


Abstract
Fenobam [N-(3-chlorophenyl)-N′-(4,5-dihydro-1-methyl-4-oxo-1H-imidazole-2-yl)urea] is an atypical anxiolytic agent with unknown molecular target that has previously been demonstrated both in rodents and human to exert anxiolytic activity. Here, we report that fenobam is a selective and potent metabotropic glutamate (mGlu)5 receptor antagonist acting at an allosteric modulatory site shared with 2-methyl-6-phenylethynyl-pyridine (MPEP), the protypical selective mGlu5 receptor antagonist. Fenobam inhibited quisqualate-evoked intracellular calcium response mediated by human mGlu5 receptor with IC50 = 58 ± 2 nM. It acted in a noncompetitive manner, similar to MPEP and demonstrated inverse agonist properties, blocking 66% of the mGlu5 receptor basal activity (in an over expressed cell line) with an IC50 = 84 ± 13 nM. [3H]Fenobam bound to rat and human recombinant receptors with Kd values of 54 ± 6 and 31 ± 4 nM, respectively. MPEP inhibited [3H]fenobam binding to human mGlu5 receptors with a Ki value of 6.7 ± 0.7 nM, indicating a common binding site shared by both allosteric antagonists. Fenobam exhibits anxiolytic activity in the stress-induced hyperthermia model, Vogel conflict test, Geller-Seifter conflict test, and conditioned emotional response with a minimum effective dose of 10 to 30 mg/kg p.o. Furthermore, fenobam is devoid of GABAergic activity, confirming previous reports that fenobam acts by a mechanism distinct from benzodiazepines. The non-GABAergic activity of fenobam, coupled with its robust anxiolytic activity and reported efficacy in human in a double blind placebo-controlled trial, supports the potential of developing mGlu5 receptor antagonists with an improved therapeutic window over benzodiazepines as novel anxiolytic agents.
Anxiety disorders are the most common mental illnesses in the Western world. Although effective treatment exists in the form of the benzodiazepines and SSRI/norepinephrine reuptake inhibitors, there is still considerable room for improvement. The benzodiazepines are generally regarded as the most efficacious agents, especially for short term use, but they suffer from dose-limiting side effects, including sedation, memory impairment, ataxia, abuse potential, an interaction with ethanol, and physical dependence (Woods et al., 1992). The SSRIs/norepinephrine reuptake inhibitor as well as other serotonergic agents (such as buspirone) are commonly prescribed for the longer term treatment of anxiety but in addition to a delayed onset of action of 3 to 6 weeks and sometimes an increased level of agitation for the first few weeks, they also suffer from side effects in terms of sexual dysfunction, nausea, anorexia, and headache (Vaswani et al., 2003). Numerous recent preclinical and clinical studies have demonstrated the involvement of metabotropic glutamate (mGlu) receptors in the anxiety and stress disorders (Swanson et al., 2005).
The mGlu5 receptor is one of eight G protein-coupled receptors (GPCRs) activated by glutamate, in addition to the well characterized ionotropic glutamate receptors (Conn and Pin, 1997). The eight mGlu receptors are divided into three groups on the basis of their sequence similarities, signal transduction, and agonist rank order of potency. Group I (mGlu1 and 5) are coupled to the activation of phospholipase C, and group II (mGlu2 and 3) and group III (mGlu4, 6, 7, and 8) are negatively coupled to cAMP production (Conn and Pin, 1997). The mGlu receptors are members of class C family of GPCRs, having a large extracellular amino-terminal domain for agonist binding (venus flytrap model) (Kunishima et al., 2000) and the 7TM helical segments that form a binding pocket for the novel class of compounds acting as allosteric modulators (Kew, 2004). Because mGlu5 receptors are highly expressed in brain regions known to be involved in emotional processes such as the limbic brain structures (prefrontal cortex, amygdala, basal ganglia, and hippocampal regions) (Shigemoto et al., 1993; Romano et al., 1995), the mGlu5 receptors have attracted considerable attention for psychiatric disorders (Spooren et al., 2001). Indeed, with the notable exception of the benzodiazepines, the prototypical noncompetitive antagonist MPEP (Gasparini et al., 1999) and its close analog MTEP (Cosford et al., 2003) (Fig. 1) have been reported to be active in the broadest range of preclinical anxiety tests, confirming a critical involvement of this receptor in mood control.
During a functional high-throughput screening (HTS) campaign (fluorometric imaging plate reader; FLIPR) of F. Hoffmann-La Roche's small-molecule library for mGlu5 modulators, we identified fenobam as a potent antagonist of mGlu5 receptors. Interestingly, fenobam (McN-3377) was originally developed as a nonbenzodiazepine anxiolytic (by McNeil Laboratories, 1978-1982), with an unknown molecular target. In preclinical studies, fenobam is reported to be active in rat models of anxiety (conflict tasks including Geller-Seifter and Vogel) (Patel et al., 1982), and this activity was not blocked by a benzodiazepine antagonist (Goldberg et al., 1983).
In a double blind placebo-controlled clinical trial, fenobam had an efficacy and onset of action comparable with diazepam (HAM-A/HSRC/HAM-D) (Pecknold et al., 1982). Additionally, fenobam has been reported to be active in outpatients with severe anxiety (Pecknold et al., 1980) and in a single blind study (Lapierre and Oyewumi, 1982). In all three studies, fenobam was reported to have a good safety profile and no oversedation, no muscle relaxant, and no interaction with ethanol, further supporting a mechanism of action that is distinct from GABA-ergic potentiation (Pecknold et al., 1980, 1982; Lapierre and Oyewumi, 1982). However, fenobam has also been reported to be inactive in one phase II outpatient trial where side effects of a psychostimulant nature were reported (Friedmann et al., 1980). At the time, fenobam was discontinued from further development as an anxiolytic, with indications that the molecule required further refinement (Friedmann et al., 1980; Wu et al., 1995).
Given the recent interest in mGlu5 receptors for anxiety and our recent discovery of fenobam's mode of action via mGlu5 receptor antagonism, we have characterized fenobam in a broad range of in vitro pharmacology tests and in vivo rodent models of anxiety. In the present study, we show that fenobam is a potent, subtype-selective, and noncompetitive antagonist of mGlu5 receptors and that fenobam has inverse agonist activity similar to MPEP (Pagano et al., 2000). Furthermore, we demonstrate that fenobam has potent antianxiety properties in rodent models of anxiety tests, including stress-induced hyperthermia, Vogel conflict, Geller-Seifter conflict, and conditioned emotional response.

Chemical structures of the noncompetitive mGlu5 antagonists (fenobam, MPEP, MTEP, SIB-1893, and SIB-1757), the competitive group I mGlu antagonist (S-4-CPG), and allosteric GABAA receptor agonist (diazepam).
Materials and Methods
Materials
Fenobam and MTEP were synthesized at F. Hoffmann-La Roche (Basel, Switzerland). SIB-1757, SIB-1893, 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester, and NPS 2390 are commercially available. [3H]Fenobam (specific activity, 24.6 Ci/mmol) was synthesized by Dr. P. Huguenin (Hoffmann-La Roche chemical and isotope laboratories). MPEP (specific activity, 36 Ci/mmol; TRK 1070), l-quisqualic acid (0188), and S-4-CPG (0323) were obtained from Tocris Cookson Inc. (Bristol, UK). MPEP was obtained from Sigma (Buchs, Switzerland). myo-[2-3H]Inositol (TRK 911) and yttrium silicate RNA binding beads (RPNQ0013) were purchased from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Glutamate pyruvate transaminase (catalog number 737 127) was from Roche Diagnostics (Rotkreuz, Switzerland).
Plasmids, Cell Culture, and Membrane Preparation
cDNAs encoding the rat mGlu1a, 2, 4a, 5a, 7a, and 8a receptors in pBlueScript II was obtained from Prof. S. Nakanishi (Kyoto University, Kyoto, Japan). The cDNA encoding human mGlu5a was isolated as described previously (Malherbe et al., 2002). The cDNA for rat high-affinity glutamate transporter EAAC1 (AC: U39555) was isolated by reverse transcription-polymerase chain reaction from a rat brain cDNA library.
HEK-293 cells were transfected as described previously. Fortyeight hours post-transfection, the cells were harvested and washed three times with ice-cold PBS and frozen at -80°C. The pellet was suspended in ice-cold 50 mM Tris-HCl buffer containing 10 mM EDTA, pH 7.4, and homogenized with a Polytron (Kinematica, Basel, Switzerland) for 10 s at 10,000 rpm. After centrifugation at 48,000g for 30 min at 4°C, the pellet was resuspended in ice-cold 50 mM Tris-HCl buffer containing 0.1 mM EDTA, pH 7.4, homogenized, and recentrifuged as described above. This pellet was further resuspended in a smaller volume of ice-cold 50 mM Tris-HCl buffer containing 0.1 mM EDTA, pH 7.4. After homogenization for 10 s at 10,000 rpm, the protein content was measured using the bicinchoninic acid method (Pierce Chemical, Socochim, Lausanne, Switzerland) with bovine serum albumin as the standard. The membrane homogenate was frozen at -80°C before use.
[3H]Fenobam and [3H]MPEP Bindings
mGlu5 receptor binding experiments were performed as described previously, with similar conditions for [3H]fenobam as for [3H]MPEP (Malherbe et al., 2003). Briefly, after thawing, the membrane homogenates resuspended and polytronized in 15 mM Tris-HCl, 120 mM NaCl, 100 mM KCl, 25 mM CaCl2, and 25 mM MgCl2 binding buffer at pH 7.4 to a final assay concentration of 20 and 30 μg protein/well for [3H]MPEP and [3H]fenobam binding, respectively. Saturation isotherms were determined by addition of 12 radioligand concentrations (0.04-100 nM) to these membranes (in a total volume of 200 μl) for 1 h at 4°C ([3H]MPEP binding) and for 1 h at RT ([3H]fenobam binding). At the end of the incubation, membranes were filtered onto unifilter (96-well white microplate with bonded GF/C filter preincubated 1 h in 0.1% polyethylenimine in wash buffer; PerkinElmer Life and Analytical Sciences, Boston, MA) with a Filtermate 196 harvester (PerkinElmer Life and Analytical Sciences) and washed three times with ice-cold 50 mM Tris-HCl, pH 7.4, buffer. Nonspecific binding was measured in the presence of 10 μM MPEP for both radioligands. At the Kd values, the nonspecific binding for [3H]MPEP and [3H]fenobam was approximately 5 and 35%, respectively, of total bound radioactivity for both radioligands. The radioactivity on the filter was counted (3 min) on a TopCount microplate scintillation counter with quenching correction after addition of 45 μlof Microscint 40 (Canberra Packard S.A., Zürich, Switzerland) and shaking for 20 min. Saturation experiments were analyzed by Prism 3.0 (GraphPad Software Inc., San Diego, CA) using the rectangular hyperbolic equation derived from the equation of a bimolecular reaction and the law of mass action, B = (Bmax × [F])/(Kd + [F]), where B is the amount of ligand bound at equilibrium, Bmax is the maximum number of binding sites, [F] is the concentration of free ligand, and Kd is the ligand dissociation constant. For inhibition experiments, membranes were incubated with 2 nM [3H]MPEP or 20 nM [3H]fenobam and 10 concentrations of the inhibitory compound. IC50 values were derived from the inhibition curve and Ki values were calculated according to the equation Ki = IC50/(1 + [L]/Kd), where [L] is the concentration of radioligand and Kd is its dissociation constant at the receptor, derived from the saturation isotherm.
[3H]Inositol Phosphates Accumulation Assay
[3H]Inositol phosphates accumulation was measured using the methodology of Brandish et al. (2003) with the following modifications. HEK-293 cells stably expressing hmGlu5 receptors were transfected with the high-affinity glutamate transporter EAAC1, plated at 70,000 cells/well in a polylysine-coated 96-well plate with the addition of 5 μCi of myo-[2-3H]inositol at the time of plating (24 h before assay) in an inositol/glutamate-free DMEM with 10% dialyzed fetal bovine serum, 1% penicillin/streptomycin, and 1 mM glutamate. On the day of assay, cells were washed three times, incubated in the presence of glutamate pyruvate transaminase and sodium pyruvate for 1 h before the addition of agonists or antagonists in assay buffer (Hanks' balanced salt solution in 20 mM HEPES plus 8 mM LiCl). When present, antagonists were incubated for 5 min before agonist addition. After 45-min incubation with agonist, the assay was terminated by the aspiration of the assay buffer and the addition of 100 μl of 20 mM formic acid to the cells. After 20-min incubation, a 20-μl aliquot was mixed with 80 μl of yttrium silicate beads that bind to the inositol phosphates (but not inositol). The plates were counted on a TopCount, and data were expressed as a percentage of the basal signal, i.e., cells in the absence of added drugs.
Intracellular Ca2+ ([Ca2+]i) Mobilization Assay (FLIPR)
HEK-293-hmGlu5 stable cell line was seeded at 6 × 104 cells/well in the poly-d-lysine-treated, 96-well, black/clear-bottomed plates (BD Biosciences, Palo Alto, CA). After 24 h, the cells were loaded for 1 h at 37°C with 4 μM Flou-4AM (Molecular Probes, Eugene, OR) in loading buffer (1× Hanks' balanced salt solution and 20 mM HEPES). The cells were washed five times with loading buffer to remove excess dye, and [Ca2+]i mobilization was measured using FLIPR (Molecular Devices, Menlo Park, CA) as described previously (Malherbe et al., 2003). All antagonists were dissolved in 100% DMSO, and diluted in assay buffer to a 5× stock (2.5% DMSO). This stock was then applied to the cells at a final DMSO concentration of 0.5%. The antagonist potency of various compounds on the hmGlu5 receptors was determined using 10 to 20 nM quisqualate as agonist (a concentration that gave 60 to 80% of maximum agonist response, determined daily). The antagonists were applied 5 min before the application of the agonist. Responses were measured as peak increase in fluorescence minus basal, normalized to the maximal stimulatory effect induced by 10 nM quisqualate. Inhibition curves were fitted according to the Hill equation: y = 100/(1 + (x/IC50)nH), where nH is slope factor using prism 3.0 (GraphPad Software Inc.).
[35S]GTPγS Binding Assay
This assay was performed for selectivity test of fenobam toward mGlu4a and 7a receptors using membranes from Chinese hamster ovary (CHO) cells expressing transiently mGlu4a and stably mGlu7a receptors. After thawing, the membranes were washed once and resuspended in ice-cold 20 mM HEPES-NaOH buffer containing 10 mM MgCl2, 2 mM EGTA, and 100 mM NaCl, pH 8.0. Wheat germ agglutinin SPA beads (RPNQ0001; GE Healthcare) were suspended in the same buffer (40 mg of beads/ml). Membranes and beads were mixed (beads, 13 mg/ml; membranes, 200 μg protein/ml) and incubated with 10 μM GDP at RT for 1 h, with mild agitation. [35S]GTPγS binding was performed in 96-well microplates (Picoplate; PerkinElmer Life and Analytical Sciences) in a total volume of 180 μl with 30 μg (mGlu4a) and 5 μg (mGlu7a) membrane proteins and 0.3 nM [35S]GTPγS. Nonspecific binding was measured in the presence of 10 μM ice-cold GTPγS. For mGlu4a selectivity test, [35S]GTPγS binding was stimulated with various concentrations of L-AP4 (3 nM-30 μM) ± 10 μM fenobam. For mGlu7a selectivity test, fenobam concentration response curves (3 nM-100 μM) were performed in the presence of 0.1 mM (EC20) and 0.7 mM (EC80) L-AP4. Plates were sealed and agitated at RT for 3 h. The beads were then allowed to settle and the plate counted in a TopCount (PerkinElmer Life and Analytical Sciences) using quench correction.
cAMP Accumulation Assay
For selectivity test of fenobam toward mGlu2, measurement of cAMP accumulation assay was performed with a stable CHO cell line expressing mGlu2 receptor using the radioimmunoassay (RIA) kit (RPA559; GE Healthcare). The cells were plated at 40,000 cells/well in a 96-well plate 24 h before assay. On the day of assay, cells were washed in PBS and incubated at RT for 30 min in a total volume of 200 μl with PBS buffer containing 1 mM 3-isobutyl-1-methylxanthine (Fluka, Buchs, Switzerland), 1-aminocyclopentane-1,3-dicarboxylic acid (10 nM-30 μM) ± 10 μM fenobam, and the water-soluble forskolin analog 7-deacetyl-7-(O-N-methylpiperazino)-γ-butyryl-forskolin dihydrochloride (10 μM) (Calbiochem, Lucerne, Switzerland). The amount of cAMP formed was quantified by RIA following the cAMP SPA Biotrak direct screening assay system protocol from GE Healthcare: at the end of the incubation period, the plate was centrifuged and the supernatant was removed, and then 50 μl of lysis reagent (containing 10% dodecyltrimethylammonium bromide) was added to each well. After 5 min of shaking, 150 μl of immunoreagent solution (containing rabbit anti-succinyl cAMP serum, donkey anti-rabbit IgG second antibodies coated at the surface of SPA beads, and adenosine 3′,5′-cyclic phosphoric acid 2′-O-succinyl-3-[125I]iodotyrosine methyl ester as a tracer) was added to each sample. The plates were shaking for 18 h, allowed to settle for 3 h at RT, and then counted in a TopCount. The quantization of the cAMP formed was performed with the aid of an appropriate RIA cAMP standard curve.
Behavioral Profiling of Fenobam
Animals and Drug Treatment. Adult male Sprague-Dawley rats supplied from Charles River (Margate, Kent, UK) and NMRI mice (stress-induced hyperthermia) supplied from Iffa Credo (L'Arbresele, France) were used. All rats were group housed and mice were singly housed in separate holding rooms at controlled temperature (20-22°C) and 12-h light/dark cycle (lights on 6:00 AM). All animals were allowed ad libitum access to food and water, with the exception of those used in the operant-conditioning tests [Geller-Seifter, conditioned emotional response (CER)], where food was limited to that earned in the test session and 12 to 15 g/rat at the end of the day. The experimental procedures used in the present investigation received prior approval from the City of Basel Cantonal Animal Protection Committee based on adherence to federal and local regulations.
All compounds were prepared immediately before use in vehicle, 0.3% Tween 80 (v/v) saline. The volume of administration was 1 ml/kg (Geller-Seifter, CER) or 5 ml/kg (Vogel) body weight for rats and 10 ml/kg for mice. Fenobam or vehicle was administered p.o. 60 min before testing. All doses were calculated as weight of base.
Stress-Induced Hyperthermia in Mice. A detailed description of the stress-induced hyperthermia (SIH) test procedure can be found in Spooren et al. (2002). Briefly, drug naive individually housed (macrolon cage, 26 × 21 × 14 cm) male NMRI mice were used. Body temperature was measured in each mouse twice at t = 0 (T1) and t = +15 min (T2). T1 served as the handling stressor. The difference in temperature (T2 - T1) was considered to reflect stress-induced hyperthermia. Mice received treatment with fenobam (3, 10, or 30 mg/kg p.o.) or vehicle 60 min before T1 and subsequently underwent the SIH procedure.
Operant Conditioning Tests. The operant conditioning boxes were obtained from MED Associates (St. Albans, VT), and the protocols were run by the Kestrel Control System from Conclusive Marketing Ltd. (Harlow, UK) operating on an IBM-compatible computer.
Vogel Conflict Drinking. Male Sprague-Dawley rats (around 200 g) were water-restricted for three consecutive 24-h periods. At the end of the first 24-h period, animals were allowed access to water in their home cage for 60 min. At the end of the second 24-h period, they were placed into the test chamber containing a drinking bottle and were allowed to drink freely for 15 min, after which they received drinking water in their home cage for another 60 min. The drinking time in the test chamber was used to randomize the animals over the different treatments. The drinking time (in seconds) was determined as the time spent by the rat at the drinking spout. An optical sensor (MED Associates) was used to detect the presence of the rat's snout (i.e., not tongue movements). At the end of the third 24-h period, they were administered the test compound p.o. and isolated in a holding chamber until testing 60 min later. During the 10-min test, they were allowed to drink freely for a cumulative time of 5 s, after which drinking was punished. That is, an electrical shock between the grid floor and the drinking spout (0.5 mA; 250 ms) was triggered every second of cumulative drinking time. This shock level strongly suppresses drinking to approximately 10% of normal levels. Fenobam was tested at 3, 10, and 30 mg/kg p.o. (n = 12/dose).
Geller-Seifter Conflict. For further details regarding the training schedule and criteria for baseline performance, see Martin et al. (2002). This procedure was divided into three stages (one lever was extended throughout the whole session): 1) unpunished responding (5 min) during which the house light was illuminated and animals received food reward to reinforce lever pressing on a schedule of variable interval of 30 s (VI30); 2) nonrewarded responding (time-out period of 2 min) during which the house light was extinguished and animals were able to press the lever, but they did not receive a food reward; and 3) punished responding (8 min) during which the house light remained off and a cue light above the lever was illuminated. In the latter stage, a fixed ratio of 10 (FR10) was used so that, on every tenth lever press, the rat received simultaneously a food reward and a foot shock (0.6 mA; 0.5 s). These three stages were repeated in the same order so that the total daily session duration would be 30 min.
Eight trained rats were included in the experiment to evaluate fenobam and were tested using a Latin-squares design, twice weekly with at least a 2-day interval between test sessions. Rats were trained between test days to maintain baseline performance. Fenobam was tested at 10, 30, and 100 mg/kg p.o.
CER. For further details regarding the training schedule and criteria for baseline performance, see Martin et al. (2002). The 60-min session consisted of a VI60 schedule with two randomized presentations of a conditioned stimulus (CS) for 2 min. The CS consisted of a 2.9-kHz tone delivered from a Sonalert system and a cue light above the lever. During the final 0.5 s of the CS, animals received a foot shock (0.6-1.0 mA; 0.5 s). The number of lever presses during the CS period and during the 2 min before this period were recorded and used to calculate suppression ratios (SRs): number of lever presses during CS/total number of lever presses before and during CS. An SR value of 0 indicates that animals have suppressed responding during the tone presentation due to a conditioned fear response. An SR value of 0.5 indicates that the animals are responding equivalently during the conditioning period and 2 min immediately before this period.
Twelve trained rats were included in the experiment to evaluate fenobam and were tested using a Latin-squares design, twice weekly with at least a 2-day interval between test sessions. During a test session, the animals did not receive a foot shock after the conditioning periods. However, animals always received foot shock during intervening baseline days. Rats were trained between test days to maintain baseline performance. Fenobam was tested at 10, 30, and 100 mg/kg p.o.
Behavior Data Analysis. SIH data were statistically evaluated using a Dunnett multiple comparison test (two-tailed) after a one-way ANOVA. Geller-Seifter data were analyzed using a repeated measures ANOVA followed by a paired t test. For the CER test, the SR data were analyzed using nonparametric statistics because this is a ratio with a cut-off value of 0.5; i.e., a Friedman's test followed by a Wilcoxon rank sum test. The total number of lever presses made during the 1-h session was analyzed using repeated measures ANOVA followed by paired t tests. For the Vogel test, drinking time was analyzed by Kruskal-Wallis test, because the data are not normally distributed, followed by a Mann-Whitney U test. In all studies, the accepted level of significance was p < 0.05. For uniformity, all of the data are expressed as mean ± S.E.M. in the figures.
Results
Discovery of Fenobam as HTS Screening Hit against mGlu5 Receptor
A series of urea derivatives were previously patented within F. Hoffmann-La Roche as anxiolytics (Hunkeler and Kyburz, 1980) that did not act via GABAA receptors but via a molecular target, which was unknown. During an HTS functional (FLIPR) screening of F. Hoffmann-La Roche random small-molecule library against rat mGlu5 receptors, we identified urea derivatives, including fenobam, as HTS-hits. Fenobam has a chemical structure that is different from the benzodiazepines (diazepam), competitive group I mGlu antagonist (S-4-CPG), and noncompetitive mGlu5 receptor antagonists (MPEP and its analog MTEP) (Fig. 1).
[3H]MPEP and [3H]Fenobam Binding and Displacement Studies
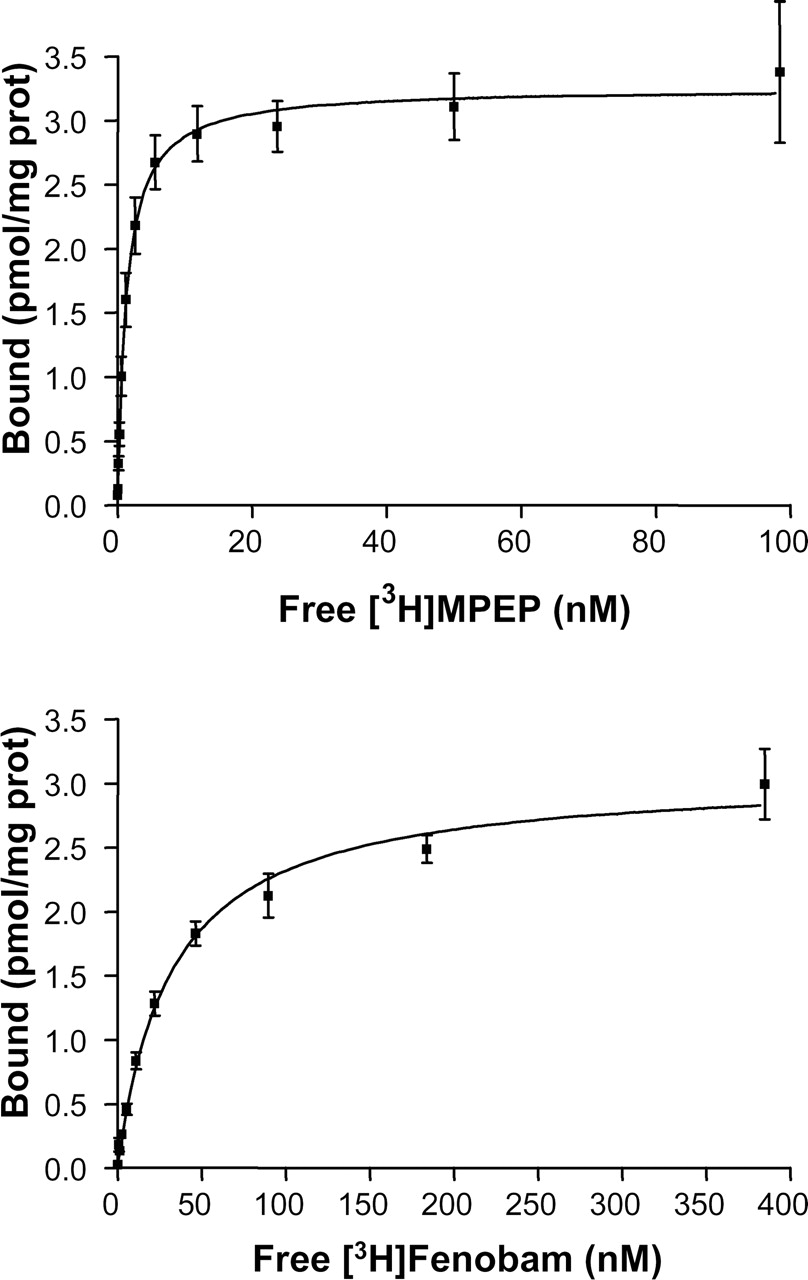
To characterize the in vitro binding of [3H]MPEP and [3H]fenobam, saturation binding analyses were performed at binding equilibrium (1-h incubation at 4°C and room temperature, respectively), as outlined under Materials and Methods on membranes isolated from the HEK-293 transiently transfected with the rat and human mGlu5 receptors. The saturation isotherm was monophasic ([3H]MPEP concentrations from 0.04 to 100 nM and [3H]fenobam from 0.04 to 400 nM) and best fitted to a one-site model for both radioligands (Fig. 2). The dissociation constants (Kd) and the maximum number of binding sites (Bmax) derived from the saturation isotherms are given in Table 1. [3H]Fenobam bound to recombinantly expressed human and rat mGlu5 receptors with a Kd of 31.14 ± 4.10 nM and 53.60 ± 5.58, respectively. The Ki values for [3H]fenobam displacement studies with various noncompetitive mGlu5 antagonists, glutamate, and quisqualate using membranes containing recombinant human and rat mGlu5 receptors are given in Table 2. For comparison, the data for in vitro binding and displacement of [3H]MPEP are also given in Tables 1 and 2. Although fenobam is less potent than [3H]MPEP (Kd of 1.5 nM at rat and human receptors), the pharmacological profile of standard mGlu5 ligands was similar at displacing either fenobam or MPEP (Table 2). We have also investigated the displacement of [3H]fenobam binding to recombinant rmGlu5a membrane by the compounds that are phenylglycine (PG) derivatives (S-4C3HPG, S-DHPG, S-3HPG, 2Me4CPG, -4-CPG, (+)-MCPG, and CHPG) or rigidified analogs of glutamate, including 2-(carboxycyclopropyl)glycine, (2S,2′R3′R′)-2-(2′,3′-dicarboxycyclopropyl)glycine, 1-aminocyclopentane-1,3-dicarboxyclic acid, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid, L-AP4, kainate, and 1-aminoindan-1,5-dicarboxylic acid. All of these compounds were inactive (Ki of >10,000 μM). Furthermore, the noncompetitive mGlu1 antagonists 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester and NPS 2390 inhibited poorly [3H]fenobam binding to recombinant rmGlu5a membrane with Ki of 50 ± 5.5 and 3.66 ± 1.4 μM, respectively.
[3H]fenobam and [3H]MPEP binding properties at human and rat mGlu5 Saturation binding isotherms of [3H]fenobam and [3H]MPEP were performed on membrane preparations from HEK-293 cells transiently transfected with the human and rat mGlu5 receptors as described under Materials and Methods. Values are mean ± S.E.M. of the Kd and Bmax values, calculated from three independent experiments (each performed in triplicate).
Ki values for inhibition of [3H]MPEP (2 nM) and [3H]fenobam (10 nM) bindings to human and rat mGlu5 receptors transfected HEK-293 cell membranes by various compounds Values are mean ± S.E.M. of the Ki calculated from three independent experiments, each performed in duplicate.
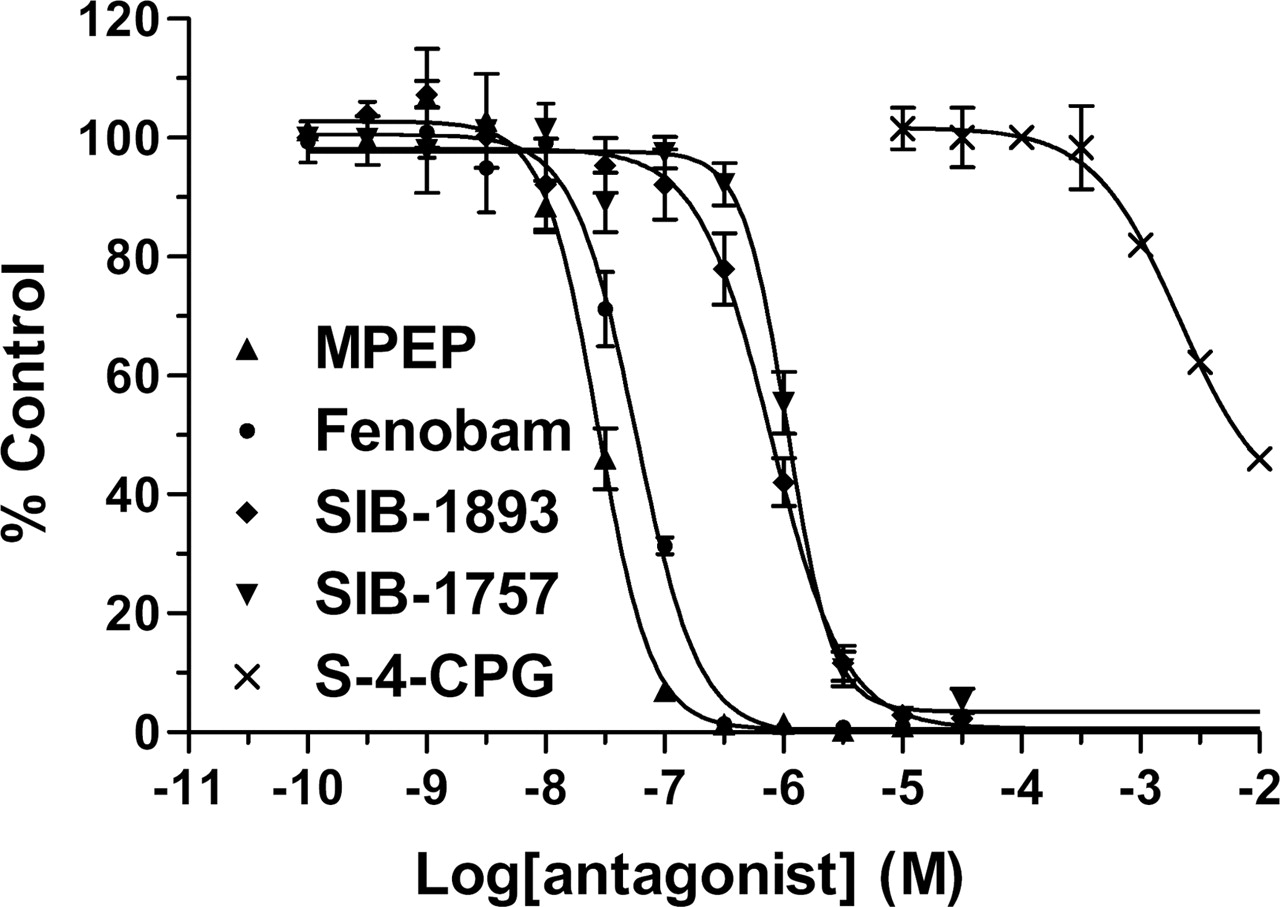
Fenobam Is a Potent and Noncompetitive Antagonist of mGlu5 Receptor with Inverse Agonist Activity. In HEK-293 cells stably expressing human mGlu5 receptors, fenobam inhibits quisqualate-evoked [Ca2+]i response with IC50 = 58 ± 2 nM, nH = 1.7 ± 0.1. For comparison, the concentration-dependent inhibition of quisqualate-evoked increases in [Ca2+]i by noncompetitive (SIB-1757, SIB-1893, and MPEP) and competitive (S-4-CPG) hmGlu5 antagonists; their derived IC50 values are shown in Fig. 3 and Table 3.
Potencies of various mGlu5 antagonists on inhibition of quisqualate-induced [Ca2+]i response IC50 and Hill coefficient (nH) values for the inhibition of quisqualate (20 nM)-evoked [Ca2+]i response in the HEK-293 cells stably expressing the hmGlu5a receptor. Data are means ± S.E. of the three to six dose-response measurements (each performed in triplicate).

Saturation bindings of [3H]MPEP and [3H]fenobam to membranes from HEK-293 cell transfected transiently with hmGlu5 receptor. Each data point is mean ± S.E. (bars) of three individual experiments performed in triplicate. The data were analyzed by nonlinear regression analysis using Prism 3.0 software and a single-site binding model.
To characterize the inhibition mode of fenobam, the concentration-response curves for [3H]IP formation stimulated by quisqualate in the presence of 0, 100 nM, 300 nM, and 1 μM fenobam in HEK-293-hmGlu5 stable cell line are shown in Fig. 4. As seen, fenobam behaves as a noncompetitive antagonist shifting the agonist concentration-response curve to the right with a concomitant decrease in maximal response consistent with a noncompetitive mode of action.
MPEP has previously been shown to inhibit the mGlu5 constitutive activity (Pagano et al., 2000). Similarly, we have tested the inverse agonist activity of fenobam using IP accumulation assay. To optimize the condition for detection of mGlu5 constitutive activity, HEK-293-hmGlu5 stable cell line expressing a high level of recombinant receptor was transiently transfected with a plasmid containing high-affinity glutamate transporter EAAC1 cDNA and the basal level of [3H]IP formation was then measured in the presence of glutamate pyruvate transaminase (a glutamate-degrading enzyme). As seen in Fig. 5, fenobam inhibited the basal activity dose dependently with an IC50 = 84 ± 13 nM, nH = 1 ± 0.2, this is compared with IC50 = 21 ± 3 nM, nH = 1 ± 0.1, for MPEP under identical conditions. The basal levels of [3H]IP formation were reduced to 32 and 34% by MPEP and fenobam at 10 μM, respectively (Fig. 5). Note that under identical experimental conditions, the competitive antagonist S-4-CPG did not exhibit any inverse agonist activity (A. Mühlemann, data not shown).

Comparison of group I mGlu antagonism on quisqualate-evoked [Ca2+]i in the HEK-293 cells stably expressing mGlu5 receptor. Concentration-dependent inhibition quisqualate-stimulated increases in [Ca2+]i by various antagonists as assayed using the Ca2+-sensitive dye Flou-4 and a fluorometric imaging plate reader. Responses are normalized to the first control response. Each curve represents mean ± S.E. (bars) of the three to six dose-response measurements (each performed in triplicate).

Schild analysis showing that the noncompetitive mode of antagonism by fenobam at mGlu5 receptors. Quisqualate concentration responses in the absence (control) and presence of various concentrations of fenobam were generated using the IP accumulation assay and HEK-293-hmGlu5 stable cells that transiently express the high-affinity glutamate transporter EAAC1. Each data point is mean ± S.E. (bars) of three individual experiments performed in quadruplet.
Selectivity. To evaluate the selectivity of fenobam relative to other mGlu receptors, functional assays evaluating both potential antagonist and agonist/enhancer activity against mGlu1a transiently expressed in HEK-293 using FLIPR assay (Fig. 6, A and B), mGlu2 stably expressed in CHO using cAMP accumulation assay (Fig. 6C), mGlu4a transiently expressed in CHO using [35S]GTPγS binding assay (Fig. 6D), mGlu7a stably expressed in CHO using [35S]GTPγS binding assay (Fig. 6E), and mGlu8a stably expressed in CHO-Gα15 using FLIPR assay (Fig. 6F) were established. When tested at concentrations up to 10 μM, fenobam was not active in assay conditions that did detect known modulators. Furthermore, the pharmacological specificity of fenobam was confirmed by testing in the diversity profile at Cerep (Paris, France) (www.cerep.fr). Fenobam was considered inactive (<50% activity at 10 μM) at all targets tested with the exception of the adenosine A3 receptor, where it caused a 65% displacement of specific binding at 10 μM (Table 4).
Broad Cerep screen undertaken to determine the pharmacological activity of fenobam

Inhibition of mGlu5 basal activity by fenobam and MPEP. The basal activity was measured using the IP accumulation assay and HEK-293-hmGlu5 stable cells that transiently expressing the high-affinity glutamate transporter EAAC1. Each data point is mean ± S.E. (bars) of three individual experiments performed in quadruplet.
Behavioral Profiling of Fenobam
Effect of Fenobam on SIH in Mice. Fenobam (3, 10, or 30 mg/kg p.o.) at doses 10 (p < 0.001) and 30 mg/kg (p < 0.001) p.o. significantly reversed SIH (Fig. 7). Fenobam had no effect on T1 (data not shown).
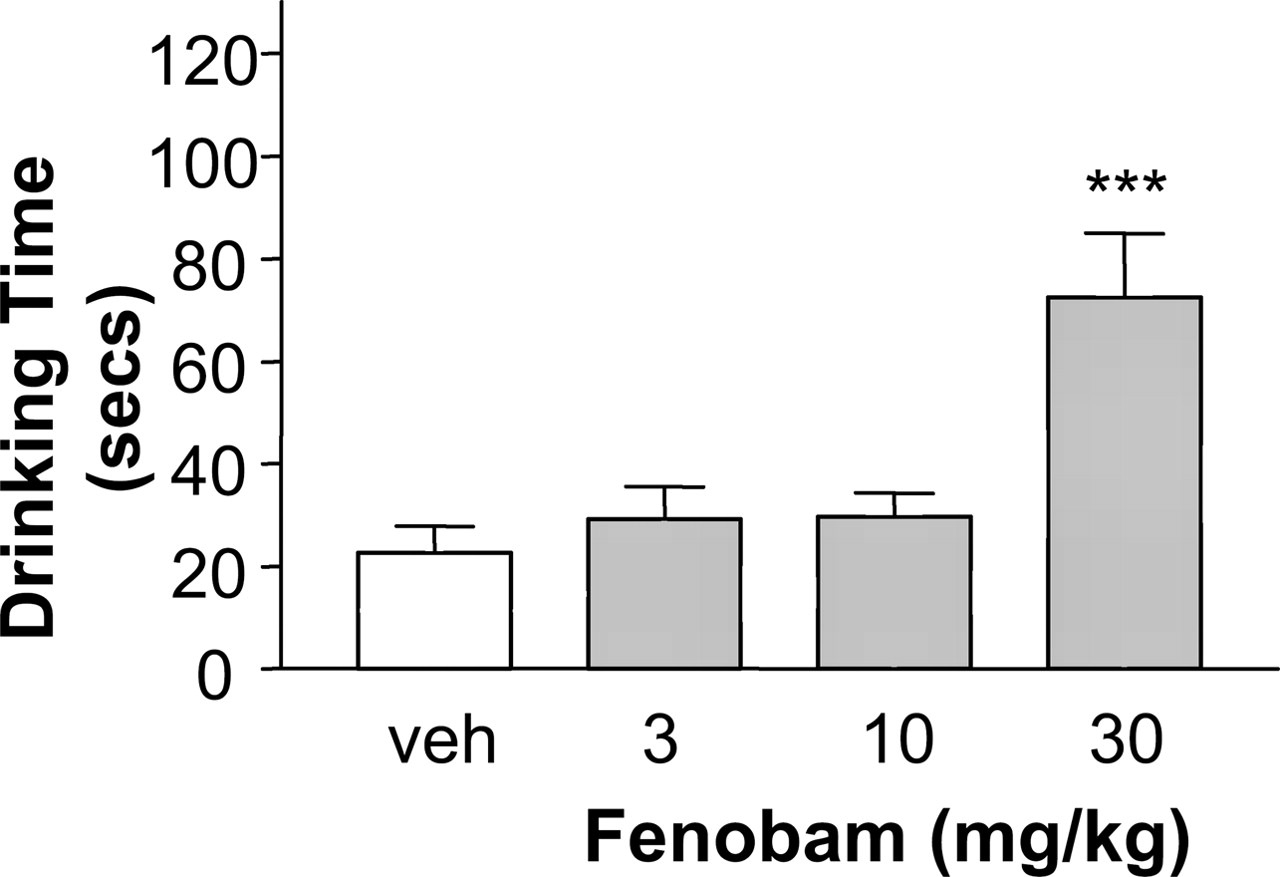
The Vogel Test as a Measure of Conflict Behavior in Rats. Fenobam (3, 10, and 30 mg/kg p.o.) increased the drinking time in the Vogel conflict test (Kruskal-Wallis, p < 0.01; Fig. 8). Although there was no significant effect at the two lower doses (p > 0.1 versus vehicle, as determined by the Mann-Whitney U test), at the dose of 30 mg/kg, the effect of fenobam in increasing drinking time was highly significant (p < 0.001).
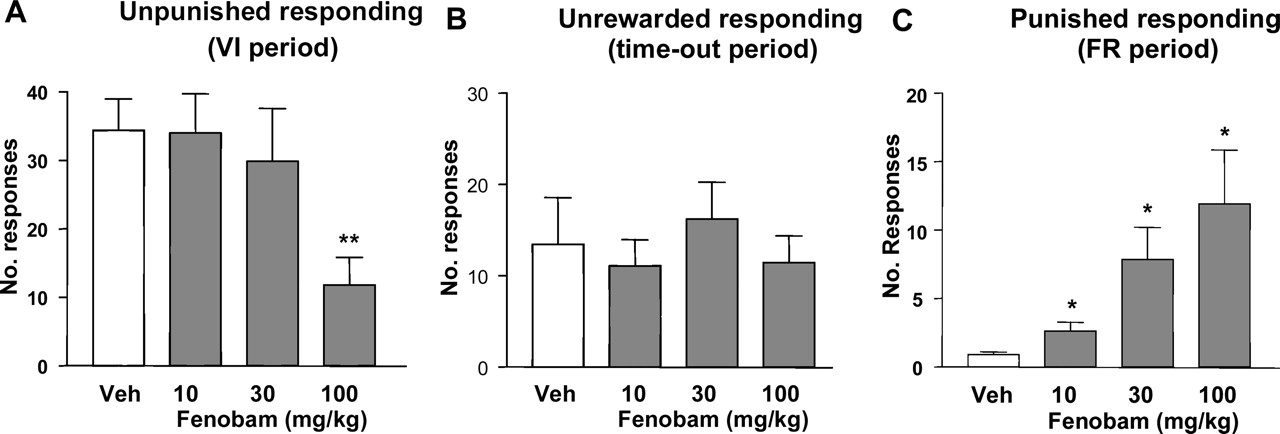
Geller-Seifter Conflict Test in Rats. Fenobam (10, 30, and 100 mg/kg p.o.) dose dependently increased punished responding [F(3,27) = 3.7; p < 0.05] in the rat conflict test with an MED of 10 mg/kg p.o. (Fig. 9C). Unpunished responding was decreased [F(3,27) = 8.9; p < 0.001], with a significant reduction at 100 mg/kg compared with vehicle (Fig. 9A). None of the doses affected unrewarded responding (Fig. 9B).
Conditioned Emotional Response Test in Rats. Fenobam produced a significant dose-dependent increase in the SR (Friedman test, p < 0.01), which reached statistical significance at 30 and 100 mg/kg p.o. (Fig. 10A). Fenobam at 30 and 100 mg/kg significantly reduced the total number of lever presses made during the 1-h session (Fig. 10B).
Discussion
The G protein-coupled metabotropic glutamate receptor mGlu5 is important in the modulation of neuronal function. The identification of MPEP, as a highly potent, systemically active, and allosteric antagonist of mGlu5 receptor (Gasparini et al., 1999), has greatly contributed to our understanding of physiological role played by this receptor in central nervous system processes. In numerous preclinical studies, MPEP and MTEP are active in animal models of anxiety and depression (Spooren et al., 2000; Tatarczynska et al., 2001; Pilc et al., 2002; Wieronska et al., 2002, 2004; Busse et al., 2004; Ballard et al., 2005), Parkinson's disease (Breysse et al., 2002), pain (Zhu et al., 2004), and addiction (McGeehan et al., 2004). The discovery in the present studies that mGlu5 is the molecular target of fenobam, a clinically validated nonbenzodiazepine anxiolytic, demonstrates further the pivotal function of mGlu5 in mood and anxiety.
SIB-1757 and SIB-1893 were the first novel nonamino acid, selective, and noncompetitive antagonists of mGlu5 receptor (IC50 values of 3.1 and 2.3 μM at hmGlu5, respectively, in IP accumulation assay) to be reported (Varney et al., 1999). MPEP, a novel derivative of SIB-1757, was the first highly potent mGlu5-selective antagonist (IC50 = 36 nM at hmGlu5a in the IP accumulation assay) to be described (Gasparini et al., 1999). Pagano et al. (2000) elucidated the site of action of [3H]M-MPEP, which binds within the 7TM domain of hmGlu5 in close contact with the residues Ala810 in TM7 and Pro655 and Ser658 in TM3. Furthermore, the homology modeling of 7TM helices of mGlu5 receptors resulted in identification of additional residues in the TM3, 5, 6, and 7 regions that are important molecular determinants of the high-affinity binding site of [3H]MPEP (Malherbe et al., 2003). The structure-activity relationship analysis of MPEP has later led to MTEP (Cosford et al., 2003) and similar close analogs.
Fenobam was discovered as an mGlu5 receptor antagonist serendipitously in a screening campaign. Interestingly, the structure of fenobam belongs to a chemical class different from MPEP and its analogs. [3H]Fenobam binds to a single saturable site on recombinantly expressed human and rat mGlu5 receptors (Bmax of 2951 and 5301 fmol/mg protein, respectively) with high affinity (Kd of 31.1 and 53.6 nM, respectively). Binding of [3H]fenobam was temperature-dependent and increased substantially at 25°C. Interestingly, fenobam did consistently exhibit a more potent profile at human than at rat receptors. The noncompetitive mGlu5 antagonists displaced [3H]fenobam binding with the following rank order of potency: MPEP > MTEP > Fenobam > SIB-1893 > SIB-1757, which is consistent with the rank order potency of these ligands in inhibiting quisqualate-evoked [Ca2+]i response at mGlu5 receptors. Moreover, glutamate, quisqualate, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, kainate, and a series of phenylglycine derivatives or rigidified analogs of glutamate (acting as competitive agonist or antagonists) were unable to displace the [3H]fenobam binding to rmGlu5a receptors expressed in HEK-293 cell membranes. Because MPEP and its analog MTEP strongly inhibited the [3H]fenobam binding with Ki values of 6.7 ± 0.7 and 14.1 ± 2.0 nM, respectively, we conclude that fenobam shares a common binding pocket in the transmembrane region of the receptor, at least overlapping with that of MPEP.
In functional studies, fenobam was able to inhibit quisqualate-induced [Ca2+]i response in a HEK-293-hmGlu5 stable cell line with an IC50 value of 58 nM (similar in potency to MPEP). The investigation of the antagonism mechanism by fenobam revealed that it behaved as a noncompetitive antagonist, very similar to MPEP, shifting the quisqualate dose-response curves to the right in the presence of increasing fenobam concentrations with a concomitant decrease in the maximal effect of quisqualate using IP accumulation assay. In the present study, using a high receptor-expressing cell line, fenobam was found to be an inverse agonist comparable with MPEP. Moreover, fenobam displayed no activity toward rat mGlu1a, 2, 4a, 7a, and 8a receptors in a functional assay at concentration up to 10 μM. With the exception of the A3 receptor where it displaced 65% of specific binding at 10 μM, it did not bind to a battery of GPCRs in a broad CEREP profiling.
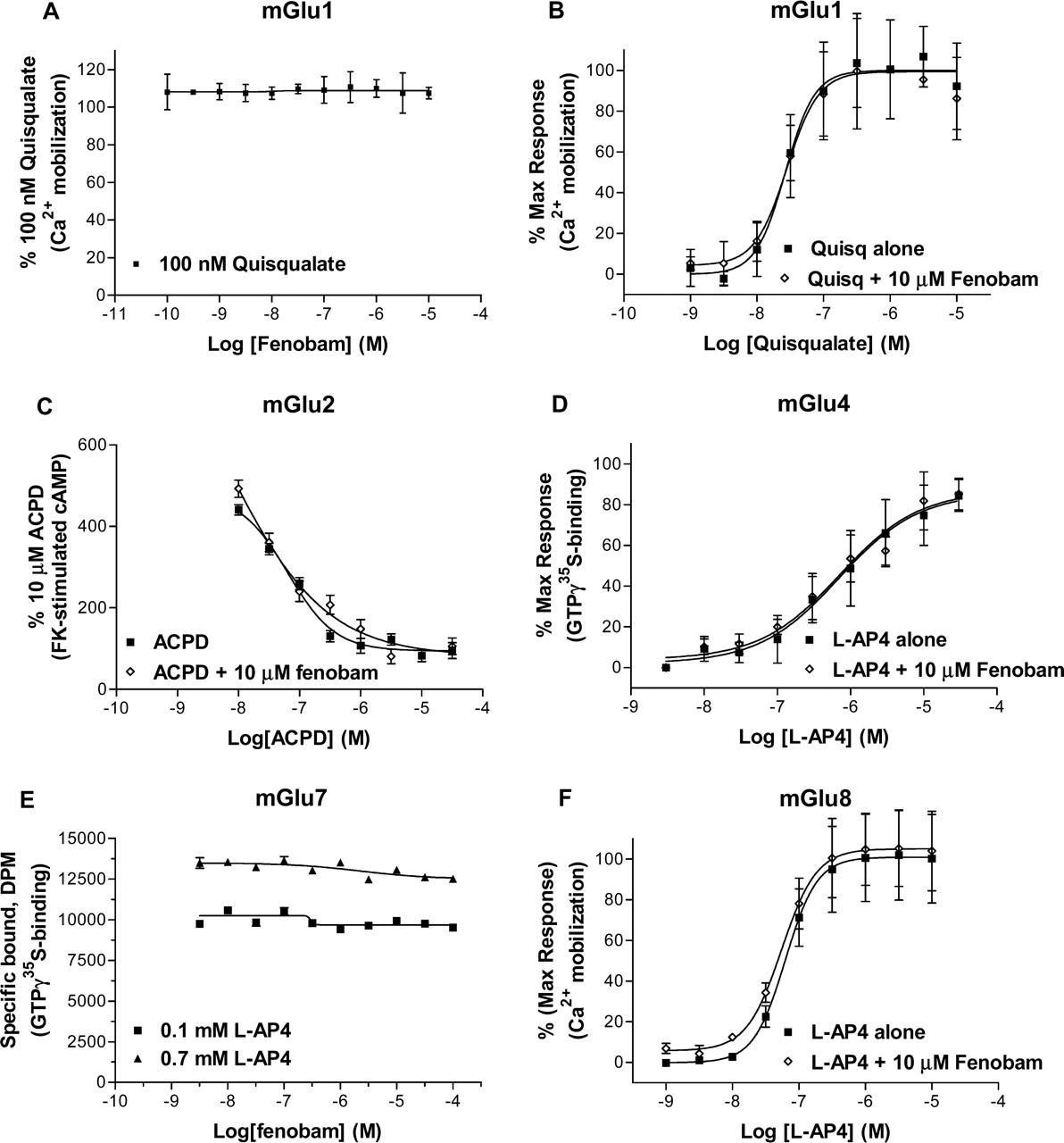
Fenobam displayed no effect toward rat mGlu1a, 2, 4a, 7a, and 8a receptors. A, fenobam (1 nM-10 μM) was not able to inhibit 100 nM quisqualate in HEK-293 cell transiently expressing mGlu1a. B, quisqualate concentration response in absence and presence of 10 μM fenobam. C, 1-aminocyclopentane-1,3-dicarboxylic acid dose-response curves in absence and presence of 10 μM fenobam in CHO-mGlu2 cell line. Effect of 10 μM fenobam on L-AP4 concentration responses in CHO cell transiently expressing mGlu4a (D) and in CHO-Gα15 cell stably expressing mGlu8a (F). The fenobam concentration responses in the presence 0.1 mM (EC20) and 0.7 mM (EC80) in CHO-mGlu7a stable line are shown in E. Each data point is mean ± S.E. (bars) of two to four experiments performed in triplicate or quadruplet.

Reversal of stress-induced hyperthermia in mice. Male NMRI mice received fenobam at doses of 3, 10, and 30 mg/kg p.o., 1 h before T1. Data are mean ± S.E.M. based on eight animals per group. veh, vehicle (0.3% Tween 80 in NaCl 0.9%). **, p < 0.01; ***, p < 0.001 versus vehicle, Dunnett multiple comparison test (two-tailed) following a one-way ANOVA.

Increase in drinking time in the Vogel conflict test in rats. Male naive Sprague-Dawley rats received fenobam at doses of 3, 10, and 30 mg/kg p.o. (1-h pretreatment time) and were tested in the Vogel conflict test. Data are mean ± S.E.M. based on 12 animals per group. Vehicle, 0.3% Tween 80 in 0.9% NaCl. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle, as determined by the Mann-Whitney U test.
The behavioral data indicate that fenobam exhibits anxiolytic-like activity in all four anxiety tests included in the present study; three rat conflict tests (Geller-Seifter, CER, and Vogel conflict) and a mouse model of autonomic hyperreactivity in anxiety/stress. Fenobam was active in a comparable dose range after oral administration in all tests (MED 10-30 mg/kg p.o.). These data confirm the previously reported preclinical studies with fenobam (i.e., rat conflict tasks including Geller-Seifter and Vogel) (Patel et al., 1982; Goldberg et al., 1983). The data are also in line with previous findings with selective mGlu5 receptor antagonists, particularly the prototypical mGlu5 receptor antagonist MPEP (for review, see Spooren et al., 2001). MPEP and MTEP have previously been shown to be active in the Vogel conflict test (Tatarczynska et al., 2001; E. Prinssen, unpublished observations) and the Geller-Seifter conflict test (Brodkin et al., 2002; Busse et al., 2004). Furthermore, we have recently shown that MPEP produces a robust anxiolytic-like effect in all three rat conflict tests (Geller-Seifter, CER, and Vogel) at an equivalent dose range after oral administration, with a minimum effective dose of 10 mg/kg p.o. (Ballard et al., 2005). Moreover, the anxiolytic effect of MPEP was comparable with that of diazepam, which was used as a positive control in the same tests.
In the current study, fenobam differs from MPEP and MTEP in that it has a greater effect on nonspecific aspects of the task; i.e., significant reduction in lever pressing in CER and in unpunished responding in Geller-Seifter (Busse et al., 2004; Ballard et al., 2005). The greater behavioral disruptive properties of fenobam, in contrast to that observed with MPEP and MTEP in the animal models, may have some consistency with the clinical data reporting of side effects of fenobam and may be due to the very variable interindividual oral bioavailability of fenobam (Itil et al., 1978). Interestingly, analogs of fenobam (Hunkeler and Kyburz, 1980) or the more structurally diverse mGlu5 receptor antagonists MPEP and MTEP do not cause the same behavioral disruption as fenobam in animals. Consequently, we cannot rule out an additional site of action of fenobam or possibly the formation of a biologically active metabolite because fenobam is extensively metabolized in vivo (Wu et al., 1995).
Antagonism of mGlu5 receptors by MPEP (Gasparini et al., 1999) and the anxiolytic-like effects of mGlu5 receptor antagonists have been shown to be independent of any benzodiazepine-like mechanism (Klodzinska et al., 2002; Schwarz et al., 2002) and likely to be independent of GABAergic potentiation (Wieronska et al., 2004; Ballard et al., 2005). Moreover, in contrast to diazepam, MPEP did not impair working memory or spatial learning at anxiolytic doses (Ballard et al., 2005). Fenobam has also been shown to be devoid of side effects such as muscle relaxant, sedation, and interaction with ethanol, which are associated with benzodiazepines, confirming a mechanism of activity by fenobam distinct from GABAergic activity (Goldberg et al., 1983). The non-GABAergic activity of fenobam, coupled with its anxiolytic activity in rodents and human (Itil et al., 1978; Lapierre et al., 1982; Pecknold et al., 1982), supports further the development of mGlu5 antagonists as novel anxiolytic agents with the potential for improved efficacy and side effect profiles over the current mainstay treatments of benzodiazepines and SSRIs.

Rat Geller-Seifter conflict test. Effect of fenobam (10, 30, and 100 mg/kg p.o.) on responding in the Geller-Seifter conflict test. The data are presented according to each component of the test schedule: unpunished (VI30) period (A), nonrewarded (time-out) period (B), and punished (FR10) period (C). n = 8 male Sprague-Dawley rats were used in this study. Data are expressed as the mean ± S.E.M. number of lever presses per minute. *, p < 0.05 versus vehicle (V).

Rat CER test. Effect of fenobam (10, 30, and 100 mg/kg p.o.) on responding in CER test. Data are expressed as mean ± S.E.M. A, suppression ratio. B, total number of lever responses recorded over the test session. n = 12 male Sprague-Dawley rats were used in this study. *, p < 0.05 versus vehicle (V).
Acknowledgments
The excellent technical assistance of Christophe Fisher, Catherine Diener, Jenny Piussi, Marie-Thérèse Zenner, Severine Weil-Bandinelli, and Sean Durkin is acknowledged.
Footnotes
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.105.089839.
-
ABBREVIATIONS: SSRI, serotonin reuptake inhibitor; mGlu, metabotropic glutamate; GPCR, G protein-coupled receptor; TM, transmembrane; MPEP, 2-methyl-6-(phenylethynyl)-pyridine; HTS, high-throughput screening; FLIPR, fluorometric imaging plate reader; fenobam, N-(3-chlorophenyl)-N′-(4,5-dihydro-1-methyl-4-oxo-1H-imidazole-2-yl)urea; MTEP, 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pridine; SIB-1757, 6-methyl-2-(phenylazo)-3-pyridinol); SIB-1893, (E)-2-methyl-6-(2-phenylethenyl)pyridine; NPS 2390, N-adamantan-1-yl-2-quinoxaline-carboxamide-2-quinoxaline-carboxamide-N-adamantan-1-yl; S-4-CPG, S-4-carboxyphenylglycine; HEK, human embryonic kidney; PBS, phosphate-buffered saline; RT, room temperature; IP, inositol phosphates; hGlu, human metabotropic glutamate; [Ca2+]i, intracellular calcium; DMSO, dimethyl sulfoxide; GTPγS, guanosine 5′-O-(3-thio)triphosphate; S-4C3HPG, S-4-carboxy-3-hydrophenylglycine; S-DHPG, S-dihydrophenylglycine; S-3HPG, 3-hydroxyphenylglycine; 2Me4CPG, 2-methyl-4-carboxyphenylglycine; -4-CPG, S-4-carboxyphenylglycine; (+)-MCPG, (+)-α-methyl-4-carboxyphenylglycine; CHPG, RS-2-chloro-5-hydroxyphenylglycine; L-AP4, 2-amino-4-phsophonobutyric acid; CHO, Chinese hamster ovary; RIA, radioimmunoassay; CER, conditioned emotional response; SR, suppression ratio; ANOVA, analysis of variance; PG, phenylglycine; MED, minimum effective dose.
-
↵1 Current address: Addex Pharmaceuticals S.A., Plan Les Ouates, Switzerland.
- Received May 19, 2005.
- Accepted July 19, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}