


Abstract
Targeted drug delivery can significantly influence the efficacy of a drug. In the past decades, diverse drug-delivery technologies, including nano- and microparticles, co-crystals, and microneedles have been developed to maximize therapeutic efficacy and minimize undesired side effects of therapeutics. Nanoparticles—submicron-sized drug carriers—have been actively investigated for the delivery of antibiotics, nucleic acids, peptide/proteins, and chemotherapeutics. Recently, nanoparticles have gained attention as a vaccine delivery platform for tumor-associated antigens (TAAs) and/or vaccine adjuvants. Agonists of imidazoquinoline-based Toll-like receptor (TLR) 7/8 are potent cytokine inducers that are used as cancer vaccine adjuvants to elicit robust T-cell response by activating dendritic cells (DCs). Despite their in vitro potency, the translation of TLR7 agonists as cancer vaccine adjuvants in the clinic has been limited by their poor retention at the injection site. Therefore, a formulation that could improve the availability of TLR7/8 agonists to DCs via conventional vaccine administration routes (subcutaneous, intramuscular) can broaden the application of TLR7/8 agonists for cancer immunotherapy. Polymeric nanoparticles fabricated with poly(d,l-lactide-co-glycolide) (PLGA) can be an efficient TLR7/8 agonist delivery platform. PLGA is a biocompatible polymer, and nanoparticles prepared from this polymer are stable in saline and are small enough to be administered by subcutaneous or intramuscular injections. Furthermore, nanoparticulate TLR7/8 delivery can enhance DC uptake and facilitate lymphatic drainage, both of which can enhance the adjuvanticity of TLR7/8 agonists compared with soluble forms. In this review, we discuss the use of PLGA nanoparticles with TLR7/8 agonists for improving cancer immunotherapy.
Introduction
Cancer immunotherapy is a therapeutic strategy that utilizes the immune cells of the host to kill malignant tumor cells by stimulating immature immune cells and/or providing immune components that are deficient in the host (Rosenberg et al., 2004). Cancer immunotherapy, including monoclonal antibodies, adoptive cell transfer, checkpoint inhibitors, and cancer vaccines, have shown promising results in preclinical and clinical studies (Berzofsky et al., 2004). For example, cancer vaccines gained attention with their ability to mobilize natural killer (NK) cells and CD8 T cells to kill malignant tumor cells (Kawarada et al., 2001). However, vaccines based on tumor-associated antigens (TAAs) alone often failed to elicit NK cell and CD8 T-cell responses (Mellman et al., 2011), pointing to the importance of vaccine adjuvants for enhancing the immunogenicity of cancer vaccines.
Toll-like receptor (TLR) agonists have been examined as vaccine adjuvants in a number of clinical trials, as TLR ligation in dendritic cells (DCs) facilitates TAA presentation and immunostimulatory signals to prime TAA-specific CD8 T cells (Smith et al., 2018). Despite their excellent in vitro adjuvanticity, the in vivo efficacy of TLR agonists is often hampered by rapid clearance from the injection site (Wang et al., 2012), suggesting the need for a formulation that can efficiently deliver the TLR agonist to the optimal anatomic locations (e.g., lymph nodes) to elicit antitumor responses. Polymeric nanoparticles (NPs) have the potential to fulfill this need as the polymeric matrix provides protection from rapid clearance for encapsulated adjuvants (Silva et al., 2013).
In this review, we will introduce current cancer immunotherapeutics, focusing on cancer vaccines. We will also discuss the potential of TLR agonists, especially imidazoquinoline-based synthetic TLR7/8 agonists, as immune adjuvants to potentiate immune response. In addition, limitations of TLR agonists and approaches to overcome these will be covered. Furthermore, the advantages of nanoparticulate vaccine delivery system will be covered, with specific emphasis on poly(lactide-co-glycolide) polymer nanoparticles.
Cancer Immunotherapeutics
Monoclonal Antibodies.
Monoclonal antibodies can play multiple roles in cancer immunotherapy with their ability to block tumor-promoting receptors, activating or inhibiting other immune cells, and inducing antibody-dependent cellular cytotoxicity (ADCC) responses (Weiner et al., 2010). Monoclonal antibodies that target tumor-specific receptors include epidermal growth factor receptor targeting cetuximab and human epidermal growth factor receptor 2 targeting trastuzumab (Robert et al., 2006; Kurai et al., 2007). These antibodies block tumor cell signaling and also elicit ADCC responses to kill the tumor cells. Tumor-angiogenic growth factors, including vascular endothelial growth factors and platelet-derived growth factor, can be inhibited by bevacizumab (Willett et al., 2004). Monoclonal antibodies that target leukemia/lymphoma-related molecules, including CD20, CD30, CD33, and CD52, are also approved therapeutics (Scott et al., 2012).
Checkpoint Inhibitors.
Checkpoint inhibitors promote antitumor responses by neutralizing the inhibitory signals that deactivate T cells and NK cells (Zou and Chen, 2008). Cytotoxic T-lymphocyte antigen 4 (CTLA4) is a homolog of T-cell-activating molecule CD28 (Buchbinder and Desai, 2016). CTLA4 is expressed on CD8 T cells and can be activated via costimulatory molecules CD80 and CD86 of DCs and macrophages. Therefore, anti-CTLA4 antibodies can block the inhibitory CTLA4 and enhance CD8 T-cell-stimulatory CD28-CD80/86 signaling, which leads to activation of effector CD8 T cells. The anti-CTLA4 antibody ipilimumab is approved for melanoma (Margolin et al., 2012) and is being evaluated for other cancers, including hormone-refractory prostate cancer and lung cancer (Small et al., 2007; Reck et al., 2013).
Programmed death-1 (PD-1) and programmed death ligand-1 (PD-L1) are also major checkpoint proteins that suppress T-cell activation. PD-L1 is expressed on tumor cells, monocytes, macrophages, and myeloid-derived suppressive cells (MDSCs). PD-L1 ligates PD-1 expressed on CD8 T cells and NK cells, and causes exhaustion of these cells (Zhao and Subramanian, 2017). Upregulation of PD-L1 on solid tumors reduces tumor infiltration by T cells and is associated with poor prognosis for solid tumors, which has led to the use of PD-L1 as a biomarker for cancer immunotherapy (Chen and Han, 2015; Patel and Kurzrock, 2015). By 2018, the anti-PD-1 antibodies nivolumab and pembrolizumab were approved for the treatment of melanoma, bladder cancer, and non–small-cell lung cancer (Rizvi et al., 2015; Rosenberg et al., 2016; Sharma et al., 2017). Anti-PD-L1 antibodies avelumab and durvalumab were approved as bladder cancer treatments (Apolo et al., 2017; Balar et al., 2017).
Adoptive Cell Transfer.
Adoptive cell transfer (ACT) therapies use ex vivo-stimulated effector cells to fight tumors. The first ACT to be approved for cancer therapy was sipuleucel-T (Provenge). DCs are purified from the blood of the cancer patient and then stimulated in vitro with granulocyte-macrophage colony stimulating factor and pulsed with prostatic acid phosphatase (PAP) antigen. The prostate tumor antigen-laden DCs are then returned to the patient to trigger a PAP-specific CD8 T-cell response to kill hormone-refractory prostate cancer cells (Small et al., 2006). Other ACT strategies include infusion of ex vivo-stimulated T cells isolated from the tumors of patients (Restifo et al., 2012). Engineered T cells that express a chimeric antigen receptor (CAR) or defined T-cell receptor are also used in ACT-based cancer immunotherapy (Rosenberg et al., 2008). Currently, CAR T cells specific for CD19 expressed on B-cell cancers have been approved for treatment of acute lymphoblastic leukemia and diffuse large B-cell lymphoma (Kochenderfer et al., 2015; Lee et al., 2015).
Cancer Vaccine Strategies and Challenges
Cancer vaccines composed of TAAs and vaccine adjuvant aim to mobilize cytotoxic CD8 T cells to kill malignant tumor cells. To elicit a tumor-specific CD8 T-cell response, maturation of DCs is the required first step (Lanzavecchia and Sallusto, 2001). DCs process and present the TAA to CD8 T cells, along with the necessary co-stimulatory molecules and proinflammatory cytokines that trigger the activation of TAA-specific CD8 T cells and NK cells (Kawarada et al., 2001). Activated CD8 T cells and NK cells can kill tumor cells via perforin/granzyme, tumor necrosis factor alpha–related apoptosis-inducing ligand (TRAIL), and Fas ligand; they also secrete proinflammatory cytokines that further activate other DCs, T cells, and NK cells (Martínez-Lostao et al., 2015). The mechanism of action for a typical cancer vaccine is illustrated in Fig. 1.

Cancer vaccine strategy scheme. Combination of TAA and vaccine adjuvant triggers DC activation. DCs present the TAA epitope to CD8 T cells via major histocompatibility complex I and provide co-stimulatory molecule signaling (CD40, CD80/86) and proinflammatory cytokines (IL-2,12,15,18, IFNα, β). Activated T cells further activate other effector immune cells to directly kill the tumor cells.
Autologous tumor cells collected from patients were used in the first cancer vaccine. One benefit of using autologous tumor cells is the broad repertoire of TAAs specific for the individual patient (Berd et al., 1990). Allogeneic tumor cell vaccines, which are mixtures of established immunogenic tumor cell lines, are also being evaluated for the treatment of melanoma and non–small-cell lung cancer (Sondak and Sosman, 2003). However, owing to the limited availability of patients’ tissue samples and the intricate procedures needed to prepare tumor cell vaccines, TAA-derived peptide/proteins are widely investigated. TAA peptide/proteins can be classified to several types, including oncofetal antigens (McClintock et al., 2015), oncoviral antigens, antigens that are overexpressed or accumulated in the tumor (de Vries et al., 1997), mutated antigens (Scanlan et al., 2002), and shared tumor-specific antigens (Shinozaki et al., 2004). TAA peptide/proteins are cost-effective and can elicit TAA-specific T-cell response compared with tumor cell-based vaccines, which make them a promising antigen source for cancer immunotherapy (Nestle et al., 1998).
However, tumor cells and peptide/protein TAAs are often not immunogenic enough to trigger a T-cell response, pointing to the necessity of including an immunostimulant to enhance the immunogenicity of a cancer vaccine (Pullen et al., 1989). Several approaches, including irradiation of the tumor cells, and coincubation with stimulatory cytokines IL-12 (Lehner et al., 2007) and granulocyte-macrophage colony-stimulating factor (Simons and Sacks, 2006), have been investigated to enhance the immunogenicity of cancer vaccines. Additionally, TLR agonists (Napolitani et al., 2005) have also been actively examined in the clinical trials as vaccine adjuvants for peptide/protein vaccines since they can potentiate the expression of co-stimulatory molecules and proinflammatory cytokines by the DCs.
TLR Signaling Promotes Activation of Antigen-Presenting Cells
The host immune system detects pathogens via several mechanisms. TLRs are pattern-recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) (Reis e Sousa, 2004). TLRs are mainly expressed by antigen-presenting cells, including DCs, macrophages, and B cells, but mast cells, monocytes, and epithelial cells can also express TLRs. Expression of TLRs varies among species and TLRs 1 through 10 are reported in humans (Reis e Sousa, 2004). TLRs 1, 2, 4, 6, and 10 are located on the cell membrane, while TLRs 3, 7, 8, and 9 are located in the intracellular endo/lysosomes. Ligands for TLRs can be divided to three categories: TLRs 1, 2, 4, and 6 recognize lipids, TLR5 recognizes proteins, and TLRs 3, 7, 8, and 9 recognize nucleic acids.
Upon ligation, TLRs 1, 2, 5, 6, 7, 8, and 9 activate myeloid differentiation primary-response gene 88 (MyD88), and TLRs 3 and 4 activate Toll/IL-1R (TIR) domain-containing adaptor protein inducing IFNβ (TRIF) signaling pathways (Gilliet et al., 2008). MyD88 signaling activates interferon-regulatory factor (IRF)7, nuclear factor-kappa B (NF-κB), and mitogen-activated protein kinases (MAPKs) that result in the production of multiple interferon (IFN) type I signals, including IFNα, IFNβ, IFNλ, and IFNω. Furthermore, MyD88 signaling upregulates co-stimulatory molecules CD40, CD80, and CD86 and triggers the secretion of proinflammatory cytokines such as IL-1β, IL-12, IL-18, tumor necrosis factor (TNF)-α, and IFN-γ. In addition to NF-κB and MAPKs, TRIF signaling activates IRF3 to trigger IFN and co-stimulatory responses.
With these aspects, several TLR agonists have been examined as immunostimulatory adjuvants. TLR2 agonist (polysaccharide krestin), TLR3 agonist (polyriboinosinic-polyribocytidylic acid–polylysine carboxymethylcellulose), TLR4 agonist (lipopolysaccharide), and TLR9 agonist (CpG oligodeoxynucleotide) combined with TAAs, including NY-SEO-1, MUC-1, and MART1, have been investigated in preclinical studies and clinical trials (Torisu et al., 1990; Didierlaurent et al., 2009; Sabbatini et al., 2012; Zent et al., 2012). In these studies, TLR agonist-based cancer vaccine-treated groups showed enhanced IFN-γ + CD8 T-cell responses and antigen-specific cytotoxic T-cell responses. Upregulation of innate immune response was also observed, which demonstrates the potent adjuvanticity of TLR agonists for cancer immunotherapy.
TLR7/8 Agonists for Cancer Immunotherapy
TLRs 7 and 8 are intracellular receptors and recognize single-stranded RNA and activate NF-κB signaling. However, recent studies report that although TLR7 and TLR8 recognize similar molecular patterns, they are functionally different (Larange et al., 2009). Whereas TLR7 is mainly expressed on plasmacytoid DCs (pDCs) and induce type I IFN secretion, TLR8 is expressed mainly on myeloid DCs (mDCs) and potentiates TNF-α and IFN-γ response, suggesting a TLR7/8 mixed agonist can stimulate stronger Th1 immunity than either TLR7- or 8-specific agonists. Previous studies report that stimulating both plasmacytoid and CD8α DCs is required to elicit strong CD8 T-cell responses (James et al., 2014), which further implies the advantage of utilizing TLR7/8 agonist as immunostimulatory adjuvants for cancer immunotherapy.
Synthetic imidazoquinoline derivatives are potent TLR7- or 8-specific or 7/8 bi-specific agonists (Wang et al., 2010). The TLR7-selective agonist imiquimod was first approved by the US Food and Drug Administration (FDA) in 1997 for the treatment of basal and squamous cell carcinoma and genital warts as a single agent (Stanley, 2002). Imiquimod activates pDCs and macrophages to promote proinflammatory cytokine production, including type I IFN, TNF-α, and IL-12 (Gibson et al., 2002). These cytokines activate CD4 T cells, CD8 T cells, and NK cells and promote their effector function, such as killing tumor cells (Durand et al., 2004; Iwasaki and Medzhitov, 2004). Imiquimod can also directly induce apoptosis of TLR7-expressing tumors (Schön et al., 2003). These findings suggest that imidazoquinoline-based small molecules can foster a potent adaptive immune response, which is important for cancer treatment.
Several groups have reported the synthesis of imidazoquinoline derivatives and demonstrated that the TLR7 and/or 8 activities are associated with the C2-alyl chain length of the imidazoquinoline structure, where butyl and pentyl derivatives potentiate TLRs 7 and 8, respectively (Schiaffo et al., 2014). Recently, it was shown that TLRs 7 and 8 activities can be selectively achieved by N1 modification of imidazoquinoline compounds (Larson et al., 2017). TLR7/8 agonists induce the production of type 1 interferons and proinflammatory cytokines from both murine BMDCs and human PBMCs.
Nanoparticulate Vaccine Delivery
Nanoparticles have emerged as a suitable drug delivery platform for several conventional drugs to overcome limitations in pharmacokinetics and target bioavailability (Blanco et al., 2015). Polymeric NPs and liposomes have been investigated to encapsulate payloads, including drugs, proteins, vectors, and nucleic acids (Kumari et al., 2010). Discovery of the enhanced permeability and retention effect (Fang et al., 2011), which explains enhanced accumulation of macromolecules via extravasation through leaky blood vessels in the tumor, triggered extensive investigation of nanoparticles for passive targeting of solid tumors. These studies led to the development of FDA-approved nanomedicines liposomal doxorubicin (Doxil) and albumin-bound paclitaxel (Abraxane) (Micha et al., 2006; Barenholz, 2012).
Recent studies suggest that NPs are also an efficient vaccine delivery platform that can enhance the efficacy of cancer vaccines compared with conventional delivery of antigens and/or vaccine adjuvants. Encapsulation of peptide antigens in polymeric nanoparticles (Akagi et al., 2011; Xiang et al., 2015), liposomes (Chen and Huang, 2008; Vasievich et al., 2012), and dendrimers (Lu et al., 2015) results in enhanced antibody and T-cell responses compared with soluble forms of antigens. Inorganic materials, including iron oxide (Shen et al., 2011), graphene oxide (Xu et al., 2016), aluminum hydroxide (Li et al., 2014), zinc oxide (Afroz et al., 2017), and silicon dioxide (Mahony et al., 2013), have also been examined as peptide delivery platforms and have shown enhanced immune responses with negligible toxicity. In addition to peptide-based antigens, whole tumor cell lysates encapsulated in chitosan (Shi et al., 2017a) and other polymeric NPs (Liu et al., 2013; Huang et al., 2015) trigger stronger antitumor efficacy compared with soluble form of cell lysates. Interestingly, coating NPs with the cell lysates, rather than encapsulating them within, has also been shown to be effective (Fang et al., 2014; Liu et al., 2016). In addition to antigen delivery, NPs have been examined for adjuvant delivery. NPs encapsulating TLR agonists, including poly I:C (TLR3) (Bocanegra Gondan et al., 2018), lipopolysaccharide (TLR4) (Sarti et al., 2011; Mulens-Arias et al., 2015), imidazoquinoline compounds (TLR7/8) (Lynn et al., 2015; Kim et al., 2018a), and CpG ODN (TLR9) (Wei et al., 2012) were examined to determine the advantages of nanoparticulate delivery of TLR agonists. De Titta et al. (2013) reported that encapsulation of CpG ODN in an NP formulation resulted in greater DC uptake and maturation, in vivo T-cell activation, and enhanced antitumor therapeutic efficacy compared with soluble CpG ODN (Fig. 2).

Nanoparticulate vaccine delivery provokes stronger immune response than soluble vaccine. (A) CpG ODN nanoparticle (NP-CpG-B) formulations enhance DC uptake of CpG ODN and enhance CD86 expression on DCs. (B) CpG-NPs augmented cytotoxic (CD107a+/IFN-γ +) CD8 T cells more than CpG-B (free) (C) CpG-NP immunization showed antitumor efficacy superior to soluble CpG. Reprinted from de Titta et al. (2013) with permission from National Academy of Sciences. **P < 0.01; ***P < 0.001.
Other immunostimulants, including stimulator of interferon gene agonists (An et al., 2018; Collier et al., 2018) and small-interfering RNA (Jadidi-Niaragh et al., 2017), have demonstrated stronger antitumor responses in NP formulations. Furthermore, it has been reported that co-delivery of antigen and adjuvant to DCs simultaneously is critical to triggering CD8 T-cell response (Berzofsky et al., 2004; Shi et al., 2017b). NPs that can co-deliver antigen and adjuvants in a single formulation significantly enhanced antigen-specific T-cell responses compared with the separate delivery of antigen and adjuvants (Tao et al., 2015; Neek et al., 2018). These studies suggest encapsulation of antigens and/or adjuvants in NPs can protect the encapsulated payload from degradation and enhance delivery to DCs, which results in improved T-cell response.
In addition to their protective function, NPs can be modified to target DCs. NPs targeting receptors highly expressed on DCs, including CD40 (Rosalia et al., 2015), mannose receptor (Yang et al., 2018), DC-SIGN (Arosio et al., 2014), and DEC-205 (Saluja et al., 2014), have shown stronger DC activation potency than nontargeted nanoparticles in preclinical studies. pH-responsive NPs that enhance endo/lysosomal delivery of the antigen (Liu et al., 2015) and TLR agonist (Kim et al., 2018b) are also efficient vaccine delivery platforms that can elicit stronger T-cell responses than conventional NPs.
Although most NP formulations tested to date have been delivered via subcutaneous and intramuscular routes, some studies have also reported the application of NPs for intranasal (Bae et al., 2016; Bailey et al., 2017) or oral vaccine delivery (Borges et al., 2006; Garinot et al., 2007; Jiang et al., 2014).
PLGA NPs as a Delivery Platform for TLR7/8 Agonists
PLGA is an FDA-approved polymer that has a well established biocompatibility and safety profile (Dinarvand et al., 2011). Upon contact with aqueous solutions, PLGA degrades by hydrolysis into lactic acid and glycolic acid, which can be further metabolized to carbon dioxide and water (Makadia and Siegel, 2011). Particle size and drug release kinetics of PLGA NPs can be fine-tuned by modulating the properties of PLGA, including the molar ratio of lactide to glycolide and molecular weight, and/or addition of various terminal functional groups (Astete and Sabliov, 2006). Additionally, targeting moieties can be easily incorporated onto PLGA NPs by surface modification using terminal functional groups. Nano- (Kim et al., 2018a) and microparticles (Ke et al., 2011) fabricated with PLGA are generally spherical in shape as shown at Fig. 3.

Nano- and microparticles fabricated with PLGA. (A) Transmission electron microscopy image of nanoparticles fabricated with PLGA. Scale bar, 50 nm. Reprinted from Kim et al (2018a) “Polymeric nanoparticles encapsulating novel TLR7/8 agonists as immunostimulatory adjuvants for enhanced cancer immunotherapy.” Biomaterials 164 (2018): 38–53. with permission from Elsevier. (B) Scanning electron microscopy (SEM) image of microparticles fabricated with PLGA. Scale bar, 5 μm. Reprinted from Ke et al. (2011) with permission from John Wiley and Sons.
PLGA NP Enhances DC Uptake of Antigen and Adjuvant.
DC uptake of antigens and/or immunostimulatory adjuvant is critical to eliciting T-cell immunity. Previous studies report that size, surface charge, hydrophobicity, and shape significantly influence DC uptake of macromolecules (Benne et al., 2016). Among the various factors, the size of the particles plays a critical role as NPs have shown superior DC internalization compared with microparticles (Hamdy et al., 2011). Submicron (20–200 nm)-sized NPs can be efficiently internalized by DCs via clathrin- and caveolae-dependent endocytosis pathways (Bachmann and Jennings, 2010). However, endocytosis of micron-sized particles is mainly facilitated via phagocytosis. PLGA NPs have been shown to be internalized by in vitro-generated DCs, including human peripheral blood monocyte-derived DCs, human cord-blood CD34+ DCs, and murine bone marrow-derived DCs (BMDCs) (Foged et al., 2005). Efficient DC uptake of PLGA NPs is particularly advantageous for delivery of endosomal TLR agonists. TLRs 3, 7, 8, and 9 are located at the luminal side of endo/lysosome, which implies that these TLR agonists have to cross the cellular membrane and internalize into endo/lysosomes to ligate the TLR. Upon endocytosis, PLGA NPs enter endo/lysosome (Panyam and Labhasetwar, 2003), the target site for TLRs 3, 7, 8, and 9 ligation. These features suggest PLGA NPs are efficient delivery vehicles for TLR agonists.
PLGA NP Is an Efficient In Vivo Drug Delivery Platform.
The in vivo efficacy of cancer vaccines can be hampered by rapid clearance of the immunogen from the injection site (Swartz, 2001; Wang et al., 2012), leading to insufficient recognition by immune cells in lymphoid organs and suboptimal therapeutic efficacy. Subcutaneous (SC) injection is a conventional vaccine administration route, as vaccine components need to migrate to draining lymph nodes to stimulate antigen-presenting cells and cytotoxic cells (Swartz, 2001). SC administration delivers the vaccine components to the interstitial space of hypodermis composed of adipocytes, fibroblast, collagen and, glycosaminoglycans (Supersaxo et al., 1990). PLGA NPs can protect encapsulated payloads from biodegradation at the injection site, which is particularly critical for peptide/protein-based vaccines. Previous studies also report that peptide/proteins encapsulated in polymeric NPs maintain their activity in biologic fluids longer than the soluble form (Panyam et al., 2003; Kumari et al., 2010).
Upon SC injection, vaccine components can enter the blood circulation or the lymphatic system. Although blood capillaries are tightly structured, lymphatic vessels are relatively more permeable as they lack interendothelial tight junctions (Bachmann and Jennings, 2010). Owing to the permeability difference between capillaries and lymphatics, large molecules and NPs have limited capacity to enter the systemic circulation directly, and preferably enter the lymphatic system. Molecules with a molecular weight of ≤16 kDa and/or a size of ≤10 nm preferentially enter the systemic circulation via blood capillaries following SC injection (Supersaxo et al., 1990; Wang et al., 2012). In a previous study (Manolova et al., 2008), NPs (∼20 nm in diameter) were detected in the popliteal lymph nodes within 2 hours of SC injection into the footpad, whereas a negligible concentration of microparticles (∼1000 nm) was detected (Fig. 4A).
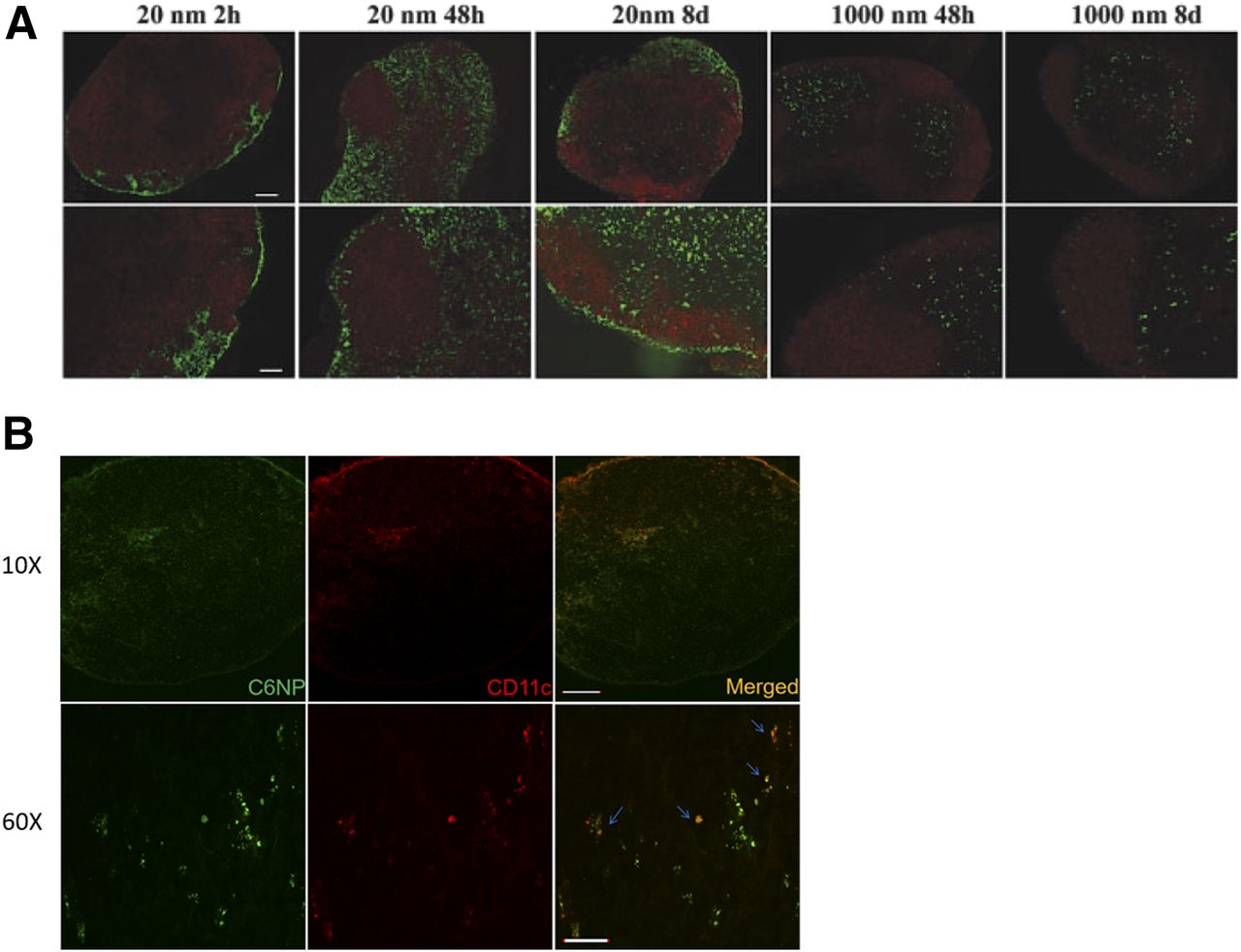
Nanoparticles are an efficient lymphatic delivery platform. (A) Confocal images of popliteal lymph nodes after footpad injection of particles. Green fluorescent polystyrene nanoparticles (20 nm) and microparticles (1000 nm) are shown with red fluorescent B-cell follicles (B220+ cells). Scale bar, 100 μm. Reprinted from Manolova et al. (2008) with permission from John Wiley and Sons. (B) Confocal images of inguinal lymph nodes after subcutaneous injection of nanoparticles in the abdomen of mice are shown. Green fluorescence, nanoparticles; red fluorescence, CD11c+ DCs. Scale bar, 200 μm (original magnification, 10×), 20 μm (original magnification, 60×). Reprinted from Kim et al (2018a) “Polymeric nanoparticles encapsulating novel TLR7/8 agonists as immunostimulatory adjuvants for enhanced cancer immunotherapy.” Biomaterials 164 (2018): 38–53. with permission from Elsevier.
Previous studies report that SC-injected macromolecules can directly migrate to lymph nodes via paracellular uptake or a DC-meditated mechanism (Manolova et al., 2008; Jeanbart et al., 2014). Macromolecules with a size of 100–200 nm can directly enter the lymphatic system within hours of injection (Manolova et al., 2008). On the other hand, macromolecules with a size of 200–500 nm can be transported by DCs to the lymphatic system. PLGA NPs with a diameter of 200 nm efficiently traffic to the inguinal lymph nodes and co-localized with CD11c+ DCs (Fig. 4B) (Kim et al., 2018b). These findings suggest rapid clearance into systemic circulation following SC injection can be reduced by nanoparticulate delivery of vaccine components.
PLGA NP Allows Co-Delivery of Antigen and TLR7/8 Agonist.
Simultaneous co-stimulatory molecule signaling and antigen-presentation by DCs is required to elicit an antigen-specific T-cell response, as absence of co-stimulatory signaling fails to activate T cells (Kovacsovics-Bankowski et al., 1993). Therefore, co-delivery of antigen and vaccine adjuvant is critical to eliciting an antigen-specific T-cell response (Diwan et al., 2002). Co-delivery of TAA and synthetic TLR7/8 agonist, however, is challenging. Although many TLR agonists (including polyIC:LC and CpG ODN are soluble in aqueous buffers, synthetic TLR7/8 agonists are soluble in organic solvents. This solubility issue limits TLR7/8 agonist formulations to topical creams and gels (Duong et al., 2013; Rook et al., 2015). Consequently, application of synthetic TLR7/8 agonists is limited to topical treatments (ClinicalTrials.gov, NCT01676831, NCT01808950) and intratumoral injection (NCT02556463). In clinical studies where topical treatment of TLR7/8 agonists were used as vaccine adjuvants (NCT01748747, NCT00960752), peptide/protein-based TAAs were administered separately via SC injection. These features demonstrate the need for a formulation that can deliver TLR7/8 agonists via SC or intramuscular injection together with TAA.
Encapsulation of TLR7/8 agonists in PLGA NPs can overcome this solubility limitation. PLGA NPs can encapsulate both hydrophilic and hydrophobic compounds (Makadia and Siegel, 2011). Therefore, imidazoquinoline-based synthetic TLR7/8 agonists, which are slightly hydrophobic small molecules, can be loaded in PLGA NPs using emulsification solvent evaporation methods (Astete and Sabliov, 2006). As PLGA NPs can be stably dispersed in aqueous buffers, encapsulated TLR7/8 agonists can be delivered via conventional vaccine administration routes, including SC and intramuscular injection, which allows co-delivery of peptide/protein-based TAA and TLR7/8 agonists. Furthermore, systemic delivery of nanoparticulate TLR agonists via intravenous injection can be efficient for specific tumor models (Nierkens et al., 2009; Kranz et al., 2016).
Combination Therapy with TLR7/8 Agonists
Modulating Tumor Microenvironment to Enhance TLR7/8 Agonist-Based Vaccine.
In addition to proinflammatory cytokine induction, TLR agonists also trigger the secretion of IL-10, which is a strong immunosuppressive cytokine. IL-10 can promote tumor growth by directly inhibiting the function of effector T cells and NK cells, and by inducing the activation of immunosuppressive cells, including MDSCs, regulatory T cells (Tregs), and M2 macrophages (Zou, 2005; Zitvogel et al., 2006).
Several approaches were examined previously to modulate immune-suppressive mechanisms triggered by TLR agonists. Blockade of IL-10 using anti-IL-10 antibodies can restore the CD8 T-cell functions and enhance the antitumor efficacy of TLR7 agonist-based vaccines (Lu et al., 2010; Llopiz et al., 2017). Co-treatment with agents that reduce MDSCs and Tregs, including cyclophosphamide (Dumitru et al., 2010; Dewan et al., 2012) and tyrosine kinase inhibitors sorafenib (Ho et al., 2015), lapatinib (Gao et al., 2016), and sunitinib (Xin et al., 2009; Yin et al., 2015) can enhance cancer immunotherapy. Docetaxel (Kodumudi et al., 2010), gemcitabine (Liu et al., 2010), and paclitaxel (Ramakrishnan et al., 2010) are also potential agents that can synergize with TLR7/8 agonist-based vaccines.
IFN-γ can induce the expression of programmed death ligand 1 (PD-L1) on tumor cells (Abiko et al., 2015). Thus, TLR agonist-driven expansion of IFN-γ + CD8 T cells can be simultaneously restricted by increased expression of PD-L1, pointing to the need for blocking PD-1/PD-L1 interactions. To neutralize the PD-L1/PD-1 signaling, combining checkpoint inhibitors (e.g., anti-PD-1 or -PD-L1 antibodies) with TLR7/8 agonists restored CD8 T-cell activity, resulting in effective tumor regression (Weir et al., 2016; Sato-Kaneko et al., 2017; Nishii et al., 2018). As PD-L1 is also expressed by MDSCs (Lu et al., 2016; Davis et al., 2017), combination of checkpoint inhibitors and MDSC-eliminating reagents can be expected to further enhance the therapeutic efficacy of TLR7/8 agonist vaccines.
Cotreatment of TLR7/8 Agonists and Chemotherapeutics.
In addition to potentiating T-cell responses, TLR7/8 agonists can facilitate NK cell activation by cytokine production and direct TLR ligation on NK cells (Gorski et al., 2006; Zhou et al., 2015), suggesting TLR7/8 agonists can be potent NK-cell stimulants for antibody-based cancer immunotherapies (Lu et al., 2012). The combination of TLR7 agonist and photodynamic and laser therapy has also shown synergistic effects against lentigo maligma (De Vries et al., 2013) and basal cell carcinoma (Osiecka et al., 2012). Likewise, a synergistic effect was reported for the combination of TLR7 agonist and radiation therapy against breast cancer (Dewan et al., 2012; Demaria et al., 2013) and lymphoma (Dovedi et al., 2013, 2016). The combination of TLR7 agonists with chemotherapy, including 5-fluorouracil (Smith et al., 2001b), paclitaxel (Salazar et al., 2017), and sulindac (Smith et al., 2001a), have been also examined. A common feature of these combination therapies is the presence of a cell death-inducing component (ADCC, photodynamic/laser therapy, radiation, chemotherapy) along with proinflammatory TLR7/8 agonist.
Other Features of TLR7/8 Agonists.
Several studies have reported the ability of TLR7/8 agonists to repolarize immune suppressive cells into immunostimulatory cells. TLR7/8 ligation promotes MDSC differentiation into DCs and M1 macrophages (Lee et al., 2014). Systemic administration of TLR7 agonist reduced the function of intratumoral MDSCs, leading to enhanced CD8 T-cell functions (Spinetti et al., 2016). Additionally, TLR7/8 agonist facilitated the polarization of tumor-promoting M2 macrophages into tumor-destructive M1 macrophages (Rodell et al., 2018). Together, these studies suggest that TLR7/8 agonists alone can contribute to cancer immunotherapy by modulating immune suppressive cells. Interestingly, there is also data to suggest that TLR7 agonists can inhibit tumor angiogenesis (Sidbury et al., 2003; Majewski et al., 2005). TLR7 treatment was shown to decrease tumor proliferation, increase the expression of tissue inhibitor of matrix metalloproteinase (MMP)-1, and decrease the activity of MMP-9.
Perspectives
TLR7/8 agonists can play multiple roles in cancer immunotherapy. As we have described, TLR7/8 agonists are potent immunostimulatory vaccine adjuvants for cancer vaccines and can synergize with conventional cancer therapies, including chemotherapy, radiation therapy, laser therapy, and surgery. Additionally, TLR7/8 agonist monotherapy has been shown to have anticancer effects. This review discussed the use of PLGA NPs as carriers for TLR7/8 agonists. PLGA is biocompatible, exhibits negligible toxicity, and its properties can be varied to fine-tune the physiochemical properties of NPs. NPs can efficiently migrate to the draining lymph nodes and enhance endo/lysosomal delivery of the payload, which is particularly desirable for TLR7/8 agonists. Further studies investigating mechanisms that promote immune activation and long-term memory response over immunosuppression as well those evaluating the safety of NP-encapsulated TLR7/8 agonist will advance their clinical use as effective anticancer vaccine adjuvants.
Authorship Contributions
Participated in research design: Kim, Panyam.
Wrote or contributed to the writing of the manuscript: Kim, Griffith, Panyam.
Footnotes
- Received November 17, 2018.
- Accepted January 3, 2019.
Abbreviations
- CpG ODN
- CpG Oligodeoxynucleotide
- DC
- dendritic cell
- IFN
- interferon
- MDSC
- myeloid-derived suppressor cell
- NF-κB
- nuclear factor kappa B
- NK cell
- natural killer cell
- NP
- nanoparticle
- PD-1
- programmed death-1
- PD-L1
- programmed death-ligand 1
- PLGA
- poly(d,l-lactide-co-glycolide)
- SC
- subcutaneous(ly)
- TAA
- tumor-associated antigen
- TLR
- Toll-like receptor
- TNF
- tumor necrosis factor
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Cancer Immunotherapeutics
- Cancer Vaccine Strategies and Challenges
- TLR Signaling Promotes Activation of Antigen-Presenting Cells
- TLR7/8 Agonists for Cancer Immunotherapy
- Nanoparticulate Vaccine Delivery
- PLGA NPs as a Delivery Platform for TLR7/8 Agonists
- Combination Therapy with TLR7/8 Agonists
- Perspectives
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters