


Abstract
ProSAAS is one of the most widely expressed proteins throughout the brain and was recently found to be upregulated in chronic fibromyalgia patients. BigLEN is a neuropeptide that is derived from ProSAAS and was recently discovered to be the endogenous ligand for the orphan G protein-coupled receptor GPR171. Although BigLEN-GPR171 has been found to play a role in feeding and anxiety behaviors, it has not yet been explored in pain and opioid modulation. The purpose of this study was to evaluate this novel neuropeptide-receptor system in opioid-induced antinociception. We found that GPR171 is expressed in GABAergic neurons within the periaqueductal gray, which is a key brain area involved in pain modulation and opioid functions. We also found that, although the GPR171 agonist and antagonist do not have nociceptive effects on their own, they oppositely regulate morphine-induced antinociception with the agonist enhancing and antagonist reducing antinociception. Lastly, we showed that the GPR171 antagonist or receptor knockdown decreases signaling by the mu-opioid receptor, but not the delta-opioid receptor. Taken together, these results suggest that antagonism of the GPR171 receptor reduces mu opioid receptor signaling and morphine-induced antinociception, whereas the GPR171 agonist enhances morphine antinociception, suggesting that GPR171 may be a novel target toward the development of pain therapeutics.
SIGNIFICANCE STATEMENT GPR171 is a recently deorphanized receptor that is expressed within the periaqueductal gray and can regulate mu opioid receptor signaling and antinociception. This research may contribute to the development of new therapeutics to treat pain.
Introduction
Despite many years of research, opioids remain among the best therapeutics for the treatment of chronic pain, but side effects with acute and long-term use limit their effectiveness. The development of novel therapeutics that increase opioid potency would allow for smaller prescribed doses of opioids to treat pain, and would thus decrease the detrimental side effects. A new and promising avenue of study toward developing novel pain therapeutics is targeting novel neuropeptide-receptor systems, given that many neuropeptides play important roles in many sensory modalities, including pain. ProSAAS is one of the most widely expressed proteins in the brain and has been implicated in a wide range of functions (Wei et al., 2004; Morgan et al., 2010; Hoshino et al., 2014; Berezniuk et al., 2017). It was recently found that the ProSAAS derived peptide, BigLEN, is the endogenous ligand for the previously orphan G protein-coupled receptor, GPR171 (Gomes et al., 2013), however, little is known about the physiologic functions of this peptide-receptor system. Studies from our laboratory showed that this system regulates anxiety (Bobeck et al., 2017), and feeding behaviors in mice (Wardman et al., 2016). Our studies used homology modeling to identify and characterize a small molecule agonist and antagonist. We found that administration of the GPR171 antagonist within the basolateral amygdala decreased anxiety behaviors, while in the hypothalamus the agonist increased feeding and body weight (Wardman et al., 2016; Bobeck et al., 2017).
Recently, ProSAAS was shown to be upregulated in the cerebrospinal fluid of fibromyalgia patients (Khoonsari et al., 2019); however, alterations in specific brain regions have not been assessed. Possible sites of action are within the descending pain modulatory pathway, which includes the periaqueductal gray (PAG), rostral ventromedial medulla (RVM), and the dorsal horn of the spinal cord. Opioids produce their antinociceptive effects in part by excitation of neurons projecting from the PAG to the RVM, which in turn inhibits incoming pain signals at the dorsal horn of the spinal cord (Heinricher et al., 2009; Lau and Vaughan, 2014). Many studies have highlighted the importance of the PAG in opioid antinociception (Depaulis et al., 1985; Morgan et al., 1991, 2009; Bobeck et al., 2009).
Given that the PAG is an important structure in pain modulation, the goal of this current research was to investigate GPR171 expression in PAG, the antinociceptive properties of GPR171 ligands, and GPR171 interactions with the opioid system. To do this, we used GPR171 small molecule ligands that were characterized previously (Wardman et al., 2016; Bobeck et al., 2017). We first sought to investigate GPR171 localization in neuronal subtypes within the PAG utilizing immunohistochemistry. We found GPR171 primarily in the GABAergic neurons. Next, we examined the behavioral effects of the agonist and antagonist on antinociception and morphine-induced antinociception in vivo using the hot plate and tail flick behavioral assays and found that the agonist and antagonist oppositely regulate morphine antinociception. Lastly, we examined the effect of GPR171 ligands on opioid receptor signaling in vitro utilizing a radioligand guanosine 5′-O-(3-[35S]thio)triphosphate (GTPγS) signaling assay. We found that GPR171 blockade decreased MOPr-mediated G protein activity. Taken together, these experiments give us a better understanding of how GPR171 is interacting with the opioid system to regulate signaling and behavior.
Materials and Methods
Subjects.
Eighty-seven male C57BL/6 mice (Charles River Laboratories, Wilmington, VA), 6–8 weeks old, weighing 18–26 g at the start of the experiment, were used for behavior experiments. Food and water were available ad libitum, except during testing. Mice were housed (four to five per cage) in a humidity- and temperature-controlled room with a 12:12-hour light/dark cycle (on 0700–1900). Testing took place between 0800 and 1600. All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals adopted by the National Institutes of Health and were approved by the Utah State University Institutional Care and Use Committee (Protocol #2775).
Drug Treatment.
Morphine sulfate (2 or 5 mg/kg, s.c.; Hikma, London, UK) was suspended in 0.9% saline. The GPR171 antagonist, MS21570 (5 mg/kg, i.p.; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and the GPR171 agonist, MS15203 (10 mg/kg, i.p.; ChemBridge Co., San Diego, CA) were diluted in 10% DMSO in saline (Sigma-Aldrich, St. Louis, MO). All drugs were administered at a volume of 10 ml/kg. These doses were chosen based on previous studies evaluating feeding and anxiety using these compounds (Wardman et al., 2016; Bobeck et al., 2017). Male mice (n = 87) were randomly divided into eight groups: DMSO+saline, DMSO+morphine (5 mg), MS21570+saline, MS21570+morphine (5 mg), MS15203+saline, MS15203+morphine (5 mg), DMSO+morphine (2 mg), and MS15203+morphine (2 mg).
Behavioral Testing.
Mice were habituated and handled daily for 3 days prior to each experiment. Mice were brought into the testing room for 1 hour followed by exposure to the testing apparatuses at room temperature. All behavior testing was conducted by experimenters blinded to the treatment groups. Nociception was assessed using the hot plate (Harvard Apparatus, Holliston, MA) and tail flick warm-water test (Thermo Fisher, Waltham, MA). Latency for the mice to lick the hind paw when placed on a 50°C hot plate and tail-withdrawal latency from a water bath of 52°C were measured. To avoid tissue damage, mice were removed if no response occurred within 60 seconds on hot plate and 20 seconds on tail flick test.
Mice were injected with MS21570 (5 mg/kg, i.p.), MS15203 (10 mg/kg, i.p.), or 10% DMSO (10 ml/kg, i.p.) 10 minutes prior to injections of morphine (2 or 5 mg/kg, s.c.) or saline (10 ml/kg, s.c.). All animals were tested on hot plate and tail flick tests prior to any drug administration to assess baseline scores, and then at 15, 30, 60, and 120 minutes after the second drug injection of morphine or saline.
Immunohistochemistry.
Immunohistochemistry was conducted as described previously (Bobeck et al., 2017). Briefly, five mice were deeply anesthetized with isoflurane and perfused transcardially through the ascending aorta with 4% paraformaldehyde. Brains were postfixed for 1 hour and stored in PBS. Immunohistochemistry was performed on free-floating coronal cut brain tissue sections (50 μm) containing PAG. Sections were incubated in 1% sodium borohydride in PBS for 30 minutes followed by blocking buffer (5% normal goat serum and 0.3% Triton X-100 in PBS) at room temperature for 1 hour. Tissues were incubated overnight at 4°C in primary antibodies against GPR171 (rabbit, 1:400; GeneTex, Irvine, CA), GAD67 (mouse, 1:500; Millipore, Temecula, CA), or vGlut2 (guinea pig, 1:500; Millipore, Burlington, MA) in 1% bovine serum albumin (Sigma-Aldrich) and 0.1% Triton X-100 (Sigma-Aldrich). The GPR171 antibody was previously validated in mouse brain tissue using knockdown or knockout approaches (Wardman et al., 2016). Primary antibodies were visualized with goat anti-rabbit 594, goat anti-mouse 488, goat anti-guinea pig 647, and goat anti-rabbit 488 (1:1000; Invitrogen, Carlsbad, CA) followed by 5 minutes of incubation with the nuclear stain DAPI (100 ng/ml; Invitrogen). Sections were mounted with ProLong Diamond Antifade (Invitrogen). Microscopy was performed at the Utah State University Microscopy CORE using Zeiss 710 confocal microscope.
Cell Culture.
Chinese Hamster Ovary (CHO) cells stably expressing N-terminally Flag-epitope tagged mu opioid receptors (CHO-MOPr) or N-terminally myc-epitope tagged GPR171 (CHO-GPR171) were grown in F12 media containing 10% FBS, 1X streptomycin-penicillin and 500 μg/ml geneticin (G418). Neuro 2A (N2A) cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 1× streptomycin-penicillin. N2A cells were chosen given the vast expression of many GPCRs including MOPr, DOPr, and GPR171 (Supplemental Fig. 1; Gomes et al., 2013). N2A cells stably expressing GPR171 (N2A-GPR171) were generated as described (Trapaidze et al., 2000; Gomes et al., 2013) and maintained in DMEM containing 1× streptomycin-penicillin and 500 μg/ml geneticin (G418). Neuro 2A cells stably expressing GPR171 lentiviral shRNA (N2A GPR171 KD) were generated as described (Gomes et al., 2013) and maintained in DMEM containing 1× streptomycin-penicillin and 8 μg/ml puromycin.
Membrane Preparation.
Membranes from CHO-MOPr, CHO-GPR171, N2A, N2A-GPR171, or N2A-GPR171 KD were prepared as described previously (Gomes et al., 2003, 2013). Briefly, cell pellets were homogenized in 25 volumes (1 g wet weight/25 ml) of ice-cold 20 mM Tris-Cl buffer (pH 7.4) containing 250 mM sucrose, 2 mM EGTA, and 1 mM MgCl2, followed by centrifugation at 27,000 g for 15 minutes at 4°C. The pellet was resuspended in 25 ml of the same buffer and the centrifugation step was repeated. The resulting membrane pellet was resuspended in 40 volumes (of original wet weight) of 2 mM Tris-Cl buffer (pH 7.4) containing 2 mM EGTA and 10% glycerol. The amount of protein in the cell homogenates was determined using the Pierce BCA protein assay reagent, after which they were stored in aliquots at −80°C until use.
Displacement Binding Assay.
Displacement binding assays were carried out as described previously (Gomes et al., 2003, 2013). Briefly, membranes from CHO-MOPr or CHO-GPR171 (50 μg) were incubated with [3H]DAMGO (3 nM final concentration) in the absence or presence of either 10 μM DAMGO, MS15203, or MS21570 for 1 hour at 37°C in Hepes-buffered Hank’s balanced salt solution containing protease inhibitor cocktail (Sigma-Aldrich). At the end of the incubation period, membrane bound radioactivity was separated by filtration using a Brandel filtration system and GF/B filters. Filters were washed three times with ice-cold 50 mM Tris-Cl buffer (pH 7.4), and bound radioactivity measured in a scintillation counter. To detect the presence of opioid receptors in N2A membranes, they were incubated with [3H]diprenorphine (3 nM final concentration) in the absence or presence of either (0–10 μM) the MOPr antagonist, D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP), or the DOPr antagonist, H-Tyr-Tic[CH2NH]-Phe-Phe-OH (TIPPψ), as described above.
[35S]GTPγS Binding Assay.
[35S]GTPγS binding assays were carried out as described previously (Gomes et al., 2003, 2013) using 50 mM Hepes (pH 7.4) containing 5 mM MgCl2, 100 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 0.1% bovine serum albumin, and a protease inhibitor cocktail. Briefly, membranes (20 μg) from CHO-MOPr were incubated for 1 hour at 30°C with DAMGO (0–10 μM) in the absence or presence of 10 μM MS21570 in the presence of 2 mM GD) and 0.5 nM [35S]GTPγS. Nonspecific binding was determined in the presence of 10 µM [35S]GTPγS. Basal values represent values obtained in the presence of GDP and the absence of cold ligand. At the end of the incubation period, samples were filtered using a Brandel filtration system and GF/B filters. Filters were washed three times with 3 ml of ice-cold 50 mM Tris-Cl (pH 7.4), and bound radioactivity was measured using a scintillation counter. In a separate set of experiments membranes from N2A, N2A-GPR171, or N2A GPR171 KD were incubated with either DAMGO or deltorphin II (0–10 μM) and membranes from N2A or N2A GPR171 KD were incubated with MS21570 (0–10 μM) in the absence or presence of 10 μM DAMGO.
Data and Statistical Analyses.
Statistical analyses of data were generated by one-way or two-way ANOVA (repeated measures), when appropriate, using Prism software (version 7.0; GraphPad Software). Dunnett’s or Tukey’s honestly significant difference post hoc tests were conducted to make pairwise comparisons.
Results
GPR171 Expression in the Periaqueductal Gray.
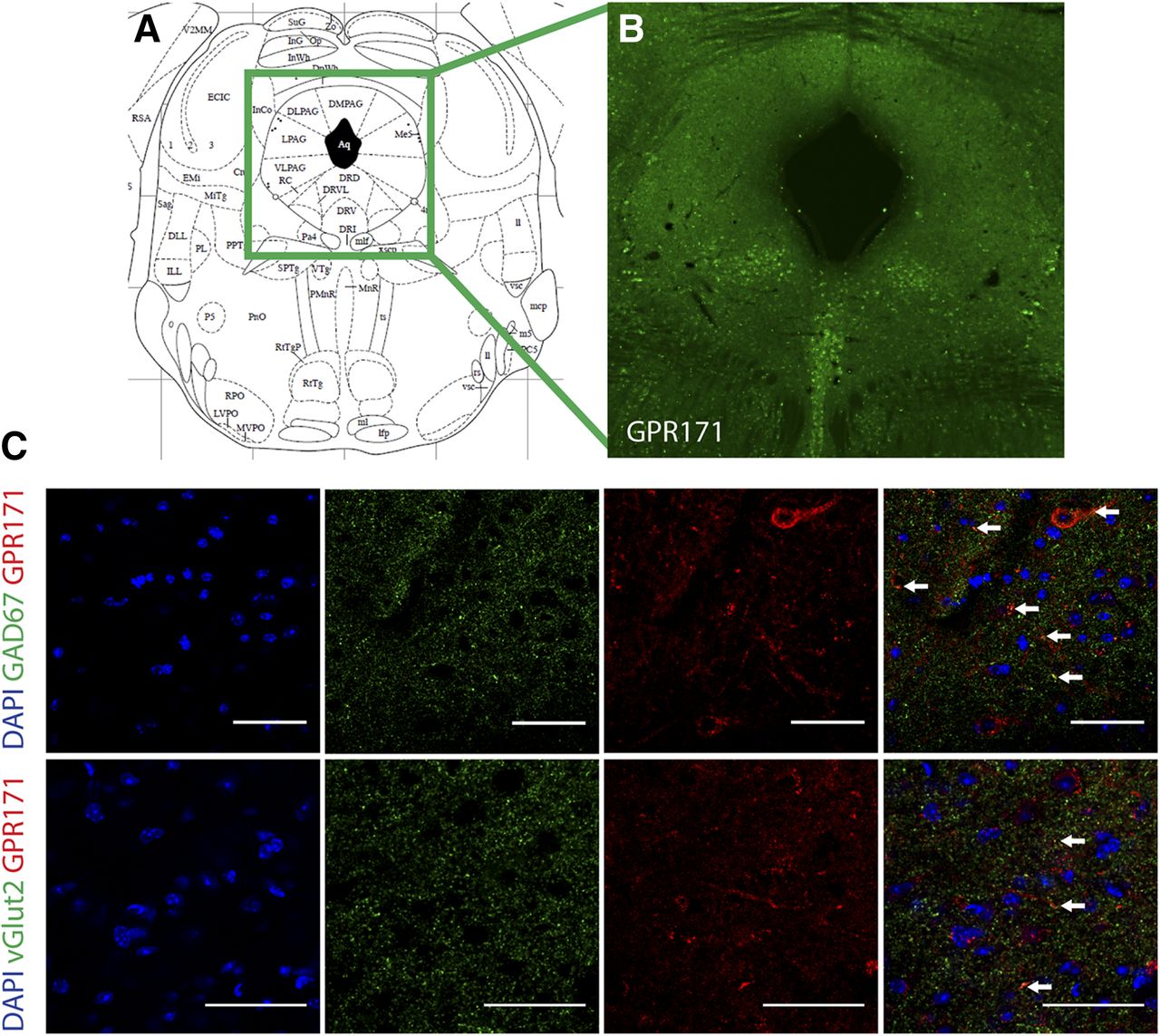
To evaluate the protein expression of GPR171 within the PAG, we used free floating immunohistochemistry. Our results show high localization in all regions of the PAG including the ventrolateral region, which is known to be involved in antinociception (adapted from Paxinos and Franklin, 2001; Fig. 1, A and B). Furthermore, GPR171 was found primarily in GABAergic neurons and, to a lesser extent, in glutamatergic neurons as indicated by colocalization of GPR171 with GAD67 and vGLUT2, respectively (Fig. 1, C and D).

GPR171 expression in different neuronal populations. (A) Image from mouse brain atlas indicating location of PAG adapted from (Paxinos and Franklin, 2001). (B) Immunohistochemistry shows high expression of GPR171 in the PAG. (C) Immunohistochemistry results show that GPR171 is colocalized with GAD67 (i.e., GABA neurons). There are fewer cells that show colocalization of GPR171 and vGlut2. Data are representative of two sections from four mice. Arrows indicate colocalization. Scale bars, 50 µm.
GPR171 Antagonist and Agonist Do Not Alter Acute Thermal Nociception.
Prior to starting the experiment, there was no statistically significant difference in baseline tail flick latencies [F(7,79) = 1.16, P = 0.338] or hot plate latencies [F(7,79) = 0.877, P = 0.528] between any group. Systemic administration of MS21570+saline (n = 8) or MS15203+saline (n = 8) were not significantly different from vehicle+saline (n = 12) controls [F(2,25) = 0.309, P = 0.737] at any time point on the tail flick test [F(4,100) = 2.91, P = 0.253, Fig. 2A]. In addition, administration of MS21570 or MS15203 did not alter nociception on the hot plate test [F(2,25) = 2.98, P = 0.069, Fig. 2B] at any time point [F(4,100) = 1.55, P = 0.194]. These data suggest that these GPR171 small molecule ligands produce neither acute pronociception nor antinociception when administered alone.

Small molecule ligands targetting GPR171 alone do not produce antinociception. GPR171 agonist (MS15203; 10 mg/kg, i.p.), GPR171 antagonist (MS21570; 5 mg/kg, i.p), or vehicle (10% DMSO in saline, i.p.) were administered to mice and then tested on the warm water (52°C) tail flick assay (A) or hot plate (50°C) assay (B) at 15, 30, 60, or 120 minute. The data reveal no change in nociception on either test. Data are the means ± S.E. of 8–12 animals/group.
GPR171 Ligands Regulate Morphine-Induced Antinociception.
As expected, all morphine-treated groups showed an increase in tail flick latencies [F(5,65) = 29.12, P < 0.0001] across all time points [F(4,260) = 122.5, P < 0.0001] compared with vehicle+saline-treated animals (Fig. 3A). There was an overall significant main effect between vehicle+morphine (5 mg), MS15203+morphine (5 mg), MS15203+morphine (2 mg), and MS21570+morphine (5 mg) compared with vehicle+saline-treated mice (Dunnett’s, P < 0.05). The agonist, MS15203+morphine (5 mg, n = 10), did not differ from morphine-induced antinociception on the tail flick test (n = 15; Tukey’s, P > 0.05). Given that the 5 mg morphine dose produced a maximal antinociceptive effect on the tail flick, 2 mg morphine (n = 13) was also administered in combination with MS15203 (n = 13). MS15203+morphine (2 mg) caused an increase in antinociception on the tail flick test compared with saline (Dunnett’s, P < 0.05), whereas the 2 mg morphine did not produce anticonception compared with saline (Fig. 3A). Interestingly, administration of the GPR171 antagonist, MS21570+morphine (5 mg; n = 8) produced a reduction in antinociception compared with morphine (5 mg) at the 120-minute time point (Tukey’s, P < 0.05).

GPR171 ligands alter morphine antinociception. GPR171 agonist (MS15203; 10 mg/kg, i.p.), GPR171 antagonist (MS21570; 5 mg/kg, i.p), or vehicle (10% DMSO in saline, i.p.) were administered 10 minutes prior to saline (10 ml/kg) or morphine (Mor; 2 or 5 mg/kg, s.c.). Antinociception was evaluated at 15, 30, 60, or 120 minutes on the warm water (52°C) tail flick assay (A) or hot plate (50°C) assay (B). GPR171 agonist, MS15203, does not alter morphine (5 mg)-induced antinociception while GPR171 antagonist decreases antinociception as measured on the tail flick assay. MS15203 increases antinociception following the lower dose of morphine (2 mg). (B) On the hot plate test, MS15203 increases 5 mg morphine-induced antinociception at 15, 30, and 60 minutes, while MS21570+Mor (5 mg) does not induce antinociception greater than saline controls. MS15203+Mor (2 mg) produced significant antinociception at 30 minutes, whereas Mor (2 mg) did not. Inset: Dunnett’s post hoc to evaluate main effect compared with saline. *P< 0.05; ***P < 0.001; ****P < 0.0001, compared with saline; #P<0.05, compared with morphine; @P<0.05, compared with morphine + agonist. n.s., Not significant; HP, hot plate; TF, tail flick. Data are the means ± S.E. of 8–15 animals/group.
Similarly we found an overall time effect [F(4,260) = 35.51, P < 0.0001] and drug effect [F(5,65) = 8.94, P < 0.0001] on the hot plate test (Fig. 3B), indicating morphine-induced antinociception. When compared with vehicle+saline-treated mice there was an overall significant main effect between vehicle+morphine (5 mg), MS15203+morphine (5 mg), and MS15203+morphine (2 mg) (Dunnett’s, P < 0.05). Vehicle+morphine (5 mg)-or MS15203+morphine (5 mg)-treated mice showed a significant increase in hot plate latencies compared with vehicle+saline-treated mice at the 15-, 30-, and 60-minute time point (Tukey’s, P < 0.05). Importantly, MS15203+morphine (5 mg) treatment produced significantly greater antinociception than vehicle+morphine (5 mg) at 30 minutes (Tukey’s, P < 0.05). However, MS21570+morphine (5 mg) was not significantly different from the vehicle+saline group at any time point (Tukey’s, P > 0.05), but was significantly different from MS15203+morphine (5 mg) at 15, 30, and 60 minutes (Tukey’s, P < 0.05). Similar to what was found on the tail flick test, 2 mgmorphine did not produce antinociception compared with saline controls (Tukey’s, P > 0.05). However, MS15203+morphine (2 mg) produced a significant increase in hot plate latencies compared with saline controls at the 30-minute time point (Fig. 3B; Dunnett’s, P < 0.05). Overall, these results indicate that the GPR171 antagonist reduces morphine-induced antinociception on the tail flick and hot plate tests and that the GPR171 agonist enhances morphine-induced antinociception on the hot plate test.
GPR171 Regulates Mu Opioid Receptor Signaling.
Given the fact that GPR171 ligands regulate morphine-induced antinociception, we hypothesized that the MS21570 decreases opioid antinociception by interfering with opioid signaling. To determine if the GPR171 compounds bind to the MOPr, displacement binding of [3H]DAMGO was conducted with membranes from CHO cells stably expressing MOPr or GPR171. As expected, DAMGO displaces [3H]DAMGO, whereas MS15203 and MS21570 do not (Fig. 4A) revealing that MS15203 and MS21570 do not bind to MOPr. DAMGO, MS15203, and MS21570 do not displace [3H]DAMGO binding using CHO cells stably expressing GPR171, indicating that DAMGO does not bind to GPR171 (Fig. 4B). In addition, we find that in CHO-MOPr cells, the GPR171 antagonist (MS21570) has no effect on DAMGO-mediated increases in [35S]GTPγS binding (Fig. 4C). Together, these results indicate that small molecule ligands to GPR171 do not bind to MOPr.

GPR171 ligands do not bind or signal at the MOPr . Displacement binding assays were carried out with membranes (50 μg protein) from CHO cells stably expressing either MOPr (A) or GPR171 (B) using [3H]DAMGO (3 nM final concentration) in the absence or presence of either 10 μM DAMGO, MS15203, or MS21570 as described in Materials and Methods. Data shows that MS15203 and MS21570 do not bind to MOPr and DAMGO does not bind to GPR171. (C) Membranes (20 μg protein) from CHO cells stably expressing MOPr were subjected to a [35S]GTPγS binding assay using DAMGO (0–10 μM) in the absence or presence of 10 μM MS21570 as described in Materials and Methods. Data shows that MS21570 does not affect signaling by DAMGO in cells only expressing MOPr. Emax was calculated at 10 μM DAMGO. Data are mean ± S.E. of three experiments in triplicate. ****P < 0.0001; one-way ANOVA; n.s., not significant.
We previously showed that N2A cells endogenously express GPR171. To confirm they also express MOPr and DOPr, we examined displacement of [3H]diprenorphine binding with the selective MOPr antagonist, CTOP, and the selective DOPr antagonist, TIPPψ. We found that both compounds displace [3H]diprenorphine with nanomolar affinity (Supplemental Fig. 1). To explore the role of GPR171 in modulating opioid receptor signaling, we used a G protein activity assay and measured agonist-induced increases in [35S]GTPγS binding assay to membranes from N2A cells, N2A cells overexpressing GPR171 (N2A-GPR171), or N2A cells with shRNA-mediated knockdown of GPR171 (N2A-GPR171 KD). In N2A cells, the MOPr agonist DAMGO caused a dose-dependent increase in signaling. There was an increase in DAMGO potency in N2A-GPR171 cells and a reduction in potency and Emax in N2A-GPR171 KD cells (Fig. 5A). Interestingly, signaling by the DOPr agonist, Deltorphin II, did not change in any of the three cell lines (Fig. 5B). To further investigate the interactions of MOPr and GPR171, we examined if the GPR171 antagonist, MS21570 could affect signaling by DAMGO. In N2A cells, MS21570 alone did not produce any change in [35S]GTPγS binding; however, MS21570 caused a dose-dependent attenuation in DAMGO-mediated increases in [35S]GTPγS binding (Fig. 5C). This decrease was not found in N2A cells with GPR171 knockdown. These results indicate that MOR signaling is modulated by levels of GPR171 and that the GPR171 antagonist can block DAMGO-mediated signaling in cells coexpressing MOPr and GPR171.

GPR171 expression alters MOPr mediated signaling, but not DOPr signaling. Membranes (20 μg protein) from Neuro 2A cells (N2A), N2A cells stably overexpressing GPR171 (N2A-GPR171) or N2A cells with shRNA-mediated knockdown of GPR171 (N2A-GPR171 KD) were subjected to a [35S]GTPγS binding assay using DAMGO (0–10 μM) (A) or Deltorphin II (0–10 μM) (B). Data shows that levels of GPR171 affect signaling by DAMGO, but not Deltorphin II. *P < 0.05; ****P < 0.0001 two-way ANOVA for graph. *P < 0.05; **P < 0.01; ****P < 0.0001, one-way ANOVA for inset (C) N2A or N2A-GPR171 KD were subjected to a [35S]GTPgS binding assay using MS21570 (0–10 μM) in the absence or presence of 10 μM DAMGO. Data shows that the GPR171 antagonist, MS21570 dose dependently decreases signaling by 10 μM DAMGO in cells coexpressing MOPr and GPR171. Emax was calculated at 10 μM DAMGO or Deltorphin II. **P < 0.01, t test for inset (MS21570 vs. +DAMGO). Data are Mean ± S.E. of three experiments in triplicate.
Discussion
In the current study, we explored the novel neuropeptide system, BigLEN-GPR171, and its ability to modulate opioid actions. We determined that GPR171 is found in a subset of GABA neurons in the PAG and is also found in a small number of glutamatergic neurons. Behaviorally, the GPR171 antagonist reduced morphine antinociception and the GPR171 agonist increased morphine antinociception, despite a lack of change in nociception with the compounds alone. The results of our in vitro signaling assays suggest that GPR171 knockdown or antagonism reduces G protein signaling mediated by the MOPr, but not DOPr. Overall, the results of these studies reveal that GPR171 may be a regulator of MOPr signaling leading to changes in antinociception. Our finding that GPR171 activation enhances opioid antinociception could have therapeutic potential in a clinical setting, where coadministration of a GPR171 agonist could lower the dose of opioids needed to alleviate pain.
GPR171 was recently identified as the receptor for the endogenous peptide, BigLEN, which is derived from the precursor ProSAAS (Gomes et al., 2013). ProSAAS-derived peptides have been shown to be involved in a wide range of functions. For example, ProSAAS-derived peptides, including BigLEN, have been implicated in body weight and feeding regulation (Wei et al., 2004; Morgan et al., 2010; Wardman et al., 2011) and have been suggested to function as an amyloid anti-aggregant against Alzheimer’s pathology (Hoshino et al., 2014). Additionally, knockout mice for ProSAAS show decreased psychomotor stimulation to cocaine compared with wild-type mice (Berezniuk et al., 2017). Although the BigLEN-GPR171 system is highly expressed throughout the brain, the physiologic functions and neural circuitry are just beginning to be understood. Recently, we found that GPR171 within the basolateral amygdala contributes to anxiety and fear-related behaviors (Bobeck et al., 2017). Furthermore, ProSAAS was recently shown to be upregulated in cerebrospinal fluid in fibromyalgia patients and was proposed as a potential biomarker for this etiologically elusive condition (Khoonsari et al., 2019). The current study is the first to look directly at the role of the BigLEN-GPR171 system in pain modulation and opioid antinociception. In vitro manipulation of GPR171 expression, where knockdown leads to a decrease in MOPr-mediated signaling and GPR171 overexpression leads to an increase in MOPr-mediated signaling, matches the in vivo regulation of morphine antinociception found with the GPR171 small molecule compounds. Thus, using a selective agonist and antagonist, we were able to explore a role for GPR171 in modulating MOPr function.
The ability of a receptor to modulate the activity of the MOPr is not unique to GPR171. It is well established that DOPr ligands can alter antinociception mediated by MOPr (Vaught and Takemori, 1979; Porreca et al., 1987; Abul-Husn et al., 2007). In addition, it has been shown that cannabinoid receptor agonists have synergistic antinociceptive effects with opioids (Cichewicz and McCarthy, 2003), perhaps as a result of coexpression of the two receptors in periaqueductal gray neurons (Wilson-Poe et al., 2012). Furthermore, opioid receptor-mediated antinociception has been observed to be regulated by other receptors, such as alpha-2 adrenergic receptor (Spaulding et al., 1979; Drasner and Fields, 1988), glutamate receptors (Nishiyama et al., 1998; Morgan et al., 2009), and orexin receptors (Azhdari-Zarmehri et al., 2013; Okumura et al., 2015; Emam et al., 2016).
One possible mechanism to explain the morphine modulatory properties of GPR171 is that it may be a result of a dimerization between GPR171 and MOPr. MOPr and GPR171 both have been shown to interact with other receptors (Fujita et al., 2015; Gomes et al., 2016; Margolis et al., 2017), but this is the first study to evaluate MOPr-GPR171 interactions directly. A MOPr-GPR171 interaction would explain the decrease in MOPr-agonist signaling in the presence of GPR171 antagonism or knockdown and the corresponding decrease in antinociception with MS21570. Another explanation is that there is crosstalk of downstream signaling pathways between MOPr and GPR171, because both are coupled to Gαi/o proteins and inhibit cAMP production (Gomes et al., 2013). It is interesting that we only find these effects with MOPr and not DOP. However, it should be noted that DOPr agonists can have markedly different signaling profiles and actions (Mitchell et al., 2014; Margolis et al., 2017), and therefore another delta agonist should be explored before ruling out a GPR171-DOPr interaction.
The PAG is an important brain region for opioid antinociception and it may be a site of action for the modulatory effects of GPR171. Our immunohistochemistry data shows that GPR171 is found in GABA neurons within the PAG. Studies have shown that disinhibition of these GABA neurons within the PAG can lead to antinociception (Bobeck et al., 2014; Lau and Vaughan, 2014). If GPR171 is also found on these neurons, then it may enhance morphine antinociception by inhibiting these GABA neurons to a greater extent than morphine alone. We hypothesize that the enhanced antinociception observed behaviorally is due to greater G protein signaling that in turn results in less GABA released, and thus an excitation of neurons projecting from the PAG to the RVM and the dorsal horn of the spinal cord.
The small molecule agonist and antagonist used in this study were previously found to have high specificity for GPR171 with less than 25% binding at any other receptors (Wardman et al., 2016; Bobeck et al., 2017). The one exception is that the GPR171 antagonist has greater binding affinity and agonist properties for the melatonin 1A receptor (Bobeck et al., 2017). Although activation of a different melatonin receptor (M2) can produce antinociception (Danilov and Kurganova, 2016), MS21570 did not show antinociception on its own and produced a reduction in morphine antinociception, therefore it is unlikely that the binding of the M2 melatonin receptor impacted our results. More importantly, it does not appear that MS15203 or MS21570 bind to MOPr given current binding data in CHO cells in addition to previous results that found minimal binding at 10 µM concentrations (Wardman et al., 2016; Bobeck et al., 2017), which is likely much greater than any receptor would see following systemic injections in vivo. In the current study, neither MS15203 nor MS21570 were able to displace the MOPr agonist, DAMGO, in CHO cells expressing only MOPr or GPR171, which supports specificity of these small molecule ligands.
In the current study, we found comparable effects on both hot plate and tail flick tests, suggesting that systemic administration of GPR171 ligands can alter MOPr-mediated antinociception in both spinal (i.e., tail flick) and supraspinal (i.e., hot plate) pain tests. However, at the higher dose of morphine, less pronounced effects were found on the tail flick test compared with hot plate test. It is possible that the site of action of GPR171 is in a higher order brain region, such as PAG, and thus is more efficient at producing antinociception on supraspinal pain tests. It is difficult to determine whether the agonist may be enhancing morphine-induced antinociception on the tail flick test due to the ceiling effect for morphine (5 mg/kg) alone and the cutoff score that was used to prevent tissue damage, which limited our ability to assess agonist enhancement of morphine antinociception above 20 seconds. The 2 mg/kg dose of morphine did not produce antinociception on its own, but when combined with MS15203 there was enhanced antinociception compared with saline on both the tail flick and hot plate tests. These data support the idea that GPR171 can regulate opioid receptor-induced antinociception on both spinal and supraspinal measures.
In conclusion, the outcomes of this study shed light on the role of GPR171 in pain and opioid modulation. These results suggest that altering the activity of GPR171 affects opioid signaling and antinociception. Therefore, this study is an important step in understanding this novel neuropeptide-receptor system and its possibility as a target in the development of pain therapeutics.
Acknowledgments
We extend our appreciation to Joseph Allred of Utah State University for his technical assistance.
Authorship Contributions
Participated in research design: McDermott, Afrose, Gomes, Devi, Bobeck.
Conducted experiments: McDermott, Afrose, Gomes, Bobeck.
Performed data analysis: McDermott, Afrose, Gomes, Bobeck.
Wrote or contributed to the writing of the manuscript: McDermott, Afrose, Gomes, Devi, Bobeck.
Footnotes
- Received April 19, 2019.
- Accepted July 10, 2019.
This work was supported by National Institutes of Health [Grants DA0886 and NS026880 to L.A.D. and by a Young Investigator Grant from Brain and Behavior Research Foundation to E.N.B.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- CHO
- Chinese hamster ovary cells
- DMEM
- Dulbecco’s modified Eagle’s medium
- DOPr
- delta opioid receptor
- GTPγS
- 5′-O-(3-[35S]thio)triphosphate
- KD
- knockdown
- MOPr
- mu opioid receptor
- N2A
- neuro 2A cells
- PAG
- periaqueductal gray
- RVM
- rostral ventromedial medulla
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}