatment (Fig. 6C). In wild-type mouse thalamic synaptosomes, norepinephrine did not significantly alter [3H]norepinephrine retention, and in the presence of SKF86466, norepinephrine induced efflux of [3H]norepinephrine by 22.2 ± 3.4%, relative to vehicle treatment (p < 0.05). In TAAR1 knockout mouse thalamic synaptosomes, norepinephrine increased [3H]norepinephrine retention by 12.3 ± 2.6%, relative to vehicle treatment, but this was not significant, and SKF86466 reversed this increase but did not result in efflux, relative to vehicle treatment (Fig. 6D).
Serotonin did not significantly alter [3H]serotonin retention in either rhesus or tamarin monkey striatal synaptosomes, but in the presence of methiothepin, serotonin induced efflux of [3H]serotonin by 29.2 ± 4.5 (p < 0.01) and 20.9 ± 4.1% (p < 0.05) in rhesus and tamarin monkey striatal synaptosomes, respectively, relative to vehicle treatment (Fig. 6E). In wild-type mouse striatal synaptosomes, serotonin increased [3H]serotonin retention by 8.1 ± 1.9%, but this was not significant, and in the presence of methiothepin, serotonin induced efflux of [3H]serotonin by 21.1 ± 4.2% relative to vehicle treatment (p < 0.05). In TAAR1 knockout mouse striatal synaptosomes, serotonin increased [3H]serotonin retention by 10.7 ± 2.8%, but this was not significant, and methiothepin reversed this increase but did not result in efflux, relative to vehicle treatment (Fig. 6F).
Discussion
In the present study, we confirmed that TAAR1 is activated by common biogenic amines in vitro and then demonstrated that TAAR1 activation by common biogenic amines can modulate DAT, NET, and SERT function. In transfected cells, TAAR1 activation by the common biogenic amines inhibited [3H]monoamine uptake and promoted [3H]monoamine efflux. In monkey and wild-type mouse brain synaptosomes, the common biogenic amines (100 nM, a concentration that activates TAAR1; Xie et al., 2007) induced temporal uptake halting in time course uptake assays or reduced uptake to a level significantly lower than that of vehicle treatment in pretreatment assays when the monoamine autoreceptors were blocked by the selective monoamine autoreceptor inhibitors. However, under the same condition, such effects of the common biogenic amines on [3H]monoamine uptake did not occur in TAAR1 knockout mouse synaptosomes. With regard to [3H]monoamine efflux in synaptosomes, when the autoreceptors were blocked, norepinephrine and serotonin reduced the retention of [3H]norepinephrine and [3H]serotonin, respectively, to a level significantly lower than that of vehicle treatment in monkey and wild-type mouse synaptosomes. However, in TAAR1 knockout mouse synaptosomes, norepinephrine and serotonin did not alter [3H]norepinephrine and [3H]serotonin retention, respectively, relative to vehicle treatment when the autoreceptors were blocked. These data suggest that the common biogenic amines interact with TAAR1 to modulate DAT, NET, and SERT function in brain. Interestingly, dopamine induced [3H]dopamine efflux in striatal synaptosomes of monkeys, wild-type mice, and also TAAR1 knockout mice in the absence of autoreceptor blockade, which may suggest that, in addition to TAAR1, dopamine may also interact with other cellular components to trigger [3H]dopamine efflux independent of TAAR1. In this respect, further study can be designed to clarify whether other trace amine-associated receptors or other dopamine receptor subtypes are involved.

Effects of common biogenic amines on [3H]monoamine efflux in monkey and mouse brain synaptosomes. The synaptosomes were preloaded with [3H]dopamine (10 nM), [3H]norepinephrine (20 nM), or [3H]serotonin (20 nM) for 20 min and then washed twice and exposed to vehicle (VEH) or 1 μM dopamine (DA), norepinephrine (NE), or serotonin (5-HT) for 30 min. To block the autoreceptors, 10 μM raclopride (RAC), SKF86466 (SKF), or methiothepin (MET) was added during the exposure to common biogenic amines following preloading. The [3H]monoamine retention values are percentage of the baseline (retention in the synaptosomes treated with efflux buffer for 30 min following preloading of [3H]monoamine). A, effect of dopamine on [3H]dopamine efflux and the influence of raclopride on the dopamine effect in rhesus and tamarin monkey striatal synaptosomes. B, effect of dopamine on [3H]dopamine efflux and the influence of raclopride on the dopamine effect in wild-type and TAAR1 knockout mouse striatal synaptosomes. C, effect of norepinephrine on [3H]norepinephrine efflux and the influence of SKF86466 on the norepinephrine effect in rhesus and tamarin monkey thalamic synaptosomes. D, effect of norepinephrine on [3H]norepinephrine efflux and the influence of SKF86466 on the norepinephrine effect in wild-type and TAAR1 knockout mouse thalamic synaptosomes. E, effect of serotonin on [3H]serotonin efflux and the influence of methiothepin on the serotonin effect in rhesus and tamarin striatal synaptosomes. F, effect of serotonin on [3H]serotonin efflux and the influence of methiothepin on the serotonin effect in wild-type and TAAR1 knockout mouse striatal synaptosomes. Data are reported as mean ± S.E.M. Experiments were performed in triplicate for three rhesus monkeys, two tamarin monkeys, and three pairs of wild-type and TAAR1 knockout mice. *, p < 0.05 and **, p < 0.01.
Although both reverse transport and vesicular release may occur in brain synaptosomes, we observed similar efflux in transfected HEK293 cells, which do not have vesicles in our in vitro experiments. Accordingly, we speculate that the efflux of [3H]monoamines is due to reverse transport and not vesicular release. This conclusion is supported by our previous studies that have demonstrated that TAAR1-mediated efflux is transporter-dependent in vitro and that [3H]monoamine efflux induced by β-PEA in synaptosomes could be blocked by specific monoamine transporter inhibitors (Xie and Miller, 2007, 2008). Therefore, the mechanism of efflux is most likely related to cAMP accumulation; consequent phosphorylation cascades resulted from receptor activation, which reversed the transport of the monoamines by monoamine transporters.
Monoamine autoreceptors are responsive to common biogenic amines released into the synaptic clefts and regulate neurotransmitter release (exocytosis). D2-like autoreceptors located in dopamine neurons modulate dopamine synthesis (Tissari et al., 1983) and release (Starke et al., 1989), 5-HT1A/1B autoreceptors located in serotoninergic neurons regulate serotonin release (Gobert et al., 1997; Yoshitake et al., 2003), and adrenergic α2A/2B autoreceptors contribute to regulation of norepinephrine release (Tavares et al., 1996; Callado and Stamford, 1999). Intriguingly, D2-like autoreceptors are also implicated in regulation of DAT activity (Meiergerd et al., 1993; Cass and Gerhardt, 1994), suggesting that the common biogenic amines may also alter monoamine transporter function via interacting with monoamine autoreceptors in the brain. Indeed, we demonstrated that D2s activation by dopamine, α2A activation by norepinephrine, and 5-HT1B activation by serotonin enhanced uptake of [3H]dopamine, [3H]norepinephrine, and [3H]serotonin, respectively, in the transfected cells. In brain synaptosomes, the common biogenic amines displayed significant inhibition of [3H]monoamine uptake and significantly induced [3H]monoamine efflux in monkey and wild-type mouse synaptosomes in the presence of specific monoamine autoreceptor inhibitors. Our interpretation of these data is that, when the autoreceptors were blocked, TAAR1 signaling in response to the common biogenic amines was liberated from inhibition by concurrent autoreceptor activation. In TAAR1 knockout mouse synaptosomes, we observed that dopamine induced a significant enhancement in [3H]dopamine uptake in the pretreatment assays, which was reversed by raclopride, and the same trend is apparent in the uptake of [3H]norepinephrine and [3H]serotonin, although it did not reach significance. Meanwhile, the data obtained from time course uptake assays showed that [3H]serotonin uptake was significantly reduced at 10 min in the presence of methiothepin in TAAR1 knockout mouse striatal synaptosomes, and the same trend is apparent in [3H]dopamine and [3H]norepinephrine time course assays. This effect may be due to blockade of autoreceptor enhancement of uptake in the TAAR1 knockout mice. These data, paralleling our in vitro observations, along with the finding that monoamine autoreceptor activation by the common biogenic amines inhibits TAAR1 signaling in vitro, strongly suggest that common biogenic amines interact with monoamine autoreceptors and consequently counteract or depress TAAR1 influence on monoamine transporter function in brain.
The present study provides the first evidence that the common biogenic amines interact with both TAAR1 and monoamine autoreceptors to modulate monoamine transporter function in the brain. The monoamine autoreceptors are known to be presynaptic release regulators (Starke et al., 1989; Tavares et al., 1996; Gobert et al., 1997; Callado and Stamford, 1999; Yoshitake et al., 2003). Although we did not investigate the influence of TAAR1 activation on the release of the common biogenic amines, it is predictable that the cross-talk between the receptor signaling of TAAR1 and the monoamine autoreceptors is influential in release regulation. In this regard, common biogenic amines released into synaptic clefts may influence their own release and transport via interacting with TAAR1 and the monoamine autoreceptors. Our parallel studies indicated that trace amines, including β-PEA, tyramine, tryptamine, and octopamine interacted with TAAR1 but not monoamine autoreceptors and that β-PEA alone inhibited uptake and promoted efflux by the monoamine transporters both in vitro and in brain synaptosomes (Xie and Miller, 2008). Trace amines are heterogeneously distributed in mammalian brain tissues, their distribution spatially parallels the origins and terminal projection areas of the monoaminergic neurons, and they are synthesized, packaged, and released along with the common biogenic amines (Durden et al., 1973; Boulton, 1976; Philips et al., 1978). Accordingly, variation in the levels of the trace amines in brain may bias receptor signaling of TAAR1 and monoamine autoreceptors and, consequently, may modify the effect of common biogenic amines on monoamine transporter function.
The synaptosomal preparations are apparently discrete monoaminergic synapses and, therefore, allow for direct evaluation of receptor-mediated effects on the monoamine transporters. Whereas the D2 receptor inhibitor raclopride altered the dopamine effect on [3H]dopamine uptake in monkey and wild-type mouse striatal synaptosomes, the 5-HT1 receptor inhibitor methiothepin did not change the dopamine effect. In the same striatal synaptosomal preparations, methiothepin but not raclopride altered the serotonin effect on [3H]serotonin uptake. In thalamic synaptosomes the α2 receptor inhibitor SKF86466, but not raclopride, altered the norepinephrine effect on [3H]norepinephrine uptake. These findings along with other evidence that TAAR1 is coexpressed with monoamine transporters in brain monoaminergic neurons (Borowsky et al., 2001; Miller et al., 2005; Xie et al., 2007b; Lindemann et al., 2008) suggest that TAAR1 is present in dopamine, norepinephrine, and serotonin neuronal terminals, is a receptor for common biogenic amines, and is a presynaptic modulator of monoamine transporter function in brain.
It is noteworthy that Wolinsky et al. (2007) recently demonstrated an enhanced sensitivity of TAAR1 knockout mice to the psychomotor-stimulating effect of amphetamine and an amphetamine-induced increase in the release of both dopamine and norepinephrine in the TAAR1 knockout mouse dorsal striatum, which the authors believed may be caused by a large increase of striatal high-affinity D2 receptors in TAAR1 knockout mice. Likewise, Lindemann et al. (2008) reported that TAAR1 knock-out mice display elevated sensitivity to amphetamine and that TAAR1 activity decreases the spontaneous firing rate of dopaminergic neurons in the ventral tegmental area. However, our data demonstrated that the change in uptake inhibition and efflux of [3H]monoamines after blockade of monoamine autoreceptors was reduced in the absence of TAAR1, suggesting that TAAR1 knockout mice may have less response to TAAR1 agonists, including amphetamines. In this regard, it is important to consider that our previous study showed that neurons scattered in the rhesus monkey substantia nigra stained positive for TAAR1 but negative for DAT (Xie et al., 2007b), and we recently have observed that TAAR1-staining neurons and fibers are also distributed in the rhesus monkey striatum, which suggests that TAAR1 may also function in nonmonoaminergic neurons and influence the function of monoamine neurons via interneuronal communication. However, because of homogenization in synaptosome preparation, interneuronal connectivity is deprived in the assays presented in this study. Accordingly, the functional profile demonstrated in the present study may represent only the presynaptic effects of TAAR1 in monoaminergic terminals.
Acknowledgments
We thank Lundbeck Research USA, Inc., for generously providing TAAR1 heterozygous founder mice for establishment of our mouse colonies. We also thank Laurie Lynch for management of the colony and Jennifer Carter for administrative support.
Footnotes
-
This work was supported by grants from the National Institute on Drug Abuse and The National Center for Research Resources: DA022323 (to G.M.M.), DA016606 (to G.M.M.), DA06303 (to G.M.M.), DA021180 (to G.M.M.), and RR00168.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.135079.
-
ABBREVIATIONS: DAT, dopamine transporter; 5-HT1A and 5-HT1B, subtype A and B of serotonin receptor 1; α2A and α2B, subtype A and B of alpha adrenal receptor 2; CRE-Luc, cAMP-response element-driven luciferase reporter; D2s, dopamine D2 receptor short isoform; DMEM, Dulbecco's modified Eagle's medium; NET, norepinephrine transporter; SERT, serotonin transporter; TAAR1, trace amine-associated receptor 1; PLB, Passive lysis buffer; RLU, relative light units; β-PEA, β-phenylethylamine; SKF86466, 6-chloro-2,3,4,5-tetrahydro-3-methyl-1H-3-benzazepine; Ro32-0432, bisindolylmaleimide tertiary amine.
- Received December 5, 2007.
- Accepted February 27, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}