


Abstract
Major depressive disorder (MDD) is a serious public health burden and a leading cause of disability. Its pharmacotherapy is currently limited to modulators of monoamine neurotransmitters and second-generation antipsychotics. Recently, glutamatergic approaches for the treatment of MDD have increasingly received attention, and preclinical research suggests that metabotropic glutamate receptor 5 (mGlu5) inhibitors have antidepressant-like properties. Basimglurant (2-chloro-4-[1-(4-fluoro-phenyl)-2,5-dimethyl-1H-imidazol-4-ylethynyl]-pyridine) is a novel mGlu5 negative allosteric modulator currently in phase 2 clinical development for MDD and fragile X syndrome. Here, the comprehensive preclinical pharmacological profile of basimglurant is presented with a focus on its therapeutic potential for MDD and drug-like properties. Basimglurant is a potent, selective, and safe mGlu5 inhibitor with good oral bioavailability and long half-life supportive of once-daily administration, good brain penetration, and high in vivo potency. It has antidepressant properties that are corroborated by its functional magnetic imaging profile as well as anxiolytic-like and antinociceptive features. In electroencephalography recordings, basimglurant shows wake-promoting effects followed by increased delta power during subsequent non–rapid eye movement sleep. In microdialysis studies, basimglurant had no effect on monoamine transmitter levels in the frontal cortex or nucleus accumbens except for a moderate increase of accumbal dopamine, which is in line with its lack of pharmacological activity on monoamine reuptake transporters. These data taken together, basimglurant has favorable drug-like properties, a differentiated molecular mechanism of action, and antidepressant-like features that suggest the possibility of also addressing important comorbidities of MDD including anxiety and pain as well as daytime sleepiness and apathy or lethargy.
Introduction
It has been 23 years since the cDNA encoding the first group 1 metabotropic glutamate receptor (mGlu) was cloned (Houamed et al., 1991; Masu et al., 1991). Thereafter, Gasparini et al. (1999) published MPEP [2-methyl-6-(phenylethynyl)pyridine], which would become a key reference compound in the mGlu5 field, followed soon by more structurally different compounds (Jaeschke et al., 2008). After that came initial clinical trials with mGlu5 antagonists.
The latest clinical trial results with mGlu5 inhibitors, however, have been mixed. The development of mavoglurant or AFQ056 [methyl (3aR,4S,7aR)-4-hydroxy-4-[(3-methylphenyl)ethynyl]octahydro-1H-indole-1-carboxylate] (Vranesic et al., 2014) for the treatment of dyskinesia induced by L-DOPA (l-3,4-dihydroxyphenylalanin) (Stocchi et al., 2013) has been discontinued in view of its reported insufficient efficacy (Petrov et al., 2014). More recently, the clinical development of mavoglurant and basimglurant for fragile X syndrome (FXS) also has been discontinued because neither drug improved the clinical phenotype of patients (FRAXA mavoglurant posting, FRAXA basimglurant posting), an unexpected result in view a strong hypothesis (Krueger and Bear, 2011; Michalon et al., 2012; Scharf et al., 2015). One therapeutic area that has not been explored at length in patients is depression. This is somewhat surprising in light of the preclinical data supporting the notion that mGlu5 antagonists may correct behaviors associated with depression and that modulation of mGlu5 receptors may promote synaptic growth and plasticity (Kubera et al., 2012; Piers et al., 2012).
Truly novel therapies for major depressive disorder have been difficult to identify. Most of the commonly used antidepressant drugs are biogenic amine inhibitors, some rather specific (e.g., citalopram) and others having multiple actions (e.g., venlafaxine). For some time now, there has been interest in developing therapies for depression based on modulation of glutamatergic tone (Covington et al., 2010; Duman, 2014). In particular, a study published by Zarate et al. (2006) showed that subanesthetic doses of ketamine, an inhibitor of NMDA (N-methyl-d-aspartate) type ion channels, has a rapid onset of action as measured by decreased scores on the Hamilton Depression Rating Scale in depressed patients deemed treatment resistant. Ketamine’s use, however, is limited to an experimental treatment in controlled clinical settings in view of its route of administration and safety issues, including the potential for drug dependence and the risk of neurotoxicity (Krystal et al., 2013). Approaching glutamatergic tone from another direction, mainly, via mGlu5, may skirt the abuse liability issue seen with ketamine and closely related compounds. For example, the prototypical mGlu5 antagonists MPEP and MTEP [3-((2-methyl-4-thiazolyl)ethynyl)pyridine] do not have reinforcing properties in rats (Swedberg et al., 2014) and can attenuate cocaine self-administration in monkeys (Platt et al., 2008), respectively. More to the point, mGlu5 antagonists correct behaviors associated with depression.
On the cellular level, mGlu5 is found on neurons mainly in the postsynaptic density (Lujan et al., 1997; Kuwajima et al., 2004), as well as on glial cells (Aronica et al., 2003). In brain, mGlu5 is expressed in multiple brain areas associated with the processing of motivation and emotion, including the frontal cortex, striatum, hippocampal formation, nucleus accumbens, and amygdala (Shigemoto and Mizuno, 2000). Thus, it was not surprising to see that mGlu5 inhibitors have behavioral effects in rodents in procedures that are used to identify potential antidepressant and related antianxiety drugs. For example, MPEP corrected escape behavior in a learned helplessness procedure while it increased levels of brain-derived neurotropic factor in the hippocampus (Liu et al., 2012); brain-derived neurotrophic factor has long been linked with affective behavior and depression (Nestler et al., 2002; Monteggia, 2011). MPEP and MTEP also decreased immobility in a mouse forced-swim test (FST); the effect of MPEP in the FST was not observed in mGlu5 knockout mice, confirming that the antidepressant-like effect was indeed mediated by mGlu5 receptors (Li et al., 2006). Also, MPEP reversed a learning deficit in olfactory bulbectomized rats (Pilc et al., 2002). For anxiety, fenobam [N-(3-chlorophenyl)-N′-(4,5-dihydro-1-methyl-4-oxo-1H-imidazole-2-yl)urea] had effects similar to diazepam in Geller-Seifter, Vogel conflict, stress-induced hyperthermia, and conditioned emotional response procedures in rodents (Porter et al., 2005). Fenobam also had a modest anxiolytic effect in a phase II clinical trial (Pecknold et al., 1982).
Herein, we report on a new mGlu5 antagonist with excellent drug-like properties. The compound basimglurant (RG7090, RO4917523) has undergone extensive pharmacologic and safety-related profiling studies and is now in clinical development for depression (NCT00809562, NCT01437657). The results of the recently completed clinical studies will be reported in detail elsewhere. In brief, basimglurant was studied in a 9-week, double-blind, placebo-controlled study (6 weeks of double-blind treatment, 3 weeks of post-treatment follow-up observation) in adult patients with DSM-IV-TR major depressive disorder (MDD) (APA, 2000; Quiroz et al., 2014). Basimglurant was administrated at two dose levels of 0.5 mg and 1.5 mg daily adjunctive to ongoing treatment with selective serotonin reuptake inhibitors (SSRIs) or serotonin-noradrenaline reuptake inhibitors (SNRIs), and the effects of treatment were evaluated using a multitude of assessments, including rating scales sensitive to the effects of antidepressants. Adjunctive basimglurant treatment at 1.5 mg daily showed a consistent antidepressant effect across end points, which warrants further investigation of the compound in depressive disorders.
Described herein is the in vitro pharmacological profile of basimglurant, showing that its mechanism of action is through negative allosteric modulation of mGlu5 receptor. Moreover, also illustrated are its general physiologic effects in vivo and, more specifically, its behavioral actions in animals, data collectively suggesting that basimglurant may be a mechanistically differentiated addition to the repertoire of antidepressant drugs.
Materials and Methods
Basimglurant [2-chloro-4-[1-(4-fluoro-phenyl)-2-methyl-1H-imidazol-4-ylethynyl]-pyridine (RO4917523, RG7090)], MPEP, and the radiolabeled compounds [3H]basimglurant, [14C]basimglurant, [3H]ABP688 [3-(6-methyl-pyridin-2-ylethynyl)-cyclohex-2-enone-O-11C-methyl-oxime] were synthesized at F. Hoffmann La-Roche AG. Antidepressants used in this study were purchased from Tocris (Bristol, UK) and Anawa Trading SA (Wangen, Switzerland). The synthesis of basimglurant and RO4623831 is described in Buettelmann et al. (2005), and the synthesis of [3H]ABP688 is described in Hintermann et al. (2007). All other radioligands, drugs, chemicals, cell culture reagents, and consumables were purchased from commercial sources, as described previously (Lindemann et al., 2011). Plasmids used in this study were described previously (Lindemann et al., 2011); in addition, a plasmid encoding human mGlu4 (GenBank accession number NM_000841.3, plasmid backbone pcDNA5/FRT/TO) was used.
Methods
Cell Culture and Membrane Preparations.
Cell culture, transfections, and preparation of membranes from cell culture and tissue material for radioligand binding experiments were conducted as previously described (Lindemann et al., 2011).
In Vitro Pharmacology and In Vitro Safety
Radioligand Binding and Selectivity Profiling.
Radioligand binding for mGlu5 and GABAA receptors was performed as described previously (Lindemann et al., 2011).
Ca2+ Mobilization Assays.
Ca2+ mobilization assays were performed essentially as described previously (Lindemann et al., 2011). In brief, the cells were seeded at a density of 5 × 104 cells/well in poly-d-lysine–treated, 96-well, black/clear-bottomed plates. After 24 hours, the cells were loaded for 1 hour at 37°C with 2.5 µM (2S)-2-amino-4-phosphonobutanoic acid (Fluo-4 AM) in loading buffer (1× Hanks’ balanced salt solution, 20 mM HEPES). The cells were washed 5 times with loading buffer to remove excess dye, and the intracellular calcium mobilization [Ca2+]i was measured using FLIPR384 (Molecular Devices, Sunnyvale, CA) and FDSS7000 (Hamamatsu, Hamamatsu City, Japan) instruments.
The potency of basimglurant was studied in the presence of an agonist (mGlu1 and mGlu2: glutamate; mGlu5: quisqualate; mGlu4, mGlu8: (2S)-2-amino-4-phosphonobutanoic acid) at a concentration triggering 60%–80% of the maximal agonist response, which was determined daily in a separate experiment. The antagonists were applied in a serial dilution with 10 different concentrations 30 minutes before the application of agonists; the potential agonist activities of basimglurant were monitored online during the 30-minute preincubation period. The responses were measured as the peak increase in fluorescence recorded after the addition of basimglurant and the agonist (testing for antagonist activity), minus the basal value (i.e., fluorescence without addition of agonist), normalized to the maximal stimulatory effect induced by a saturating concentration of the agonist measured on the same plate. Inhibition curves were fitted using XLfit software (IDBS, Surrey, UK) according to the Hill equation: y = 100/(1+(x/IC50)nH), where nH is the slope factor.
Inositol Phosphate Accumulation Assay.
Inositol phosphate (IP) accumulation assays were performed as described previously (Lindemann et al., 2011).
cAMP Accumulation Assays.
The cAMP accumulation assays using a cell line stably transfected with a cDNA encoding human mGlu7 were performed as described previously (Lindemann et al., 2011).
Selectivity Screening.
The selectivity profiling on approximately 100 targets comprised in the Broad Diversity Profile at a single concentration of 10 µM, and follow-up profiling in concentration-response on CB1 and CB2 cannabinoid receptors and on I2 imidazoline receptor were conducted at CEREP (Celle l’Evescault, France). Selectivity profiling on recombinant human mGlu3 and mGlu6 receptors were conducted in concentration-response using a cAMP assay (Bassoni et al., 2012) at DiscoverX (Fremont, CA). Profiling on mGlu1, -2, -4, -7, -8 (Lindemann et al., 2011), monoamine reuptake transporters, and GABAA (Lindemann et al., 2011) were conducted at F. Hoffmann-La Roche AG (Basel, Switzerland). Key experimental conditions for the selectivity profiling of basimglurant are summarized in Supplemental Table 1.
Monoamine Reuptake Transporter Assays.
Monoamine reuptake experiments were performed essentially as described previously (Hysek et al., 2012). In brief, HEK293 cells stably transfected with plasmids encoding human norepinephrine, 5-hydroxytryptamine (serotonin), or dopamine reuptake transporters were seeded at 3 × 104 cells/well in 96-well opaque white assay plates. After 24 hours at 37°C, the cells were washed once with 100 µl of assay buffer (Krebs-Ringer bicarbonate buffer; Sigma-Aldrich, Buchs, Switzerland) for 10 minutes while shaking. Plates were snap-inverted, and 50 µl of fresh assay buffer was added followed by 50 µl of basimglurant or indatraline [(1R,3S)-3-(3,4-dichlorophenyl)-N-methyl-2,3-dihydro-1H-inden-1-amine] (positive control) diluted in assay buffer. Cells were incubated at 37°C for 30 minutes, and 50 µl of the radioligand solution was added ([3H]norepinephrine from Anawa Trading SA; [3H]serotonin and [3H]dopamine from PerkinElmer, Waltham, MA). After incubation at 37°C for 15 minutes, the assay was terminated by washing the cells twice with assay buffer using a cell washer. The remaining assay buffer was decanted, 250 µl of Microsynth 40 (PerkinElmer) was added per well, and the radioactivity was counted on a Topcount microplate scintillation counter (PerkinElmer). The results were expressed as a percentage of the radioactivity detected in the absence of drugs, and the IC50 values were determined using XLfit software (IDBS), as described previously for radioligand competition binding.
Assessment for the Potential to Form Covalent Protein Adducts.
Assessment of the potential to form covalent protein adducts with hepatic proteins was essentially performed as described previously elsewhere (Fitch et al., 2010). In brief, [14C]-labeled basimglurant (49.2 µCi/µmol) was incubated with microsomal preparations, and radioactivity irreversibly bound to proteins after washing was measured. The incubation media (600 µl) consisted of 100 mM sodium phosphate buffer (pH 7.4), 1 mg/ml microsomal protein (human liver microsomes; Becton Dickinson Bioscience, Allschwil, Switzerland) 10 µM radiolabeled basimglurant, and 1 mM NADPH. The control incubations lacked NADPH. Additional experiments were conducted in the presence of reduced glutathione. The incubations were started by the addition of NADPH, after which the mixture was incubated at 37°C for 30 minutes. Experiments were conducted in triplicate.
The incubation was quenched by transferring 500 µl of the mixture into 650 µl of cold acetonitrile on a Multiscreen deep-well filter plate (Solvinert hydrophilic PTFE with prefilter; Millipore, Billerica, MA) to afford protein precipitation. Recovery of proteins was achieved by centrifugation at 1000g for 20 minutes at room temperature. The protein pellet was washed repeatedly with 750 µl of cold methanol containing H2SO4 (0.1%, v/v) followed by centrifugation at 1000g for 10 minutes at room temperature until background radioactivity levels were reached in the supernatants (typically 6 to 8 times). The remaining protein was dissolved in 500 µl of aqueous 1 M NaOH containing 1% (w/v) SDS at 60°C for 1 hour.
A 100-µl aliquot of the protein solution was used for protein determination (DC protein assay; Bio-Rad Laboratories, Hercules, CA), and the remaining solution was used for measuring the amount of incorporated radioactivity by liquid scintillation counting. Covalent protein binding was expressed as pmol equivalents of [14C]-labeled material bound per milligram of microsomal protein. Covalent binding to proteins was considered statistically significant if an increase as compared with the control incubation (lacking NADPH) was greater than 5-fold and exceeded background radioactivity levels. Covalent binding of >100 pmol [14C]/mg protein was considered as alert for metabolic activation under the assay conditions.
Human Ether-à-Go-Go-Related Gene Recordings.
Recording of human ether-à-go-go-related gene (hERG) currents on the recombinant human hERG channel was performed essentially as described previously (Fleury et al., 2011). In brief, the outward K+ currents were recorded in a Chinese hamster ovary cell line stably expressing recombinant hERG channels cloned from human heart (F. Hoffmann-La Roche Ltd., New York, NY) using a whole-cell configuration of the patch-voltage-clamp technique at 35–37°C using an EPC-10 triple amplifier (HEKA Elektronik GmbH, Lambrecht, Germany) and associated Patch MasterPro software (HEKA Elektronik GmbH). Cells were held at a resting voltage of −80 mV and were stimulated by a voltage pattern to activate hERG channels and to conduct outward IKhERG current (500-millisecond prepulse to +20 mV followed by a 500-millisecond test pulse to −40 mV) at a stimulation frequency of 0.1 Hz (6 beats per minute).
After the cells had stabilized for a few minutes and the currents were steady, the amplitude and kinetics of IKhERG were recorded under control conditions (vehicle control) for 3 minutes. Thereafter, basimglurant was applied at ascending concentrations for 3 minutes, each followed by a 100 nM solution of the standard IKhERG blocker E-4031 (N-[4-[1-[2-(6-methylpyridin-2-yl)ethyl]piperidine-4-carbonyl]phenyl]), which completely blocks IKhERG and is used as the positive control.
Experiments were performed in n = 3 replicates. If the drug effect on hERG currents was >20% compared with vehicle control, the concentration-response curve data were fitted using nonlinear regression analysis with FitMaster Pro software (HEKA Elektronik GmbH); if the drug effect was <20% compared with vehicle control, the inhibition was expressed as mean ± S.D.
Ames Mutagenesis Test.
The Ames mutagenesis test was performed essentially as described elsewhere (Muster et al., 2003). In brief, the Salmonella typhimurium strains TA1535, TA97, TA98, TA100, and TA102 were obtained from B. N. Ames (University of California–Berkeley). Fresh S9 rat liver mixtures were prepared for each experiment by mixing 0.1 ml of S9 preparation (Molecular Toxicology, Boone, NC), 0.2 ml of a 165 mM KCl solution, 0.2 ml of a 40 mM MgCl2 solution, 0.2 ml of 200 mM sodium phosphate-buffered saline, pH 7.4, 3.2 mg of NADP (Roche Diagnostics, Rotkreuz, Switzerland), and 1.53 mg of glucose-6-phosphate (Roche Diagnostics). Bacterial growth media and agar, supplements, and tetracycline were obtained from Sigma-Aldrich.
Cultures of the strains were grown overnight at 37°C in a shaking water bath in a nutrient broth liquid medium to which 0.3 μg/ml of tetracycline was added for strain TA102 to maintain a stable plasmid copy number (Albertini and Gocke, 1988). The bacterial density was checked photometrically, and the cultures were diluted in 0.85% NaCl as needed. The sensitivity of the S. typhimurium strains was verified using the following positive controls: NaN3 with strains TA1535 and TA100, ICR 191 with strain TA97, 2-nitrofluorene with strain TA98, and MMC with strain TA102. Moreover, 2-aminoanthracene was used with all strains with and without metabolic activation to confirm the activity of the S9 mix.
For testing basimglurant, test tubes containing 2 ml of 0.7% agar medium were autoclaved and kept in a prewarmed water bath at 42–45°C, and the following solutions were added: 1) 0.2 ml of a histidine/biotin mixture corresponding to 21 μg l-histidine and 24.4 μg biotin, 2) 0.1 ml solutions of basimglurant (20–2000 µg/plate) and positive controls, 3) 0.1 ml of bacterial overnight liquid cultures, 4) 0.5 ml of the S9 mixture where metabolic activation was needed, or 0.5 ml of 200 mM sodium phosphate-buffered saline, pH 7.4, where no metabolic activation was needed.
The contents of the tubes were mixed and poured immediately onto Vogel-Bonner minimal agar plates, allowed to solidify, and incubated at 37°C upside down for 2 days. Bacterial colonies were counted electronically using a DOMINO automatic image analysis system (Perceptive Instruments, Haverhill, UK) after inspection of the background lawn for signs of toxicity. The outcome of the test was considered positive for indicating mutagenicity when a dose-dependent increase in the number of colonies was observed that reached at least a 2-fold (strains TA1535, TA98) or 1.5-fold (strains TA97, TA100, TA102) increase over background.
In Vitro Micronucleus Test.
The in vitro micronucleus test was performed as described previously (Kirchner and Zeller, 2010). In brief, L5178Ytk+/− mouse lymphoma cells (Covance Laboratories Ltd., Harrogate, UK) in exponential growth phase in growth medium (RPMI 1640 supplemented with 10% heat-inactivated horse serum, Glutamax-I, 100 IU/ml penicillin, 100 µg/ml streptomycin, 100 µg/ml kanamycin) were seeded in 24-well cell culture plates. The S9 rat liver mixtures were prepared freshly for each experiment, as described previously (Kirchner and Zeller, 2010), and were added at a final concentration of 2% in the treatment medium.
For the micronucleus test without metabolic activation, 0.3 × 106 cells in 700 µl of growth medium were seeded per well, and basimglurant as well as positive controls were added in 7 µl as dimethylsulfoxide stock solutions; the cells were incubated for 24 hours. For the micronucleus test with metabolic activation, 0.4 × 106 cells in 700 µl of growth medium per well were incubated for 3 hours at 37°C with 140 µl of S9 mix and 8.4 µl of basimglurant and the reference compound dimethylsulfoxide stock solutions. Basimglurant was tested across a range of concentrations of up to 28 µg/ml without and 70 µg/ml with metabolic activation. The cells were then washed twice with growth medium in sterile 1.5-ml Eppendorf cups, resuspended in 700 µl of growth medium, seeded in 24-well plates, and incubated for another 21 hours at 37°C.
Preparation of slides and the fixation and staining of cells were performed as described previously (Kirchner and Zeller, 2010). The cells were inspected using fluorescence and light microscopy with phase contrast for the occurrence of micronucleation events indicative of DNA damage and cell cycle alterations. Results were considered positive for clastogenic/aneugenic if the number of micronucleated cells at any given concentration of test drug was elevated >2-fold compared with the vehicle (i.e., solvent) control.
In Vivo Pharmacology
Animals and Drug Treatment.
All experiments with animals were conducted in accordance with federal and local regulations at the respective study locations.
Rodents were maintained on a 12-hour light/dark cycle with the light period starting between 6:00 AM and 8:00 AM, with free access to chow and tap water unless specified otherwise. The room temperature (21–26°C) and humidity (30%–75%) were kept constant. The rodents used were Sprague-Dawley rats (conditioned emotional response: 350 g, male; Vogel conflict drinking test: 190–210 g, male; fear-potentiated startle: 225–287 g, male; Bennet neuropathic pain procedure: 100–250 g, female; overactive bladder procedure: 200–250 g, female; electroencephalography (EEG) recordings: 275–325 g, male; microdialysis: 250–350 g, male, in vivo receptor occupancy: 160 g, male), Wistar rats (pharmacokinetic profiles: 230–265 g, male; forced-swim test, 100–130 g, female; chronic mild stress-induced anhedonia: 350 g, male; Chung pain model: 276–340 g, male), Fischer F344 rats (functional magnetic resonance imaging [fMRI]: approximately 250 g, male), NMRI mice (stress-induced hyperthermia: ∼22 g, male; formalin-induced pain model: 24–30 g, male), and C57/Bl6J mice (in vivo receptor occupancy: 30 g, male). All experiments were performed with adult animals. The animals were housed in groups of 2 to 4 (rats) or 10 (mice), except for single-housing in the following procedures: stress-induced hyperthermia, chronic mild stress-induced anhedonia, microdialysis, and EEG. The rodents originated from Harlan/RCC (Füllinsdorf, Switzerland), Elevage Janvier (Le Genest-Saint-Isle, France), or Charles River (San Diego, CA/Margate, UK).
For recordings of pharmacokinetic profiles in nonhuman primates, adult male cynomolgus monkeys (Maccaca fascicularis) with a body weight of approximately 7.5 kg were used. The primates were maintained on a 12-hour light/dark cycle with lights on at 6:00 AM and free access to food and water unless specified otherwise.
Basimglurant and reference comparator drugs were formulated as microsuspensions in 0.9% NaCl/0.3% Tween-80, prepared and stored as previously described (Lindemann et al., 2011). They were administrated by oral (p.o.) gavage or by subcutaneous or intraperitoneal injection unless otherwise indicated.
Recording of Pharmacokinetic Profiles.
Pharmacokinetics (PK) in male rats: for intravenous PK, the compound was formulated in N-methylpyrolidone/saline (30%/70%) as the vehicle and was administered at a volume of 2 ml/kg. For p.o. gavage, the compound was administered as a suspension using gelatin/saline (7.5%/0.62% in water) at an administration volume of 4 ml/kg.
PK in male cynomolgus monkeys: For intravenous PK, the compound was formulated in cyclodextrin solution as the vehicle and was administered at a volume of 2 ml/kg. For p.o. gavage, the compound was administered in capsule form (2 mg in size-2 capsules, i.e., ∼0.3 mg/kg) to fasted or fed monkeys in a crossover design.
Processing of the blood samples and preparation of the liver microsomes and hepatocyte cultures was as described previously (Valles et al., 1995). Human liver tissue was obtained from hepatic surgical resections at the Hospital Hautepierre (Strasbourg, France) in accordance with the guidelines of the ethics committee. Hepatocyte cultures were incubated with basimglurant at a concentration of 1–10 µM for 24 hours; hepatocyte microsomes were incubated with basimglurant at a concentration of 1–10 µM for 1 hour.
All samples were analyzed using routine sample preparation techniques followed by liquid chromatography with tandem mass spectrometry analysis, as described previously (Lindemann et al., 2011). PK parameters for all studies were calculated using noncompartmental analysis.
Receptor Occupancy Measurements.
Receptor occupancy measurements were performed using a tritiated version of ABP688 (Ametamey et al., 2006; Hintermann et al., 2007) essentially as described elsewhere (Lindemann et al., 2011). Animals received p.o. doses of either vehicle or basimglurant (0.03–3 mg/kg) 60 minutes before a tail vein injection of [3H]ABP688 (0.3 mCi/kg). After 30 minutes, the animals were sacrificed, and plasma as well as brain samples were collected. Samples were processed further for mice (Lindemann et al., 2011) and rats (Michalon et al., 2014) as described previously.
[3H]Basimglurant in vivo binding was performed by intravenous injection of rats with [3H]basimglurant (0.3 mCi/kg) in the tail vein. Sixty minutes later, rats received either vehicle (0.9% saline/0.3% Tween-80) or an oral dose of 10 mg/kg RO4623831 (C16H11N3FCl; molecular weight 311.75; see Supplemental Table 2 for the pharmacological properties of RO4623831). At 60 minutes after the dose of RO4623831, the animals were sacrificed, and the brains were processed in the same manner as for the receptor occupancy measurements.
Chronic Mild Stress–Induced Anhedonia Test.
The chronic mild stress (CMS)–induced anhedonia test was performed essentially as described previously (Moreau et al., 1996). In brief, electrodes were implanted unilaterally in the mesolimbic system at the level of the ventral tegmental area of the midbrain (2 mm anterior from lambda, 0.3 mm lateral from the midline suture, and 8.5 mm ventral from the skull surface; electrode tips approximately 0.5 mm apart in the dorsoventral plane). After surgery, the animals were allowed to recover for 5 days, after which they underwent intracranial self-stimulation (ICSS) training. After a consistent ICSS baseline had been established, the rats either were subjected to 6 weeks of unpredictable CMS or were left undisturbed. From days 21 to 42, the stressed animals received once-daily doses (i.p.) of drugs (basimglurant, fluoxetine) or vehicle. The control group of animals that were not undergoing CMS received high-dose basimglurant (3 mg/kg) or vehicle. The anhedonia index was calculated as the percentage of change in the ICSS threshold from baseline.
Forced-Swim Test.
The FST was performed essentially as described previously (Cryan and Lucki, 2000). Rats were placed inside vertical plexiglas cylinders (height: 40 cm; diameter: 17.5 cm) containing 15 cm of water maintained at 23–24°C for 15 minutes. After 24 hours, the rats were again placed in the cylinder, and the total duration of immobility was measured during a 5-minute period. Rats were administered drugs (p.o.) at 24, 16, and 2 hours before the testing period.
Functional MRI in Rats.
Functional MRI imaging and data processing were performed essentially as described by Bruns et al. (2009). In brief, Fischer F344 rats received basimglurant and comparator drugs as single doses (p.o.) approximately 1 hour before the experiment. For fMRI, rats were anesthetized with isoflurane in oxygen and air (1:5) supplied via a face mask to the spontaneously breathing animals. The isoflurane concentration was adjusted between 1.8% and 2.4% to maintain stable respiration rates at 50–60 breaths per minute. The animals were placed in a cradle, and their heads were immobilized in a stereotaxic frame. Their respiratory rate, body temperature, and O2 and CO2 levels in the inhaled and exhaled air were continuously monitored on a PowerLab data acquisition system (ADInstruments, Spechbach, Germany). Body temperature was maintained at 37°C with a feedback-regulated electric heating blanket. The total time under anesthesia was 35 minutes.
Magnetic resonance imaging was performed on a 4.7 T/40 cm Bruker Biospec animal scanner (Bruker BioSpin, Ettlingen, Germany), equipped with a 72-mm bird-cage resonator for excitation and a surface receiver coil (Rapid Biomedical, Rimpar, Germany). On the scout images, the most rostral extension of the corpus callosum was used as a landmark for selection of eight coronal image planes at −10.0, −7.8, −5.3, −2.9, −1.6, −0.3, +1.0, and +2.3 mm from the bregma (Paxinos, 1986). All subsequent images were acquired from these planes, with a field of view of 4 cm × 4 cm and a slice thickness of 1 mm. First, a set of RARE T2-weighted anatomical images were obtained (TR/TEeff 1.8 seconds/39 milliseconds, RARE factor 8, matrix 256 × 256) (Hennig et al., 1986). Next, a T1 image series, required to quantitatively calibrate perfusion readouts, was obtained using an inversion-recovery snapshot FLASH sequence with eight inversion times (TR/TE 3.4 seconds/1.4 milliseconds, matrix 128 × 64) (Haase et al., 2011). Finally, perfusion-weighted images were acquired using continuous arterial spin labeling (Williams et al., 1992) with a centered RARE readout (TR/TE 3.75 seconds/5.7 milliseconds, RARE-factor 32, matrix 128 × 64, labeling pulse 2.5 seconds, postlabeling delay 0.4 seconds). Three consecutive volumes of perfusion images were acquired within 12 minutes.
The images were processed and analyzed using in-house developed software written in IDL (RSI, Boulder, CO) and MATLAB (MathWorks, Natick, MA), including the open-source software SPM5 (Wellcome Trust Centre for Neuroimaging, London, UK). For spatial normalization, the anatomical images were coregistered to a rat-brain template by affine and nonlinear transformations, which were then applied to the all functional images. T1 maps were calculated on a voxel-wise basis by fitting a three-parameter exponential to the intensities across the eight inversion times (Deichmann et al., 1999); the maps were then combined with the related continuous arterial spin labeling images to obtain quantitative absolute perfusion maps, as described elsewhere (Alsop and Detre, 1996; Bruns et al., 2009). To account for possible systemic changes affecting global brain perfusion, and to eliminate part of the interindividual variability, perfusion maps of each individual were normalized slice-wise to the brain-mean value, which was set to 100%. The perfusion values were averaged region-wise with reference to an in-house generated digital atlas, with regions of interest (ROIs) adapted from the Paxinos and Watson rat-brain atlas (Paxinos, 1986).
Statistical analysis of perfusion data was performed with JMP (SAS Institute, Cary, NC). Normalized perfusion for each dose group was compared ROI-wise with those of the pertinent control (vehicle) group using Welch’s t test. P < 0.05 was considered statistically significant. To avoid a severe drop in statistical power, the values were not corrected for multiple testing; rather, the false-discovery rate was estimated per dose group. The estimated number of false-positive results was on average 1 ± 1 (mean ± S.D. across dose groups) and never exceeded 3. The data were further characterized in a framework of pro- and antidepressant interventions by calculating the root-mean-square (RMS) of the perfusion changes across all ROIs and the scale-invariant pattern match coefficients (PMC; the dot product between the two neural activity profile vectors, each scaled to unit length) as measures of response strength and similarity between the effect of basimglurant and reference interventions, respectively.
Vogel Conflict Drinking Test.
The Vogel conflict test was performed as described elsewhere (Lindemann et al., 2011) with p.o. administration of drugs and 1 hour pretreatment time.
Stress-Induced Hyperthermia Test.
The stress-induced hyperthermia test was essentially performed as described elsewhere (Spooren et al., 2002). In brief, body temperature was measured in mice twice, 1 minute apart (T1 and T2, respectively), using a rectal probe. Measurement of T1 served as the handling stressor. The difference in body temperature (ΔT = T2 − T1) was determined. Drugs were administrated p.o. with 1 hour pretreatment time before measuring T1.
Conditioned Emotional Response.
The conditioned emotional response (CER) test was performed as described previously by Ballard et al. (2005) with p.o. administration of drugs and 1 hour of pretreatment time. In brief, the animals were tested using a Latin-square design twice weekly, with at least a 48-hour interval between the test sessions. The readout of the test, the so-called suppression ratio, is defined as the ratio of lever presses within the conditioning period to the total lever presses around this time (Lever presses 2 minutes before + Lever presses within the 2-minute conditioning period).
Fear-Potentiated Startle Response Procedure.
The fear-potentiated startle (FPS) response procedure was performed as described previously (Busse et al., 2004), with minor modifications. Rats were conditioned to associate a light stimulus with a mild foot shock (0.25 mA for 0.5 seconds) during two consecutive sessions on 2 consecutive days. Animals received drugs with p.o. administration and 1 hour of pretreatment time before the test session on the third day, in which an acoustic stimulus was either paired with the light stimulus or not. The fear-potentiated startle response was calculated by subtracting the mean startle response (unpaired) from the mean startle response (paired).
Formalin-Induced Pain (Paw Licking) Test.
The formalin-induced pain (paw licking) test was performed at Porsolt & Partners Pharmacology (Le Genest-Saint-Isle, France) as described previously elsewhere (Lopes et al., 2013), with minor modifications. In brief, mice received an intraplantar injection of 5% formalin (25 μl) into the posterior left paw. In one group of animals, the time spent on licking paws was recorded for 5 minutes, beginning immediately after injection of formalin (early phase). In a second cohort of animals receiving an identical formalin injection, the paw-licking time was recorded beginning 20 minutes after the formalin injection (late phase). NMRI mice received drugs or vehicle (p.o.) 1 hour before the start of recording paw-licking time.
Chung Model of Neuropathic Pain.
The Chung model of neuropathic pain (Kim and Chung, 1992) was performed at Porsolt & Partners Pharmacology as described previously (Basile et al., 2007). In brief, rats were anesthetized with sodium pentobarbital and the left L5 and L6 spinal nerves were ligated, followed by 2 weeks of recovery. For the thermal stimulation, a mobile infrared radiant source was focused under the nonlesioned and lesioned hind paws, and the paw-withdrawal latency was automatically recorded. The rats received drugs (p.o.) with 1 hour of pretreatment time before testing.
Bennet Model of Cold Allodynia (Chronic Sciatic Nerve Constriction Model).
The Bennett model of cold allodynia was performed as described elsewhere (Bennett and Xie, 1988; Hunter et al., 1997), with minor modifications. In brief, rats were anesthetized with isoflurane, and the sciatic nerve of the right hind limb was mildly constricted with four ligatures that were tied circumferentially around the nerve approximately 1 mm apart. After recovery for ≥4–7 days, rats were tested for cold-induced innocuous pain (allodynia) in a testing chamber filled to a depth of 1.5–2.0 cm with water at 2–4°C. After at least 1 hour of resting period, rats received drugs and vehicle subcutaneously with different pretreatment times as follows: basimglurant (0.1–10 mg/kg, 60 minutes), morphine (1 mg/kg, 30 minutes), duloxetine (3 mg/kg, 90 minutes), and vehicle (60 minutes). The inhibition rate was calculated with the following formula: Inhibition rate = (Paw lifts after treatment − Mean paw lifts after vehicle treatment))/(Sham pretreatment − Mean pretreatment vehicle) × 100. The drug-treated group was compared with the vehicle group using a two-sample t test with equal variance assumption. Basimglurant was formulated in H2O with 10% propylene glycol adjusted to pH 6.0 with 0.1 M HCl; morphine and duloxetine were dissolved in H2O.
Overactive Bladder: Measuring Threshold Volume in the Volume-Induced Micturition Reflex Model.
The volume-induced micturition reflex was performed in rats as described elsewhere (Hu et al., 2009). In brief, the urinary bladder was cannulated under anesthesia in rats and infused with saline to determine the micturition threshold. After establishing a stable baseline, the drug or vehicle doses were administrated intravenously 5 minutes before the next infusion cycle; thereafter, the change in micturition threshold volume was recorded.
Overactive Bladder: Contraction Frequency in the Isovolume Bladder Contraction Model.
Bladder contraction frequency was measured in rats as described previously (Hu et al., 2009). In brief, the urinary bladder was cannulated under anesthesia in rats and infused with saline to evoke micturition contractions. After stable isovolume bladder contractions were obtained, the infusion rate was lowered to 5 µl/min, and the system was allowed to stabilize for at least 30 minutes. Thereafter, vehicle was administered, followed 10 minutes later with single doses of basimglurant (0.003–0.03 mg/kg i.v.). The frequency of isovolume bladder contractions was recorded for 60 minutes in total.
EEG Recordings.
EEG recordings were performed at SRI International, Biosciences Division (Menlo Park, CA) as described previously (Morairty et al., 2008), with minor modifications. In brief, rats were implanted with telemetric devices for continuous recordings of EEG, electromyograph, core body temperature, and locomotor activity (F40-EET; DSI, St. Paul, MN). Animals were acclimated to the handling procedures and given 2 once-daily 1-ml doses of vehicle 7 and 3 days before start of the study. Drugs (basimglurant 0.03–0.3 mg/kg, caffeine 10 mg/kg) were formulated in standard vehicle (0.9% saline/0.3% Tween-80). Using a repeated-measure, counterbalanced design, each rat received five subchronic dosing conditions with 9 days of washout between each subchronic treatment condition. For each condition, daily dosing occurred 2 hours into the active period for 5 consecutive days (at the start of Zeitgeber time [ZT] 14). For the caffeine condition, however, vehicle was administered on days 1–4, and caffeine was only administered on the fifth day. Data were recorded and analyzed only for the fifth day of dosing starting at lights off (2 hours before dosing).
Microdialysis.
Microdialysis was performed at Renasci Ltd. (Nottingham, UK) as described elsewhere (Rowley et al., 2014), with minor modifications. In brief, rats were anesthetized with isoflurane (5% to induce, 2% to maintain) in an O2/N2O (1 l/min each) mixture and dual-probed whereby two microdialysis probes were stereotaxically implanted bilaterally into 1) the prefrontal cortex (2 mm tip, coordinates: anterior-posterior +3.2 mm; L (lateral): ±2.5 mm relative to bregma; ventral: −4.0 mm relative to the skull surface) and 2) the nucleus accumbens (2 mm tip, coordinates: anterior-posterior +2.2 mm; L: ±1.5 mm relative to bregma; ventral: −8.0 mm relative to the skull surface). After surgery, the animals were individually housed in circular chambers (dimensions 450 mm internal diameter, 320 mm wall height) with the microdialysis probes connected to a liquid swivel and a counterbalanced arm to allow unrestricted movement.
Rats were allowed a recovery period of at least 16 hours with free access to food and water before the onset of sample collection. The experiments were performed 1 day after surgery. Dialysate samples were collected from freely moving rats at 20-minute intervals, beginning 80 minutes before drug administration and continuing for 4 hours after (4 basal samples and 12 postdrug samples). Animals received p.o. injections of either basimglurant (0.1 or 1 mg/kg), paroxetine (3 or 10 mg/kg), or vehicle.
Detection and subsequent quantification of 5-HT (5-hydroxytryptamine [serotonin]), dopamine (DA), and norepinephrine (NE) in the dialysis samples were based on reverse-phase, ion-pair high-performance liquid chromatography coupled with electrochemical detection and involved the use of an ALEXYS monoamine analyzer (Antec Leyden, The Netherlands). The system consisted of two separate analytic columns that shared a dual-loop autosampler, allowing for one sample to be simultaneously analyzed by two systems optimized for different neurotransmitters. In this instance, one column separated NA (ALF-115, 150 mm × 1 mm internal diameter) while the other separated 5-HT and DA (ALF-105, 50 mm × 1 mm internal diameter). Two solvent delivery pumps (LC 110) were used to circulate the respective mobile phases (5-HT/DA: 50 mM phosphoric acid, 8 mM NaCl, 0.1 mM EDTA, 4.6 mM 1-octane sulfonic acid, 20% methanol, pH 6.0; NA: 50 mM phosphoric acid, 8 mM NaCl, 0.1 mM EDTA, 3 mM 1-octane sulfonic acid, 10% methanol, pH 3.25) at a flow rate of 50 µl/min; an Antec in-line degassing unit was used to remove air. Samples (10 µl) were injected onto the columns via an autosampler (AS 110) with a cooling tray set at 4°C.
Antec DECADE II electrochemical detectors were used with an Antec micro VT 03 cells employing a high-density, glassy carbon working electrode (+0.3 V for 5-HT and DA, +0.59 V for NA) combined with an ISAAC reference electrode. The electrode signal was integrated using Antec’s Clarity data acquisition system. Individual stock solutions of 5-HT, DA, and NA (1.0 mM) were prepared in a mixture of equal quantities of deionized water and 0.1 M perchloric acid (to prevent oxidation) and were stored at 4°C. A working solution containing all three transmitters was prepared daily by dilution in artificial cerebrospinal fluid.
At the end of the study, the rats were sacrificed, and probe placement was visually confirmed. Data were reported only from animals where probe membranes were correctly positioned.
Data Processing and Statistical Analysis
Data processing was conducted using Excel (Microsoft, Redmond, Seattle), GraphPad Prism (GraphPad Software, La Jolla, CA), and Statistica (Statsoft, Tulsa, OK). One-way analysis of variance was performed and followed either by Dunnett’s post hoc test (stress-induced hyperthermia, CER, FPS, FST) or by paired t tests (EEG: rapid eye movement [REM]/non-REM ratio and latency to REM and non-REM onset). For multidimensional data, two-way analysis of variance with repeated measures was performed followed by unpaired t tests (stress-induced anhedonia) or paired t tests (EEG: REM time, NR time, non-REM delta power, wakefulness, and locomotor activity). Microdialysis data were analyzed with analysis of covariance followed by Williams’s tests (Williams, 1971, 1972). The Mann-Whitney U test was used for the Vogel conflict test and the formalin-induced neuropathic pain test. Unpaired t tests were used for the data from the Chung and Bennett models. All models used two-tailed tests with the exception of the Vogel conflict test and the post hoc comparisons of the CER and FPS. For all statistical procedures, the alpha level was set at 0.05.
Results
Discovery of Basimglurant.
Basimglurant (RO4917523, RG7090; C18H13ClFN3, mol. wt. 325.77) (Fig. 1) was discovered in a medicinal chemistry effort conducted at F. Hoffmann-La Roche AG starting from the results of a small molecular weight compound library high-throughput screen based on a Ca2+ mobilization assay with human mGlu5a (Jaeschke et al., 2015). The high-throughput screen identified several mGlu5 antagonists such as MPEP, MTEP, and fenobam (Fig. 1).

Chemical structure of basimglurant, CTEP, MPEP, MTEP, and fenobam.
In Vitro Pharmacology, Selectivity, and Inverse Agonist Properties of Basimglurant.
The potency of basimglurant was analyzed by means of radioligand binding and functional in vitro assays. [3H]Basimglurant saturation analysis on recombinant human mGlu5 revealed monophasic saturation isotherms with dissociation constant (Kd) of 1.1 nM (Fig. 2A; Table 1). The rate of [3H]basimglurant association and dissociation on recombinant human mGlu5 in vitro at 37°C indicated that equilibrium binding was reached after approximately 2 hours at 37°C, and that the dissociation from the target in vitro at 37°C was achieved after approximately 5 to 6 hours (Supplemental Fig. 1; Table 1). In competition binding experiments on human recombinant mGlu5, basimglurant fully displaced [3H]MPEP, with an inhibition constant (Ki) = 35.6 nM (Fig. 2B; Table 1), and [3H]ABP688 (Ametamey et al., 2006, 2007; Hintermann et al., 2007), with an inhibition constant (Ki) = 1.4 nM.
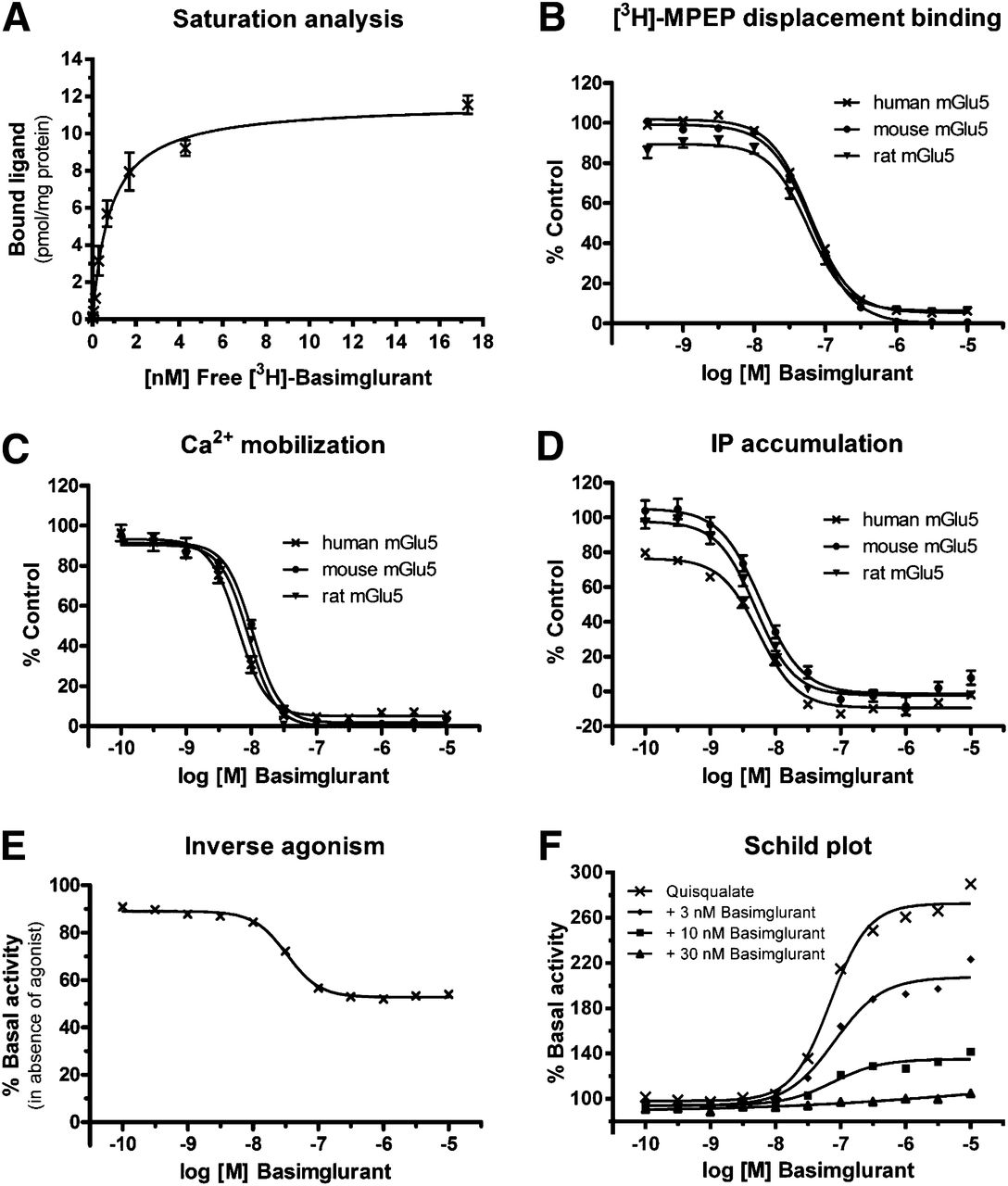
Activity of basimglurant on mGlu5 in vitro. (A) Representative saturation analysis experiment with [3H]basimglurant on membranes of HEK293 cells transiently transfected with human mGlu5. (B) [3H]MPEP displacement binding of basimglurant on membranes of HEK293 cells transiently transfected with human, mouse, and rat mGlu5. (C and D) Inhibition of quisqualate-induced Ca2+ mobilization (C) and IP accumulation (D) in monoclonal HEK293 cell lines stably expressing human, mouse, or rat mGlu5. (E) Inverse agonism of basimglurant on human mGlu5 demonstrated by suppression of constitutive receptor activity measured by IP accumulation. (F) Basimglurant dose-dependently caused a right-shift and a reduction of the maximal signal amplitude of quisqualate concentration-response curves recorded in an IP accumulation assay on human mGlu5. Data are mean ± S.E.M. of n = 4–44 except for (A) and (F), which are representative single experiments.
Basimglurant in vitro activity on recombinant mGlu5 in radioligand binding and functional assays
The potency of basimglurant on mGlu5 determined by [3H]basimglurant saturation analysis, [3H]MPEP competition binding, and Ca2+ mobilization and IP accumulation assays. Data are mean ± S.E.M., with n = 4 for saturation isotherms, n = 16–44 for [3H]MPEP binding, and n = 6–22 for IP accumulation and Ca2+ mobilization, respectively.
In HEK293 cells stably expressing human mGlu5, basimglurant inhibited quisqualate-induced Ca2+ mobilization, with an IC50 = 7.0 nM, and [3H]inositol phosphate accumulation, with an IC50 = 5.9 nM (Fig. 2, C and D; Table 1). Basimglurant showed similar potencies in the radioligand binding and functional assays on human and rodent mGlu5 receptor orthologs (Table 1).
Basimglurant acted as inverse agonist in IP accumulation assays, inhibiting constitutive receptor activity by approximately 30%, with IC50 = 38.1 nM (Fig. 2E). Basimglurant further caused a simultaneous right-shift and a reduction of the maximal signal amplitude in IP accumulation assays with recombinant human mGlu5 (Fig. 2F), demonstrating that it acts as a negative allosteric modulator.
The selectivity profiling of basimglurant on a panel of 116 radioligand binding and functional assays including all mGlu receptors revealed a more than 1000-fold selectivity for mGlu5 (Supplemental Table 1; Table 2).
Selectivity profile of basimglurant
Data generated at a single concentration of 10 μM (CEREP, DiscoverX) represent the mean of N = 2, Ki and IC50 values generated with a concentration range up to 10 μM basimglurant represent the mean of N = 2–6. Values are expressed as % control (for single concentration measurements) or as Ki or IC50 (for dose response measurements).
Pharmacokinetic and In Vitro Safety Features of Basimglurant.
After intravenous administration, basimglurant showed a low clearance and high volume of distribution, resulting in a long half-life in rats as well as in nonhuman primates of 7.5 hours and 20 hours, respectively (Fig. 3; Table 3). After single oral doses of basimglurant in rats, the compound was well absorbed, with an oral bioavailability of about 50%. In cynomolgus monkeys, the overall oral PK profiles were similar to the one seen in rats, with the same overall oral bioavailability of about 50% in nonfasted animals and approximately 100% in fasted animals.
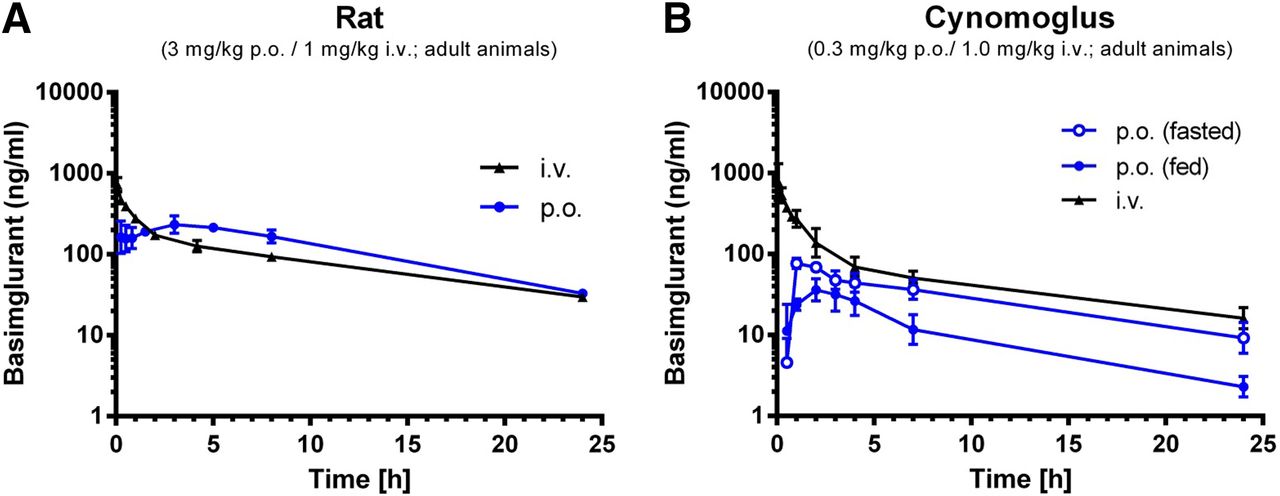
Pharmacokinetic profile of basimglurant in rat and cynomolgus. (A) Pharmacokinetic profile of basimglurant in rat after p.o. administration. (B) Pharmacokinetic profile of basimglurant in cynomolgus after p.o. administration. Data are mean ± S.D.; n = 2–4 per species and route.
Pharmacokinetic properties of basimglurant
Pharmacokinetic properties of basimglurant in male adult rat and male nonhuman primate (mean of n = 2–4 per species and route), as well as mean plasma protein binding and CLint in liver microsomes and hepatocytes from rat, nonhuman primate, and human (mean of n ≥ 2 per species).
The total brain/plasma ratio of about 2–3 in rats is in line with the good brain penetration of the drug, and the protein binding of basimglurant was consistently high across species, in the range of 97% to 99% bound to plasma proteins in rat, cynomolgus, and humans.
There was no significant retention of radioactivity to human or rat hepatic proteins when 14C-labeled basimglurant was incubated with rat and human liver microsomes in the presence of NADPH. The assessment of basimglurant in an Ames mutagenicity test showed no mutagenic potential in the absence or presence of metabolic activation with rat liver S9 extracts up to a drug concentration of 500 µM. In an in vitro micronucleation test with mouse lymphoma cells, basimglurant showed no clastogenic/aneugenic activity up to a concentration of 85 μM. Basimglurant was also tested for its potential to act on hERG. In patch-clamp recordings with recombinant human hERG at 37°C in a protein-free buffer system, basimglurant inhibited K+ currents by 11% ± 7.5% compared with vehicle controls only at the highest concentration tested (3 µM).
Brain Receptor Occupancy–Exposure Relationship of Basimglurant in Rats and Mice.
In in vivo radioligand binding experiments with a tritiated version of the positron emission tomography tracer ABP688 (Fig. 4A), basimglurant fully displaced the tracer in mice and rats at a dose of 3 mg/kg (p.o.): 50% [3H]ABP688 displacement was reached at plasma concentrations of 4.8 ng/ml in rats (Fig. 4, B–D) and 3.5 ng/ml in mice (data not shown).

Basimglurant [3H]ABP688 in receptor occupancy studies and [3H]basimglurant in vivo binding in rat. (A) Outline of the [3H]ABP688 receptor occupancy experiment with p.o. administration of basimglurant 60 minutes before intravenous administration of the tracer followed by sacrifice and further processing of the animals 30 minutes after the tracer injection. (B) Relationship between basimglurant plasma exposure (total) and brain mGlu5 receptor occupancy quantified in three brain areas. (C and D) Representative autoradiographs of parasagittal brain sections from mice receiving vehicle (C) or the highest dose of basimglurant used in the experiment achieving full tracer displacement (D). (E and F) Autoradiographs of parasagittal sections of rat brain after a bolus injection of (E) [3H]basimglurant or (F) [3H]basimglurant followed by the administration of the mGlu5 NAM RO4623831 (Supplemental Table 2) at a dose of 10 mg/kg (p.o.).
Autoradiography of parasagittal brain sections from rats receiving a bolus intravenous injection of [3H]basimglurant 60 minutes before sacrifice showed prominent labeling of multiple brain areas, including the cortex, hippocampus, striatum, amygdala, and nucleus accumbens (Fig. 4, E and F), which were previously described as expressing mGlu5 (Shigemoto and Mizuno, 2000). The binding of [3H]basimglurant was almost completely blocked when rats received an oral dose of 10 mg/kg of RO4623831, a related mGlu5 inhibitor, simultaneously with the tracer injection (see Supplemental Table 2 for the properties of RO4623831).
Antidepressant-Like Properties of Basimglurant.
The therapeutic potential of basimglurant for the treatment of depression was addressed using a combination of acute and chronic behavioral procedures as well as fMRI in rats. The quantitative effects of drug treatment described herein are expressed as the percentage of change from vehicle.
In the anhedonia model, (Moreau et al., 1992; Moreau, 2002; Holderbach et al., 2007; Hill et al., 2012), the self-stimulation behavior of rats implanted with electrodes in the ventral tegmental area was gradually disrupted by the application of unpredictable CMS. In the vehicle-treated animals, the CMS reduced the self-stimulation over the first 3 weeks, quantitatively expressed in an increased anhedonia index. Repeated once-daily drug treatment (i.p.) over 3 weeks triggered a significant gradual normalization of the anhedonia index for basimglurant at 3 mg/kg (−72%, −53%, and −64% for day 35, day 39, and day 42, respectively) and for fluoxetine at 10 mg/kg (−90%, −70%, and −97% for day 35, day 39, and day 42, respectively), indicating the antidepressant properties of basimglurant (Fig. 5A; Supplemental Table 3A).

Basimglurant activity in the chronic mild stress-induced anhedonia and the forced-swim test. (A) Chronic treatment of rats undergoing chronic mild stress with basimglurant and fluoxetine over a period of 3 weeks caused a reduction of the anhedonia index to values recorded before the chronic stress procedure. (B) Basimglurant and desipramine caused a reduction of the immobility time in the forced-swim test in rats. Drug administration route is intraperitoneal (A) or p.o. (B), respectively. Data are mean ± S.E.M. of n = 7–8 animals per group. *P < 0.05 versus vehicle based on a two-way repeated measures (A) and one-way (B) analysis of variance followed by an unpaired t test (A) and Dunnett’s post hoc test (B). See Supplemental Table 3, A and B, for related numeric information.
Basimglurant was also tested in the rat FST procedure (Porsolt et al., 1978), which is based on the principle that rodents placed in water adopt a characteristic immobile posture with only minimal movements needed to stay afloat after an initial period of vigorous activity. A reduction of the time during a test session spent in the immobile posture is considered indicative of the antidepressant potential of a particular drug. Administration (p.o.) of basimglurant caused a significant reduction of the immobility time at doses of 10 and 30 mg/kg (−19% and −16%, respectively); also the tricyclic antidepressant desipramine at a dose of 100 mg/kg (p.o.) significantly reduced immobility time (−27%) (Fig. 5B; Supplemental Table 3B).
The fMRI experiments in rats revealed that basimglurant triggered profound changes in the brain activity pattern, with increased activity in the dorsal striatum and decreased activity in the medial prefrontal cortex, dorsal hippocampus, thalamus, hypothalamus, septum, accumbens, ventral pallidum, and entorhinal piriform cortex (Fig. 6, A and B). The results were further characterized in a reference framework of various pharmacologic and nonpharmacologic interventions by means of overall response strength (RMS) and scale-invariant PMC. The effects triggered by basimglurant (10 and 30 mg/kg; the administration route for basimglurant and all reference drugs was p.o.) were comparable to those elicited by the antidepressants duloxetine, reboxetine, imipramine, and bupropione as well as by electroconvulsive treatment (ECT). The similarity of basimglurant’s response patterns to these reference treatments as gauged by the PMC was better than 71% (Fig. 6C). Also, the RMS response strength of basimglurant at 1 and 10 mg/kg surpassed that of ECT and that of standard drug treatments at 30 mg/kg (Fig. 6D). Notably, CMS led to a sizable effect opposite to that of standard treatments, with an accordingly inverted response in the neuronal activity profile.

Brain activity pattern triggered by basimglurant in comparison with antidepressants revealed by fMRI. (A and B) Brain activity pattern as revealed by fMRI upon dosing of basimglurant (1 and 10 mg/kg p.o.) to Fischer rats displayed as a bubble plot in a schematic parasagittal brain section (A) and as a spider diagram (B). For reasons of clarity not all brain (sub-)regions displayed in the spider plot are represented in (A). (C) Quantitative comparison of the brain activity pattern of basimglurant with those of prototypical antidepressants with different modes of action and nonpharmacologic interventions expressed as a scale-invariant PMC. The PMC may assume values between 1 and −1. A PMC of 1 = full agreement, 0 = no agreement; −1 identifies opposing patterns. Basimglurant at 10 mg/kg was taken as a reference. (D) Quantitative representation of the effect strength observed by fMRI for the respective interventions. The effect strength is given as root mean square (RMS) of the perfusion changes across 57 brain regions with reference to vehicle-treated animals. Drug administration was p.o. throughout; numbers in the bars (C and D) indicate drug doses administered in mg/kg. Statistical significance of drug effects in (A) is shown by the outline of the brain regions as indicated; normalized perfusion values of each dose group were compared ROI-wise to those of the vehicle group using Welch’s t test to account for nonhomogeneous variances. CA, cornu ammonis; M1, primary motor cortex; mPFC, medial prefrontal cortex; S1, primary somatosensory cortex; S2, secondary somatosensory cortex.
Anxiolytic-Like Properties of Basimglurant.
The anxiolytic-like properties of basimglurant, fenobam, and diazepam were assessed in multiple procedures sensitive to anxiolytic drugs. The quantitative effects of drug treatment (all p.o.) described in this paragraph are expressed as the percentage of change from vehicle.
In the Vogel conflict drinking test, basimglurant dose-dependently increased drinking time with a minimal effective dose of 0.03 mg/kg (+165%) up to the highest tested dose of 0.3 mg/kg (Fig. 7A; Supplemental Table 4A); fenobam and diazepam also increased drinking time, each with a minimal effective dose of 30 mg/kg (+255% and +334%, respectively). In the stress-induced hyperthermia model, basimglurant reduced the stress-induced body temperature increase with a minimal effective dose of 0.01 mg/kg (−48%) up to the highest tested dose of 1 mg/kg (−145%) (Fig. 7B; Supplemental Table 4B); fenobam and diazepam reduced the stress-induced temperature increase with minimal effective doses of 10 (−73%) and 0.1 mg/kg (−52%), respectively.

Anxiolytic-like properties of basimglurant, fenobam, and diazepam. Basimglurant, fenobam, and diazepam showed dose-dependent activities in a battery of rodent procedures sensitive to anxiolytic drugs. The minimal effective doses (i.e., lowest doses achieving a statistically significant effect) in the different tests were as follows. (A) In the Vogel conflict drinking test, the minimal effective dose was 0.03 mg/kg for basimglurant and 30 mg/kg for fenobam and diazepam. (B) In the stress-induced hyperthermia procedure, the minimal effective dose was 0.01 mg/kg for basimglurant, 10 mg/kg for fenobam, and 0.1 mg/kg for diazepam. (C) In the conditioned emotional response procedure, the minimal effective dose was 0.3 mg/kg for basimglurant and 10 mg/kg for fenobam and diazepam. (D) In the fear-potentiated startle procedure, the minimal effective dose was 0.1 mg/kg for basimglurant, and 30 mg/kg for fenobam and diazepam. Drug administration route is p.o. throughout. Data are mean ± S.E.M. of (A) n = 11, (B) n = 15–34, (C) n = 11, and (D) n = 12–24 animals per group. *P < 0.05, **P < 0.01, and ***P < 0.001 versus vehicle; (A) Mann Whitney U tests (drug groups compared individually with vehicle); (B) Dunnett’s multiple comparison test versus vehicle after a one-way analysis of variance (ANOVA); (C) ANOVA, followed by the Dunnett’s post hoc test; (D) Dunnett’s multiple comparison test versus vehicle after a one-way ANOVA. See Supplemental Table 4, A–D, for related numeric information.
Basimglurant was further examined in the CER procedure, a fear conditioning paradigm (see Materials and Methods). Basimglurant increased the suppression ratio with a minimal effective dose of 0.3 mg/kg (+567%) up to the highest dose tested of 1 mg/kg (+583%) (Fig. 7C; Supplemental Table 4C). Also, fenobam and diazepam increased the suppression ratio with a minimal effective dose of 10 mg/kg for both drugs (+400% and +483%, respectively). In the FPS test in rats, basimglurant dose-dependently reduced the startle amplitude with a minimal effective dose of 0.1 mg/kg (−53%) up to the highest tested dose (−94%) (Fig. 7D; Supplemental Table 4D). Fenobam and diazepam also were active in this procedure, each with a minimal effective dose of 30 mg/kg (−63% and −78%, respectively).
Taken together, basimglurant had robust and consistent anxiolytic-like activity in four procedures sensitive to anxiolytic drugs. Basimglurant was consistently more potent than fenobam and diazepam, with 10- to 100-fold differences in potency based on drug dose.
Effects of Basimglurant in Models of Pain and Overactive Bladder.
Glutamate is an important neurotransmitter in the context of nociception (Dickenson et al., 1997), and mGlu5 is expressed in the spinal cord and dorsal root ganglia (Shigemoto and Mizuno, 2000; Dang et al., 2002), where it has been implicated in neuropathic pain (Hu et al., 2012; Radulovic and Tronson, 2012). With this background, we studied basimglurant for its antinociceptive potential. The quantitative effects of drug treatment described herein are expressed as the percentage of change from vehicle.
In the formalin-induced pain model, formalin injected into the paw of mice induced paw-licking behavior indicative of pain. The effects of drugs on both the early and late phases of nociception were recorded. In the early phase, basimglurant had no effect at 0.1 mg/kg (p.o.) and caused a partial yet statistically nonsignificant reduction of paw licking at 1 and 10 mg/kg (−39% and −44%, respectively) whereas morphine almost completely blocked paw licking (−95%) (Fig. 8A; Supplemental Table 5A). In the late phase, basimglurant had no effect at 0.1 mg/kg and caused an almost complete block of paw-licking at 1 mg/kg (−91%, not statistically significant) and 10 mg/kg (−95%) whereas morphine completely blocked paw licking (100%) (Fig. 8B; Supplemental Table 5B).

Analgesic effects of basimglurant in rodent models of neuropathic pain. (A, B) Formalin-induced neuropathic pain. Basimglurant (0.1–10.0 mg/kg p.o.) dose-dependently inhibited formalin-induced neuropathic pain in the mouse not in (A) the early phase (i.e., basimglurant administrated 60 minutes before injection of formalin, after which recording of paw-licking behavior starts immediately) but in (B) the late phase (i.e., basimglurant administered 40 minutes before the formalin injection, recording of paw-licking behavior starts 20 minutes later). Morphine (64 mg/kg p.o.) completely blocked formalin-induced neuropathic pain in the early and late phases. (C) Chung model (spinal nerve ligature). Basimglurant (0.1–10.0 mg/kg p.o.) had no statistically significant effect on neuropathic pain induced by thermal stimulation in rats. Morphine (64 mg/kg p.o.) almost completely blocked the neuropathic pain induced by thermal stimulation. (D) Bennett model of cold allodynia. Basimglurant (0.1–10.0 mg/kg s.c.) dose-dependently inhibited cold allodynia in rats. Morphine (1 mg/kg s.c.) and duloxetine (3 mg/kg s.c.) effectively blocked paw withdrawal. Data are mean ± S.E.M. of (A and B) n = 10, (C) n = 8, and (D) n = 12 animals per group. *P < 0.05 and ** P < 0.01 versus vehicle based on (A and B) Mann-Whitney U test versus vehicle, (C) unpaired Student’s t test versus vehicle, and (D) two-sample t test. See Supplemental Table 5, A–D, for related numeric information.
In the Chung model, rats underwent unilateral spinal nerve ligations triggering neuropathic pain on the operated but not the intact side of the animal. The time to paw withdrawal after thermal or tactile stimuli was recorded as a measurement of the sensitivity to induce neuropathic pain. Basimglurant (0.1–10 mg/kg p.o.) had essentially no effect on pain induced by thermal stimuli on the lesioned side whereas morphine (64 mg/kg p.o.) almost completely restored paw-withdrawal latency of the lesioned side to that of the unlesioned side (Fig. 8C; Supplemental Table 5C). Also, when tested for antinociceptive effects using tactile stimuli, basimglurant had no apparent effects (data not shown).
In the Bennett model of cold allodynia, rats received a surgical constriction of the sciatic nerve triggering an increased sensitivity to cold. Potential analgesic effects of drug treatment were expressed as inhibition rate (see Materials and Methods). Basimglurant (0.1–10 mg/kg s.c.) dose-dependently increased the inhibition rate with a minimal effective dose of 0.3 mg/kg (36.77%) up to the highest tested dose of 10 mg/kg (58.3%) (Fig. 8D; Supplemental Table 5D). The maximal effect size reached with the highest dose of basimglurant was comparable to the effects of morphine (1 mg/kg; inhibition rate: 64.3%) and duloxetine (3 mg/kg; inhibition rate: 56.3%).
Micturition is a coordinated reflex under the control of the pontine and suprapontine centers (Blok, 2002). Glutamate signaling plays an important role in the modulation of bladder function (Kakizaki et al., 1998; Yoshiyama et al., 1999), and recently mGlu5 negative allosteric modulators (NAM) have been implicated as a potential modality for the treatment of overactive bladder (Larson et al., 2011). With this background, we assessed basimglurant in the volume-induced micturition reflex model in anesthetized rats. Basimglurant (0.01–0.3 mg/kg i.v.) dose-dependently increased the threshold of bladder filling volume, triggering the micturition reflex with a minimal effective dose of 0.03 mg/kg (+27% mean change from baseline) up to the highest dose tested of 0.3 mg/kg (+166.9%) (Supplemental Fig. 2A). In an isovolumetric bladder contraction model, basimglurant (0.003–0.03 mg/kg i.v.) dose-dependently reduced the maximum bladder intercontraction intervals, with both doses of 0.01 and 0.03 mg/kg achieving a comparable statistically significant maximal effect of −88.5% and −79.9%, respectively, of the intercontraction interval observed with vehicle (Supplemental Fig. 2B).
EEG Profile of Basimglurant in Rats.
In view of the known effects of antidepressant drugs on the EEG and sleep (Mayers and Baldwin, 2005), we studied the effect of basimglurant on sleeping and waking via telemetry in freely moving rats. Animals received five consecutive once-daily doses of vehicle or basimglurant (0.03–0.3 mg/kg p.o.) 2 hours into the dark phase (ZT 14; the active period in rats), and EEG and electromyography were continuously recorded for 22 hours after the fifth dose; the comparator caffeine (10 mg/kg p.o.) was given acutely on the day of the recordings (Fig. 9A).

EEG recordings in rats after repeated administration of basimglurant. (A) Outline of the experiment with once-daily p.o. administration of basimglurant 2 hours into the dark phase for 5 days, and recording of EEG traces after the fifth dose (the comparator caffeine was administrated as a single dose on day 5). (B) Cumulative REM/non-REM ratio calculated separately for the dark period (ZT 14–24) and the light-period (ZT 24–12). (C and D) Time spent in REM (C) and non-REM (D) activity over the entire recording period. (E) Latency to the first six continuous epochs of non-REM and the first three continuous epochs of REM sleep. (F) Hourly percentage of time spent in the waking state. (G) Time course of non-REM delta power over the entire recording period. (H) Time course of locomotor activity over the entire recording period. Drug administration is p.o. throughout. Data are mean ± S.E.M., with n = 8 rats per group. *P < 0.05 versus vehicle based on a one-way (B and F) and two-way analysis of variance (C–E and G–H) followed by a two-tailed post hoc paired t test. ET, experimental time (i.e., daytime); ZT: Zeitgeber time. The dark phase (i.e., active period) is highlighted by gray shading. See Supplemental Table 6, A and B, for related numeric information.
Basimglurant dose-dependently reduced the ratio of REM to non-REM sleep (i.e., the ratio of cumulative time spent in REM and non-REM sleep) during the dark phase (ZT 14–24) at 0.1 and 0.3 mg/kg without affecting the REM/non-REM ratio during the subsequent light phase (ZT 0–12) (Fig. 9B; Supplemental Table 6A). Basimglurant reduced the time spent in REM at 0.1 and 0.3 mg/kg (up to −100% and −97%, respectively; Fig. 9C) and non-REM at 0.1 and 0.3 mg/kg (up to −94% and −83%, respectively; Fig. 9D).
In addition, basimglurant increased the latency to the onset of both REM (up to +351%) and non-REM (up to +226%) sleep (Fig. 9E; Supplemental Table 6B) and produced a wake-promoting effect at 0.1 and 0.3 mg/kg (up to 115% and 136%, respectively; Fig. 9F) without eliciting subsequent hypersomnolence. During non-REM sleep, basimglurant caused a pronounced increase in delta power during the dark phase at all three dose levels (up to +307% at 0.3 mg/kg) that was still detectable in the subsequent light phase (Fig. 9G).
The moderate increase in locomotor activity during the dark phase in animals receiving basimglurant (up to +200% at 0.3 mg/kg; Fig. 9H) is consistent with the wake-promoting effects of the drug. Caffeine showed the expected effects: increased wakefulness (Fig. 9F) and locomotor activity (Fig. 9H) during the first half of the dark period (ZT 14–20), no effect on the REM/non-REM ratio (Fig. 9B), transient decrease (ZT 14–20) and subsequent increase (ZT 20–24) of both REM (Fig. 9C) and non-REM (Fig. 9D) sleep during the dark phase, no effect on delta power (Fig. 9G), and increased latency to REM and non-REM sleep onset (Fig. 9E).
Effects of Basimglurant on Monoamine Neurotransmitter Levels in Rats.
Monoamine neurotransmitters and their modulation play a critical role in the pathophysiology and treatment of mood disorders (Kupfer et al., 2012; Hamon and Blier, 2013). Consequently, the effects of basimglurant and paroxetine on the levels of 5-HT, DA, and NE in the frontal cortex as well as 5-HT and dopamine in the nucleus accumbens were studied by dual-probe microdialysis in freely moving rats. After we had recorded their baseline transmitter levels for 1 hour, the animals received basimglurant (0.1 and 1.0 mg/kg p.o.) or paroxetine (3.0 and 10.0 mg/kg p.o.), after which we sampled the neurotransmitter levels for 4 hours.
In the frontal cortex, basimglurant had little or no effect on the levels of 5-HT (Fig. 10A; Supplemental Table 7), DA (Fig. 10B; Supplemental Table 7), or NE (Fig. 10C; Supplemental Table 7). In contrast, paroxetine (10 mg/kg p.o.) triggered a 2-fold increase in 5-HT levels (Fig. 10A; Supplemental Table 7). Aside from a small effect at 20 minutes, paroxetine had no consistent effect on DA. In addition, paroxetine (10 mg/kg, p.o.) reduced NE during the 40- to 180-minute postdose period, with a maximum effect size of −29% compared with vehicle at 160 minutes after dosing.

Effects of basimglurant and the SSRI paroxetine on extracellular levels of monoamine transmitters in rats. Extracellular levels of monoamine neurotransmitters recorded by dual-probe microdialysis in freely moving rats (A–C) in the frontal cortex—(A) 5-HT, (B) dopamine, (C) norepinephrine—and (D and E) in the nucleus accumbens—(D) 5-HT, (E) dopamine—before and after acute administration of basimglurant (0.1 and 1.0 mg/kg) and paroxetine (3 and 10 mg/kg). Drug administration is p.o. throughout. Results are adjusted means ± S.E.M. (see Materials and Methods) of n = 5–10 per group. Dotted line indicates drug administration at t = 0 minutes. *P < 0.05, **P < 0.01, and ***P < 0.001 versus vehicle. Data analyzed by analysis of covariance with log(baseline) as covariate; multiple comparisons of each treatment to the control group by separate Williams’s tests. See Supplemental Table 7 for related numeric information.
In the nucleus accumbens, basimglurant had no significant effect on 5-HT (Fig. 10D; Supplemental Table 7) with the exception of a transient reduction by −54% compared with vehicle observed at 200 minutes after dosing. However, basimglurant caused a consistent, dose-dependent increase in DA (Fig. 10E; Supplemental Table 7), reaching statistical significance at a dose of 1.0 mg/kg, with a maximum effect size of +102% compared with the baseline at 140 minutes after the dose. Paroxetine had no statistically significant effect on extracellular 5-HT concentrations in the nucleus accumbens (Fig. 10D; Supplemental Table 7). Paroxetine caused transient elevations in DA levels at 10 mg/kg (Fig. 10E; Supplemental Table 7), reaching statistical significance at 100–140 and 180 minutes after the dose, with a maximum effect size at 140 minutes of +66% compared with vehicle.
Discussion
The mGlu5 receptor has been intensively studied as a drug target for a range of neuropsychiatric conditions, including depression, anxiety, FXS, autism, Parkinson’s disease, and pain (Gasparini et al., 2008; Jaeschke et al., 2008). Pharmacologic tools such as MPEP (Gasparini et al., 1999), MTEP (Cosford et al., 2003), fenobam (Pecknold et al., 1982; Porter et al., 2005), and CTEP [2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine] (Lindemann et al., 2011) were instrumental for understanding the target biology and evaluating the therapeutic potential of mGlu5 inhibitors in the context of disease. The pharmacology and drug-like features of basimglurant described herein made it possible to take this research one step farther into phase II clinical trials for FXS and MDD.
Basimglurant was found to act as a potent and selective mGlu5 NAM with inverse agonist properties and negligible species differences between human and rodent mGlu5 receptor orthologs. With respect to in vitro safety features, basimglurant had no relevant inhibitory activity on hERG channels, no propensity to form covalent protein adducts, no mutagenic potential in the Ames test, and no clastogenic/aneugenic activity in the in vitro micronucleus test. With respect to its pharmacokinetic properties, basimglurant had low in vitro metabolic clearance by rat, cynomolgus, and human hepatocytes; these in vitro findings are in line with the observed low in vivo clearance of the drug. The large distribution volume in combination with low clearance results in long half-lives of 7.5 hours in rats and approximately 20 hours in monkey after oral administration, suggesting a once-daily dosing regimen in humans. The high brain/plasma ratio in rats and the potent in vivo displacement of [3H]ABP688 receptor occupancy studies in rodents indicate the good brain penetration and high in vivo potency of basimglurant. Taken together, the high in vitro and in vivo potency combined with the high selectivity for mGlu5, lack of in vitro safety liabilities, excellent oral bioavailability, and long half-life constitute favorable drug-like properties for basimglurant.
Basimglurant showed robust antidepressant-like activity in the CMS-induced anhedonia procedure. These results are particularly relevant as the CMS method is considered a disease model for depression with good face-validity (Moreau, 2002; Nestler and Hyman, 2010), reflecting several key aspects of the human condition including a reduced hedonic drive, altered sleep/wake pattern, and endocrine changes in the hypothalamic-pituitary-adrenal axis (Moreau et al., 1995; Grippo et al., 2005). The dose-dependent reduction of immobility time by basimglurant in the FST, a screening procedure used for the profiling of antidepressants (Nestler and Hyman, 2010), is in agreement of what has been reported for other mGlu5 NAMs (Liu et al., 2012; Hughes et al., 2013). Moreover, fMRI recordings revealed insights into the brain regions engaged in basimglurant’s in vivo effects: many of the brain regions with altered neural activity upon basimglurant treatment have been recognized as critical parts of the neurocircuitry in depression (Russo and Nestler, 2013). The match between the changes of brain activity pattern induced by basimglurant on the one hand, and a broad range of antidepressant drugs and ECT on the other hand fit well to the antidepressant-like pharmacology of basimglurant.
In addition to its antidepressant-like properties, basimglurant had consistent anxiolytic-like activity with comparable efficacy and higher potency than fenobam and diazepam across the methods used to detect anxiolytic-like activity. Furthermore, basimglurant showed antinociceptive activity in the formalin pain and cold allodynia procedures. Basimglurant also normalized the urine bladder function in rodent models of overactive bladder, confirming previous work with other mGlu5 NAMs (Crock et al., 2012) and suggesting the possibility of benefits in the context of visceral pain. Taken together, these results indicate that basimglurant has the potential to address two important comorbidities of depression: anxiety and somatic pain.
The mechanistic underpinning of basimglurant’s in vivo pharmacology was further explored via EEG and measures of the drug’s effect on monoamine neurotransmitter levels by microdialysis in rats. EEG recordings revealed that repeated once-daily administration of basimglurant decreased the REM/non-REM ratio during the active period, and increased wakefulness and subsequent non-REM delta power. These observations are in agreement with EEG data reported for single-dose studies in rats with mavoglurant and GRN-529 [4-difluoromethoxy-3-(pyridine-2-ylethynyl)phenyl)5H-pyrrolo[3,4-b]pyridine-6(7H)-yl methanone] (Hughes et al., 2013). Disturbances of the sleep-wake pattern and EEG activity are a hallmark of MDD, contributing to daytime sleepiness as well as apathy and lethargy (Ahmadi et al., 2010). Several classes of clinically used antidepressants shift the REM/non-REM ratio in a similar way as observed with basimglurant (Beitinger and Fulda, 2010; Staner et al., 2010), lending further support to basimglurant’s antidepressant-like profile (O’Donnel and Shelton, 2011). The increase in the delta frequency spectrum suggests increased sleep pressure during the active phase and improved sleep quality during the subsequent inactive period (Tobler and Borbely, 1986; Borbely and Tobler, 2011).
Of note, no indications of sleep deprivation were detected during the subchronic basimglurant treatment. The wake-promoting features of basimglurant, which occur in a consolidated manner, hold potential benefits in addressing the daytime sleepiness, lethargy, and apathy characteristic of MDD. The effects of mGlu5 inhibition on the EEG profile in rats were studied previously with a range of compounds, including MPEP, MTEP, GRN-529, and mavoglurant (Cavas et al., 2013; Harvey et al., 2013; Ahnaou et al., 2014). These studies collectively showed a significant increase of REM after drug administration. Further, both Harvey et al. (2013) and Ahnaou et al. (2014) reported an increase of time spent in wakefulness and increased sleep consolidation. In contrast to our results, however, Ahnaou et al. (2014) reported a rebound in REM during the dark period after administration of MTEP; nonetheless, Ahnaou et al. (2014) concluded that mGlu5 inhibition has overall sleep consolidating effects, which they speculate might indirectly have beneficial effects on cognitive and memory performance.
The source of the discrepancies between our observations with basimglurant and the reported effects for MTEP are unclear and could be based on methodologic differences, including the single-drug doses for the published studies compared with subchronic drug administration for basimglurant, drug administration during the light period for the published studies as opposed to during the dark phase for basimglurant, and the substantially shorter half-life reported for MPEP and mavoglurant (Vranesic et al., 2014) as well as MTEP (Lindemann et al., 2011) compared with basimglurant.
Microdialysis studies revealed that basimglurant had no significant effect on DA, 5-HT, or NE in the frontal cortex and nucleus accumbens with the exception of moderate DA elevations in the nucleus accumbens. Contributions of direct monoamine reuptake transporter inhibition to the accumbal DA elevation can be excluded in view of basimglurant’s lack of activity on these transporters. Previous studies showed that MPEP had no effect on DA levels in the nucleus accumbens, but it blocked nicotine-evoked accumbal DA release (Tronci and Balfour, 2011) and dampened amphetamine-triggered DA outflow in the striatum (Tokunaga et al., 2009). The discrepancy between the current study and the previous report on accumbal DA levels might be due to the higher in vivo potency and longer half-life of basimglurant compared with MTEP. The microdialysis results demonstrate that the anxiolytic- and antidepressant-like activity of basimglurant is neurochemically distinct from current antidepressants, which cause a pronounced modulation of extracellular monoamine levels.
The work on novel glutamatergic antidepressant drug candidates also has investigated ligands to other mGlu receptors, most notably mGlu2/3 antagonists (Sanacora et al., 2012; Krystal et al., 2013; Celanire et al., 2015). The literature and our own work suggest that mGlu2/3 inhibitors have procognitive properties in a range of preclinical models of short-term and long-term memory, executive function, and impulse control (Higgins et al., 2004; Spinelli et al., 2005; Marek, 2010; Goeldner et al., 2013), which makes them interesting candidates to address cognitive deficits in MDD (Goeldner et al., 2013). For mGlu5, the situation is different because studies conducted in wild-type animals suggest that mGlu5 NAMs in general have no procognitive properties (Petersen et al., 2002; Ballard et al., 2005; Ahnaou et al., 2014); however, some reports have suggested that a high level of mGlu5 receptor inhibition can cause cognitive impairment, at least in the context of certain diseases (Hsieh et al., 2012). In a disease context outside of MDD, however, in FXS and autism procognitive effects of mGlu5 inhibition have been reported (Michalon et al., 2014; Seese et al., 2014).
It currently remains an open question whether the effective reduction of depressive symptoms, and of apathy/lethargy and lack of motivation (e.g., by an mGlu5 inhibitor) could result in improved cognitive functioning in MDD patients. Cognitive deficits in MDD in general have received little attention to date, which is in part due to the fact that commonly used rating scales such as the Montgomery-Åsberg Depression Rating Scale (MADRS) and Quick Inventory of Depressive Symptomatology (QIDS) do not capture cognitive function very well. The picture is further complicated by the discrepancy between self-reported and objectively measured cognitive deficits in depressed patients, and by an apparent disconnect between the severity of depressive symptoms and cognitive functioning (Goeldner et al., 2013). Here, the possible interplay, especially between attention and executive function on the one side and symptoms of apathy/lethargy and motivation in depression on the other, warrants further investigation.
When comparing the profile of basimglurant to conventional antidepressants, we found similar antidepressant-like activity in the rodent behavioral procedures employed in our studies, including in CMS-induced anhedonia and the FST as well as in the rat fMRI profile. Basimglurant showed consistent anxiolytic-like properties in four different rodent procedures that are sensitive to anxiolytic drugs such as benzodiazepines. By comparison, conventional antidepressants are reported to be mostly inactive in rodent behavioral models of anxiety (Bespalov et al., 2010), although they are effectively used clinically for the treatment of various forms of anxiety, often transiently combined with other anxiolytic drugs such as benzodiazepines during the initial phase of antidepressant treatment (O’Donnel and Shelton, 2011).
Basimglurant shows antinociceptive properties in some procedures, and conventional antidepressants such as duloxetine also show antinociceptive properties in preclinical models of pain (e.g., Bennett model of cold allodynia; Fig. 8D). Several classes of antidepressants including tricyclic antidepressants, SSRIs, SNRIs, and SSRI/SNRIs are indeed used clinically for the treatment of pain (Dharmshaktu et al., 2012; Mika et al., 2013).
With respect to sleep and sleepiness, conventional antidepressants typically do not show wake-promoting properties, and sedation is a major side effect of many antidepressant drugs (O’Donnel and Shelton, 2011). In contrast, basimglurant showed sustained wake-promoting properties, which occurred in a consolidated fashion without induction of rebound somnolence.
In light of the complexity of MDD symptoms, it is conceivable that the pharmacotherapy of choice might differ considerably, depending on the combination of symptoms presented by the individual patient. It is expected that the outcome of the recently concluded MARIGOLD (Modulating Receptors of Glutamate for Alleviating Depression) trial in depression (Quiroz et al., 2014) and future clinical research will reveal how basimglurant compares clinically to antidepressant drugs currently in use.
Collectively, the preclinical profile presented here demonstrates that basimglurant is a potent and selective mGlu5 NAM with excellent drug-like properties supportive of once-daily dosing. The in vivo pharmacology, including its antidepressant-like and anxiolytic-like properties combined with its antinociceptive and wake-promoting activity, make basimglurant a promising, mechanistically differentiated antidepressant drug candidate with the potential to address important comorbidities of MDD including anxiety and pain as well as daytime sleepiness and apathy or lethargy.
Acknowledgments
The authors thank Catherine Diener and Christophe Fischer (in vitro pharmacology); Daniel Rüher, Antonio Ricci, and Daniel Tännler (chemical synthesis of basimglurant); Roland Degen, Karina Müller, and Mathias Müller ([3H]- and [14C]basimglurant, and [3H]-ABP688 radiosynthesis); Patricia Glaentzlin and Céline Sutter (in vivo binding); Marius Hoener, Sylvie Chabos, Daniele Buchy, Paricher Malherbe, Anne Marcuz, Christoph Ullmer, Anja Osterwald, Silvia Gatti, Monique Dellenbach, and Jennifer Beck (contributions to the in vitro selectivity profiling); Roland Mory, Patrick Mortas, Martine Maco, Marie Haman, Roger Wyler, Brigitte Algeyer, Severine Bandinelli, Francoise Kahn, Sean Durkin, and Audrey Genet (behavioral pharmacology); Stephanie Schöppenthau and Thomas Bilser (fMRI imaging and image processing); Thomas Thelly and Regina Moog (formulations); Stefan Kirchner (in vitro micronucleation test); Quan-Ming Zhu, Anthony Ford, and their colleagues (Bennet model of cold allodynia, models of overactive bladder); Vincent Castagne, Karelle Davoust, Paul Moser, and their colleagues at Porsolt & Partners Pharmacology, France (formalin-induced pain procedure, Chung model of neuropathic pain); Helen Rowley, Sharon Chetam, Richard Brammer, and their colleagues at Renasci Ltd., United Kingdom (microdialysis) for their excellent contributions and technical assistance. The authors also thank Luca Santarelli, Anirvan Ghosh, and Paulo Fontoura for their continued invaluable support of the project.
Authorship Contributions
Participated in research design: Lindemann, Porter, Kuennecke, Schneider, Niederhauser, Morairty, Kilduff, Kolczewski, Borroni, Moreau, Prinssen, Spooren, Wettstein, Jaeschke.
Conducted experiments: Lindemann, Porter, Kuennecke, Harrison, Paehler, Funk, Gloge, Polonchuk, Niederhauser, Morairty, Vieira, Kolczewski, Hartung, Honer, Borroni, Moreau, Spooren, Jaeschke.
Contributed new reagents or analytic tools: Morairty, Kilduff, Kolczewski, Wichmann, Hartung, Jaeschke.
Performed data analysis: Lindemann, Porter, Scharf, Kuennecke, Bruns, Harrison, Paehler, Funk, Schneider, Parrott, Polonchuk, Niederhauser, Morairty, Kilduff, Borroni, Moreau, Spooren.
Wrote or contributed to the writing of the manuscript: Lindemann, Scharf, Kuennecke, Bruns, von Kienlin, Schneider, Kilduff, Spooren, Wettstein, Jaeschke.
Footnotes
- Received December 30, 2014.
- Accepted February 6, 2015.
S.R.M. and T.S.K. received payments from F. Hoffmann-La Roche AG. All other authors are full-time employees of F. Hoffman-La Roche AG.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ABP688
- 3-(6-methyl-pyridin-2-ylethynyl)-cyclohex-2-enone-O-11C-methyl-oxime
- AFQ056
- mavoglurant, methyl (3aR,4S,7aR)-4-hydroxy-4-[(3-methylphenyl)ethynyl]octahydro-1H-indole-1-carboxylate
- ANOVA
- analysis of variance
- CER
- conditioned emotional response
- CMS
- chronic mild stress
- CTEP
- 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine
- DA
- dopamine
- E-4031
- N-[4-[1-[2-(6-methylpyridin-2-yl)ethyl]piperidine-4-carbonyl]phenyl]
- ECT
- electroconvulsive treatment
- EEG
- electroencephalography
- fMRI
- functional magnetic resonance imaging
- FPS
- fear-potentiated startle
- FST
- forced-swim test
- FXS
- fragile X syndrome
- GRN-529
- 4-difluoromethoxy-3-(pyridine-2-ylethynyl)phenyl)5H-pyrrolo[3,4-b]pyridine-6(7H)-yl methanone
- hERG
- human ether-à-go-go related gene
- 5-HT
- serotonin, 5-hydroxytryptamine
- ICSS
- intracranial self-stimulation
- IP
- inositol phosphate
- MDD
- major depressive disorder
- mGlu
- metabotropic glutamate receptor
- MPEP
- 2-methyl-6-(phenylethynyl)pyridine
- MTEP
- 3-((2-methyl-4-thiazolyl)ethynyl)pyridine
- NAM
- negative allosteric modulator
- NE
- norepinephrine
- PK
- pharmacokinetic
- PMC
- pattern match coefficient
- REM
- rapid eye movement
- RMS
- root-mean-square
- ROI
- region of interest
- SNRI
- serotonin-noradrenaline reuptake inhibitor
- SSRI
- selective serotonin reuptake inhibitor
- ZT
- Zeitgeber time
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}