


Abstract
Cellular vesicles (CVs) have been proposed as alternatives to exosomes for targeted drug delivery. CVs, prepared from human embryonic kidney 293 cells (HEK-293), C57BL/6 mouse B16F10 skin melanoma cells (B16F10), and immortalized human cerebral microvascular endothelial cells (hCMEC/D3) by liposome technology methods, were characterized for morphology, cytotoxicity, and cell uptake properties. CV brain-targeting potential was evaluated in vitro on the hCMEC/D3 blood-brain barrier (BBB) model, and in vivo/ex vivo. CV sizes were between 135 and 285 nm, and the ζ-potential was negative. The dehydration-rehydration method conferred highest calcein loading and latency to CVs compared with other methods. The increased calcein leakage from CVs when compared with liposomes indicated their poor integrity, which was increased by pegylation. The in vivo results confirmed lower liver uptake by PEG-CVs (compared with nonpegylated) proving that the calcein integrity test is useful for prediction of CV biodistribution, as used for liposomes. The cell uptake of homologous origin CVs was not always higher compared with that of non-homologous. Nevertheless, CVs from hCMEC/D3 demonstrated the highest BBB permeability (in vitro) compared with OX-26 targeted liposomes, and brain localization (in vivo). CVs from hCMEC/D3 cells grown in different media demonstrated decreased interaction with brain cells and brain localization. Significant differences in proteome of the two latter CV types were identified by proteomics, suggesting a potential methodology for identification of organotropism-determining CV components.
Introduction
Liposomes are known for their applications as efficient drug carriers (Allen and Cullis, 2013; Farjadian et al., 2019). Currently several liposomal drug products are available for clinical use, and others are undergoing clinical testing (Akhter et al., 2018; Belfiore et al., 2018; Rosenblum et al., 2018). The therapeutic advantages of all liposomal drugs currently available in the clinic (compared with corresponding free drugs) are attributed to their modified pharmacokinetics; indeed, although active targeting of liposomes has been under extensive exploration for more than 30 years, no ligand-targeted liposome (or other nanoparticle) product has been realized to date, posing a persistent bottleneck in drug delivery.
Recent knowledge about extracellular vesicles in regards to their intercellular communication pathways and their specific organotropic behavior has opened new and exciting horizons (Kooijmans et al., 2012; Van Dommelen et al., 2012; Aryani and Denecke, 2016). Besides local cell-to-cell communication (Maia et al., 2018; Meldolesi, 2018), exosomes play key roles in interactions between cells located far apart (Hoshino et al., 2015; Becker et al., 2016; Fu et al., 2016; Peinado et al., 2017). The tremendously high organotropism of specific exosome types is in fact the major unmet goal in the ligand-targeted liposome field (Rosenblum et al., 2018). The complementarily of these two systems has initiated a novel, fast-growing research field concerned with extracellular vesicles for drug delivery. This field involves the design of targeted drug carriers after identifying the key elements that dictate the biologic fate of extracellular vesicles and, more specifically, their ability to preferably interact with specific cells (Johnsen et al., 2014; Vader et al., 2016; Antimisiaris et al., 2018).
However, one of the limitations toward rapid evolution and translation of exosomes into therapeutic products is the highly cumbersome and low-yield methodology for their isolation (Heinemann et al., 2014; Zeringer et al., 2015). To overcome the later drawback, the use of whole cells has been proposed as an alternative. In fact, cellular vesicles (CVs) are reported to possess several advantages compared with exosomes, the main advantage being their significantly higher production yield (Jang et al., 2013; Jo et al., 2014; Yoon et al., 2015; Lunavat et al., 2016; Goh et al., 2017a,b; Wu et al., 2018).
Several key points, currently unresolved, will potentially contribute to and accelerate the exploitation of CVs towards development of more efficient targeted (artificial or biologic) nanocarriers. Our current study explored the following aspects:
The particular role of parental cells used for CV isolation on their potential to facilitate delivery of their contents into cells (compared with liposomes). Although numerous studies have reported significantly higher cell uptake of CV- or exosome-associated drugs compared with liposome-associated ones, it is not clear whether CVs derived from specific parent cells will always demonstrate increased interaction toward the same cells compared with other cell types.
The potential to load CVs with drugs by applying the dehydration-rehydration vesicle (DRV) technique. This method achieves high loading of sensitive materials into liposomes (Antimisiaris, 2017) but has never been tested for exosome or CV loading.
The utility of the calcein-latency method for evaluation of CV integrity. This method, which provides important information about liposome integrity during their incubation in any medium (Kokkona et al., 2000), could perhaps serve the same purpose for CVs by acting as a useful tool for prediction of their in vivo fate.
The potential of brain endothelial cell–derived CVs to target the brain (Poller et al., 2008), as compared with CVs from other cells and targeted liposomes (Johnsen et al., 2018).
To explore these points, we constructed CVs from three cell types: C57BL/6 mouse B16F10 skin melanoma cells (B16F10), human embryonic kidney 293 cells (HEK-293), and immortalized human cerebral microvascular endothelial cells (hCMEC/D3).
Materials and Methods
We purchased 1,2-distearoyl-sn-glycerol-3-phosphatidylcholine (PC), 1,2-distearoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt) (PG), 1,2-distearoyl-sn-glycerol-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (PEG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (PEG-MAL), and lissamine rhodamine B phosphatidylethanolamine (RHO) from Avanti Polar Lipids (Alabaster, AL). Cholesterol (99%) (Chol), Triton X-100, calcein, fluorescein-isothiocyanate-dextran-4000 (FITC), calcein, lucifer yellow-CH dilithium salt (LY), and Sephadex G-50 were obtained from Sigma-Aldrich (Darmstadt, Germany). Lipophilic tracer, 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR), which was used as the lipid-label in CVs for live animal imaging, was form Molecular Probes (Eugene, OR). Mouse anti-rat CD71 IgG2a (clone OX-26) was obtained from Serotec (Kidlington, United Kingdom). Protein concentrations were measured by Bradford Micro Assay (Bio-Rad Laboratories, Hercules, CA). All other chemicals were of analytical quality and were purchased from Sigma-Aldrich or Merck (Darmstadt, Germany).
The fluorescence intensity (FI) of samples was measured with a Shimatzu RF-1501 spectrofluorometer (Shimatzu, Kyoto, Japan) using EX-540/EM-590 nm for RHO detection or EX-490 nm/EM-525 nm for FITC or calcein detection; in all cases 5-nm slits were used. A bath sonicator (Branson; Thermo Fisher Scientific, Waltham, MA) and microtip-probe sonicator (Sonics and Materials, Harborough, United Kingdom) were used for liposome and for CV preparation.
Preparation of Liposomes
Liposomes (LIP) composed of PC/Chol (2/1 mol/mol) and PC/PG/Chol (8.5/1.5/5 mol/mol), and pegylated liposomes (PEG-LIP) composed of PC/PG/Chol/PEG (7.9/1.5/5/0.6 mol/mol) were prepared by the thin-film hydration method (Markoutsa et al., 2014). The thin lipid film was hydrated with PBS, pH 7.40, or FITC-dextran (36 mM) or calcein (100 mM; osmolarity was adjusted to 300 mOsm). After initial formation of the liposome dispersions, their size was reduced by probe sonication (Sonics & Materials).
Free FITC-dextran or calcein was separated from liposomes by ultracentrifugation (40 minutes at 40,000 rpm on a Sorvall WX90 Ultra; Thermo Scientific). Targeted liposomes (t-LIP) were also prepared, consisting of PC/Chol/PEG/PEG-MAL (20:10:1.6: 0.04 mol/mol). Attachment of anti-transferrin monoclonal antibody OX-26, at 0.1 mol% density (compared with lipid), was performed by a MAL-thioether reaction, as described in detail elsewhere (Markoutsa et al., 2014). In brief, PEG-MAL containing LIPs was mixed with thiolated–OX-26 and incubated at room temperature for 4 hours and then at 4°C overnight. Nonattached antibody was removed by gel filtration (Sepharose 4B-CL), and the yield of Mab attachment was calculated via an ELISA technique as described elsewhere (Markoutsa et al., 2014). The yield was found to be 74% of the added amount of Mab, in agreement with previous reported yields (Markoutsa et al., 2014; Papadia et al., 2017).
Cell Culture and CV Formation
Three types of cells were used in this study: HEK-293 (American Type Culture Collection, Manassas, VA), B16F10 (National Cancer Institute Tumor Repository, Frederick, MD), and hCMEC/D3 (passage 25–35; obtained under license from Institut National de la Sante et de la Recherche Medicale, INSERM, Paris, France).
HEK-293and B16F10 cells were grown in RPMI 1640 medium supplemented with 10% FBS and 1% antibiotic-antimycotic solution (Invitrogen, Carlsbad, CA). The cells were cultured at 37°C in 5% CO2/saturated humidity. The medium was changed every 2 to 3 days.
The hCMEC/D3 cells were grown in EndoGro medium (Merck) supplemented with 10 mM HEPES, 1 ng/ml basic fibroblast growth factor, 1.4 μM hydrocortisone, 5 μg/ml ascorbic acid, penicillin-streptomycin, chemically defined lipid concentrate, and 5% ultralow IgG FBS. In some cases the hCMEC/D3 cells were grown in RPMI (as mentioned previously). All cultureware was coated with 0.1 mg/ml rat tail collagen type I (BD Biosciences, Franklin Lakes, NJ).
CVs were derived from hCMEC/D3, HEK-293, or B16F10 cells were cultured as mentioned previously. In some studies, CVs were isolated for hCMEC/D3 cells grown in RPMI instead of EndoGro. Cells were incubated in T175 flasks until confluency, detached from the flasks with trypsin, and washed 3 times with ice-cold PBS. Dispersions were probe sonicated (Sonics & Materials), for up to 1 minute, and the CVs were isolated by ultracentrifugation (Thermo Sorvall WX90 Ultra; Thermo Scientific) at 60,000 rpm for 2 hours at 4°C and carefully resuspended (to break aggregates) in PBS, pH 7.40.
In some cases, CVs were enriched with Chol and coated with PEG. For Chol enrichment, CVs (dispersed in H2O) were incubated with Chol (10% w/w) at 37°C for 30 minutes, followed by sonication (3 minutes) and subsequent exchange of H2O with PBS (Zhang et al., 2017; Ying et al., 2018). CVs were then pegylated by incubation with PEG micelles (which were prepared by formation of a thin film of PEG, hydration with PBS, and incubation at 65°C for 30 minutes) for 2 hours at 60°C and then overnight at 4°C (Kooijmans et al., 2016). PEG was used at a 10 mol% (compared with CV lipid content).
CVs and liposomes were loaded with hydrophilic fluorescent dyes for evaluation of CV loading efficiency (EE%) and integrity (calcein, 100 mM in PBS), and vesicle/cell interaction (FITC, 36 mM in PBS). The CV and liposome phospholipid content of was quantified by a method routinely used to measure liposomal lipid concentration (Stewart, 1980). The protein content of CVs was quantified by Bradford assay.
Size Distribution and Zeta-Potential Measurements
The particle size distribution (mean hydrodynamic diameter and polydispersity index) of CVs and liposomes in 10 mM PBS, pH 7.4 (at 0.4 mg/ml lipid), was measured by dynamic light scattering (Malvern Nano-Zs; Malvern Instruments, Malvern, United Kingdom) at 25°C and a 173° angle. The ζ-potential was measured in the same dispersions at 25°C by use of the Doppler electrophoresis technique.
Transmission Electron Microscopy (TEM)
CVs (0.5–1 mg/ml) were resuspended in 10 mM HEPES (to eliminate potential artifacts from phosphate) and then negatively stained with different staining solutions such as 2% ammonium molybdate and 1% neutral phosphotungstic acid, washed 3 times with dH2O, drained with a tissue paper tip, and observed at 100,000 eV with JEM-2100 (Jeol, Tokyo, Japan) transmission electron microscopy (TEM) (Franken et al., 2017).
Methods for Loading of CVs with Hydrophilic Dyes
CVs were initially extruded through polycarbonate filters with decreasing pore diameters, starting from 1000, then 400 nm, and finally 100 nm. Afterward, they were divided in 1-ml parts to apply different methods for calcein encapsulation as described here.
Incubation Method.
CVs were incubated at 37°C for 1 hour with 1 ml of calcein solution (100 mM), applying vortex every 15 minutes.
Sonication Method.
Calcein solution was mixed with CVs, and the mixture was probe sonicated (at 28% intensity) 4 times for 30 seconds each time (2-minute intervals).
Freeze Thaw Method.
We mixed 1 ml of calcein (100 mM) with CVs for 10 cycles of freezing (30 minutes at −80°C) and thawing (room temperature).
DRV Method.
CVs (suspended in 1 ml H2O) and 1 ml of calcein (100 mM) was mixed, freeze-dried and rehydrated with dH2O and PBS, as reported (Markoutsa et al., 2011).
After each loading method, the CVs remained at room temperature for 1 hour (to anneal any structural defects) and were then extruded through 100-nm polycarbonate filters to compare vesicles of the same size for loading efficiency. Nonassociated calcein was separated from CVs by size exclusion chromatography (1 × 35 cm column, Sephadex G-50; Sigma-Aldrich), eluted with PBS, pH 7.40. The FI of CV-loaded calcein was measured (EX-470 nm/EM-520 nm) after disruption of vesicles with Triton X-100. Calcein loading was calculated on the basis of a calibration curve (calcein concentration/FI).
Cytotoxicity Assay
Cytotoxicity assays were performed to evaluate whether CVs exacerbated toxicity to cells under the conditions applied in the cell culture studies (concentration, incubation time). Cells were seeded and grown until confluent. The medium was replaced, and the CVs (6, 12, and 24 nmol [of lipid] per 3 × 104 cells), LIP, or PEG-LIP (at the same lipid concentration) were incubated with cells for 4 hours at 37°C (5% CO2/saturated humidity). The cell number was measured with a hemocytometer. We also performed 24-hour incubations in some cases.
After incubation, the medium was removed, and the cells were washed with PBS. Fresh medium containing 0.5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added. The cells were then incubated for 2 hours, the medium was removed, and DMSO was added (at 37°C for 30 minutes) to dissolve the formazan crystals that formed.
Viable cells (%) were calculated based on the following equation: (A570 sample − A570 background)/(A570 control − A570 background) × 100 where A570 control is the OD-570 nm of untreated cells, and A570 background is the OD-570 nm of MTT without cells.
CV Integrity (In Vitro)
The integrity of calcein-loaded CVs was studied by measuring the latency of CV-entrapped calcein during incubation in the absence or presence of serum proteins (80% fetal calf serum [FCS] v/v) for 24 hours at 37°C. Liposomes were also studied under identical conditions for comparison. Calcein latency (percent) was calculated as reported elsewhere (Kokkona et al., 2000).
Cell-Uptake Studies
For evaluation of uptake of CV-entrapped drugs by cells, FITC-labeled CVs, LIPs, and PEG-LIPs were incubated with confluent monolayer’s of HEK, B16F10 and hCMEC/D3 cells (200 or 400 nmol liposomal or CV lipid/106 cells) in medium (containing 10% FBS (v/v)) at 37°C, for 4 hours. Cells were then washed 2 times with ice-cold PBS, detached from plates by scraping, resuspended in 1 ml of PBS, and assayed for FI (EX-490 nm/EM-525 nm, 5-nm slits) after cell lysis in 2% Triton X-100. Cell autofluorescence was always subtracted.
In some cases, cell-uptake experiments were also performed in medium containing 20% (v/v) FCS to investigate the effect of increased serum protein levels on cell/CV interactions (Markoutsa et al., 2014). Sample protein content was measured by Bradford assay, and FITC uptake was normalized to protein concentration.
Flow Cytometry
FITC-labeled CVs derived from B16F10 and hCMEC/D3 cells were prepared as mentioned earlier (both hCMEC/D3EndoGro and hCMEC/D3RPMI were used for CV isolation), and their uptake by cells was evaluated after incubation of 200 nmol lipid with 106 cells for 4 hours. Flow cytometry was performed on a BD FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). At least 20,000 events were acquired. FITC-positive cells were identified and their median fluorescent intensity (MFI) estimated using FlowJo v.10 software (Tree Star, Ashland, OR).
Confocal Fluorescence Microscopy
Cells were grown on collagen-covered coverslips and incubated with FITC (aqueous phase label) and RHO (membrane label) labeled CVs for 4 hours. Cells then were fixed in 4% paraformaldehyde for 10 minutes, stained with Hoechst for 5 minutes, and mounted on microscopy slides with Mowiol. Slides were observed using fluorescence microscopy on a SP5 confocal microscope (Leica, Heidelberg, Germany) to visualize their subcellular distribution.
Cell-Monolayer Permeation Studies
We seeded the hCMEC/D3 on type I collagen precoated Transwell filters (polycarbonate six-well, pore size 0.4 μm; Millipore Merck) at 5 × 104 cells/cm2. The assay medium was changed every 4 days, and the transport assays were performed 10 to 12 days after seeding. Twenty-four hours before each transport experiment, the medium was replaced with fresh medium containing 1 nM simvastatin.
For confirmation of cell junction formation, monolayers were periodically inspected by microscope and by transendothelial electrical resistance (TEER) monitoring with Millicell ERS-2 Epithelial VoltOhm meter (Millipore Merck). Monolayer quality was verified by measuring the permeability of the highly hydrophilic, low-molecular-weight compound lucifer yellow (LY). Current values were then compared with reported ones (Poller et al., 2008; Markoutsa et al., 2011).
Transport experiments were conducted in Hanks’ balanced salt solution supplemented with 10 mM HEPES and 1 mM sodium pyruvate in the cell culture medium. Transport was estimated by placing FITC-labeled CVs on the upper side of monolayer (200 nmol lipid per well) and measuring FITC FI in the lower side at various time periods (10, 20, 30, 60, 90, and 120 minutes). Lucifer yellow (LY) was also added in each well, and its permeability was calculated to verify that vesicles did not modify the barrier. Control liposomes (LIP) and targeted liposomes (t-LIP) were also evaluated for comparison.
Proteomic Analysis
The biologic replica of the vesicles generated from hCMEC/D3 cells cultured in RPMI medium or in EndoGro medium were incubated in 1 ml of water to burst followed by dialysis and ultracentrifugation (2 × 100,000g). The pellet was lysed in 4% SDS/0.1 M dithiothreitol at 98°C, incubated for 30 minutes in a sonicating water bath, and subjected to Sp3-mediated tryptic/LysC digestion according to the standard protocol (Hughes et al., 2019).
For liquid chromatography with tandem mass spectrometry (MS), supernatant containing peptides was collected, dried down, reconstituted in 2% (v/v) acetonitrile (ACN), and 0.1% (v/v) formic acid, and incubated for 3 minutes in a sonication water bath. The peptide concentration was determined by Nanodrop absorbance at 280 nm. Three micrograms peptides were preconcentrated with a flow of 3 μl/min for 10 minutes using a C18 trap column (Acclaim PepMap100, 100 μm × 2 cm; Thermo Scientific) and then loaded onto a heated at 35°C C18 column (50 cm,75 μm ID, particle size 2 μm, 100 Å, Acclaim PepMap100 RSLC; Thermo Scientific). The binary pumps of the high-pressure liquid chromatography (RSLCnano; Thermo Scientific) consisted of solution A (2% (v/v) ACN in 0.1% (v/v) formic acid) and solution B (80% [v/v] ACN in 0.1% [v/v] formic acid). Peptides were separated using a linear gradient of 4% B up to 40% B in 210 minutes (flow rate 300 nl/min).
Eluted peptides were ionized by a nanospray source and detected by an LTQ Orbitrap XL MS (Thermo Fisher Scientific) operating in data dependent mode. Full-scan MS spectra were acquired in the Orbitrap (m/z 300–1600) in profile mode with the resolution set to 60,000 at m/z 400 and automatic gain control target at 106 ions.
The six most intense ions were sequentially isolated for collision-induced MS/MS fragmentation and detection in the linear ion trap. Dynamic exclusion was set to 1 minute and activated for 90 seconds. Ions with single charge states were excluded. Lockmass of m/z 445,120,025 was used for continuous internal calibration. XCalibur (Thermo Scientific) was used to control the system and acquire the raw files.
For protein identification/quantification, the mass spectral files (.RAW files) were processed using MaxQuant software (1.6.3.3) (Cox and Mann, 2008; Cox et al., 2014). Default parameters were used for protein identification and quantification. Trypsin specificity with two missed cleavages was allowed and set, and the minimum peptide length was set to seven amino acids. Cysteine carbamidomethylation was set as fixed, and methionine oxidation, deamidation of asparagine and glutamine, and N-terminal acetylation were set as variable modifications (maximum of five modifications per peptide). The false-discovery rate both for peptide and protein was set to 1%. For calculation of protein abundances, label-free quantification (LFQ) was performed with both “second peptide” and “match between run” options enabled. The complete human database was downloaded from Uniprot (94731 entries, 05_11_18).
In Vivo Studies: Biofluorescence Imaging
In vivo live animal imaging experiments were performed to estimate the pharmacokinetics and ex vivo organ distribution of CVs. DiR-labeled-CVs were used because free DiR is rapidly eliminated from mice after injection, as previously verified elsewhere (Markoutsa et al., 2014; Papadia et al., 2017). CVs from hCMEC/D3 cells (grown in RPMI and EndoGro) and B16F10 cells were evaluated, as well as CVs that were enriched with Chol and PEG (as described earlier).
FVB (Friend leukemia virus B) albino mice and C57BL/6 mice, purchased from Hellenic Pasteur Institute (Athens, Greece), were bred at the Center for Animal Models of Disease, University of Patras, Faculty of Medicine (Rio, Greece). FVB mice were chosen for their white skin and fur that permits enhanced light penetration; C57BL/6 mice were used as syngeneic to the B16F10-derived CVs Animal care and experimental procedures were approved by the Veterinary Administration Bureau of the Prefecture of Achaia, Greece (protocol approval numbers 3741/16.11.2010, 60291/3035/19.03.2012, and 118018/578/30.04.2014) and were conducted according to Directive 2010/63/EU (European Union 2010) and European Union Directive 86/609/EEC for animal experiments.
The mice were matched for sex (male-female), weight (20–25 g), and age (6–12 weeks). Biofluorescence imaging of living mice and explanted murine organs was done on an IVIS Lumina II imager (Perkin Elmer, Santa Clara, CA). The mice were anesthetized using isoflurane and were serially imaged at various time points after injection of DiR-labeled CVs (200 μg lipid/mouse). Retro-orbital venous sinus injection, which is equally effective to tail-vein injection, was used to avoid potential animal distress and/or retention of significant amounts of the dose in the tail. Standard excitation/emission wavelengths for DiR were applied as follows: excitation 710–760 nm; emission 810–875 nm. The images were acquired and analyzed using Living Image v4.2 software (Perkin Elmer). In detail, specific bodily area or explanted organ regions of interest were created and were superimposed over all images acquired in a uniform fashion, and the photon flux within these regions were measured.
Statistical Analysis
All results are expressed as mean ± S.D. from at least four independent experiments. Most data were analyzed by using one-way ANOVA followed by Bonferroni post hoc test. P = 0.05 was considered statistically significant for all comparisons. When more factors were compared, two-way ANOVA was performed. The significance of comparisons is presented in the graphs.
For the proteomic analysis results, statistical analysis was performed using Perseus (version 1.6.2.2) (Tyanova and Cox, 2018). Proteins identified as “contaminants,” “reverse,” and “only identified by site” and with “less than two peptides identified” were filtered out. The LFQ intensities were transformed to logarithmic. Zero intensities were imputed (replaced by normal distribution). The replicas were grouped for each set of conditions—that is, for “RPMI medium” and “EndoGro Medium” a two-sided Student’s t test of the grouped proteins was performed using P values for truncation (P < 0.05).
Results
Role of Parental Cells of CVs on Their Potential to Facilitate Delivery of Their Contents into Cells.
TEM studies revealed round-shape morphology of CVs (Fig. 1A). CVs prepared by processing HEK-293and B16F10 cells had mean hydrodynamic diameters between 276 and 286 nm, more than 2 times higher than the 100 nm membranes from which they were extruded as the final step of their preparation procedure. Oppositely, the liposomes that were formulated by the same methodology had mean diameters close to 100 nm; the polydispersity index (PDI) values of CVs were also much larger compared with those measured for the liposomes (Table 1). The ζ-potential of the CVs was similar to that of the negatively charged (PC/PG/Chol) liposomes in accordance with previous reports (Jang et al., 2013; Lunavat et al., 2016).
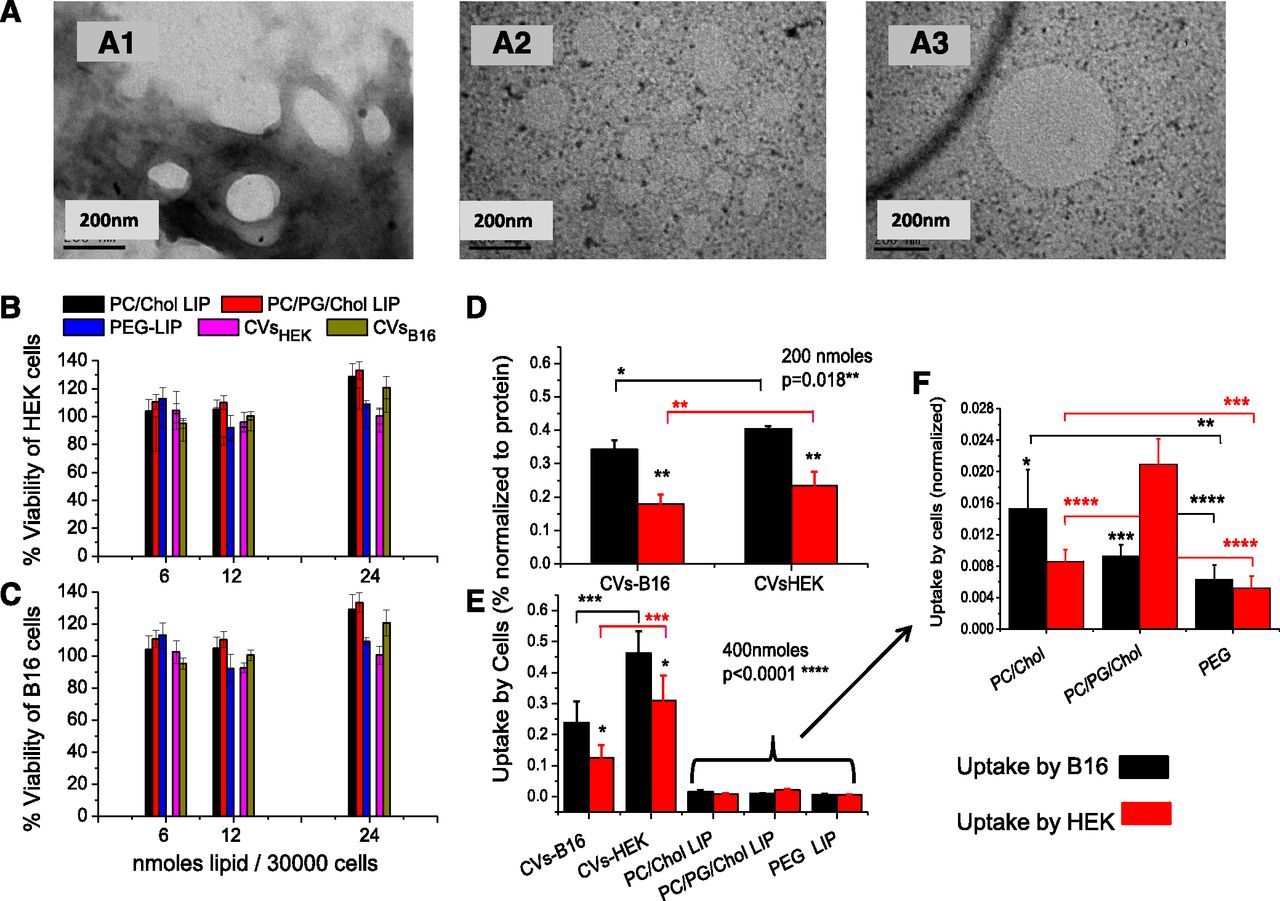
Results of preliminary studies: Representative negative stain TEM micrographs of CVB16F10 (A) (with ammonium molybdate as stain [A1], or phosphotungstic acid 1% [A2, A3]). Cytotoxicity of various concentrations of CVs from HEK-293 and B16F10 cells, and three different liposome types (6, 12, and 24 nmol of lipid/30,000 cells), expressed as % viability of control, after 4 hours of coincubation at 37°C toward HEK-293 cells (B) and B16F10 cells (C). Uptake of CV- or liposome-associated FITC by B16F10 cells and HEK-293 cells after 4 hours of incubation at 37°C of (D) 200 nmol lipid/106 cells or (E) 400 nmol lipid/106 cells. (F) The part of graph E that corresponds to the liposome formulation uptake presented in magnified form. The statistical significance of individual differences of CV/liposomes between the two cell types is presented as asterisks on the top of the bars, and other individual differences are presented by connecting lines. One-way ANOVA P values (for the effect of CV type) are shown.
Physicochemical properties—mean diameter, polydispersity index (PDI), and ζ-potential—of cellular vesicle from HEK-293(CVHEK-293) and B16F10 (CVB16F10) cells, and various liposome types
Mean value (n = 3 preparations) ± S.D.
CVs from HEK-293and B16F10 cells were loaded with FITC (by the DRV method) to evaluate their potential to facilitate delivery of their contents into cells. Three liposome types with different lipid compositions were tested in parallel for comparison. CVs and liposomes were initially demonstrated to be noncytotoxic toward the cells under the conditions applying in the cell uptake study, as seen in Fig. 1, B and C. Cell-uptake study results showed that B16F10 cells take up CVHEK-293 at larger amounts compared with the (homologous origin) CVB16F10 at both doses used, 200 nmol (Fig. 1D) and 400 nmol (Fig. 1E), indicating that CVs do not always demonstrate increased interaction with homologous (parental) cells, compared with vesicles from nonhomologous origin.
When the results of the two sets of experiments are compared (Fig. 1, D and E), it can be seen that while the uptake of CVB16F10 by both cell types is saturated at the higher dose (% uptake values are lower at the higher dose), this is not the case for the CVHEK-293, indicating different uptake mechanisms of the two CV types. Furthermore, CV uptake by B16F10 cells is higher than that by HEK-293 cells for both CVs, irrespective of the amount of CV-lipid incubated with the cells (200 or 400 nmol).
Interestingly, the uptake of FITC by both cell types is substantially higher (from 9 to 23 times) from the CVs when compared with all the liposome types evaluated (Fig. 1E). Considering the uptake of the liposomes by the cells (seen in Fig. 1F in magnification), most liposome types are taken up at higher amounts by B16F10 cells compared with HEK-293 cells (as observed also for CVs), with the exception of the negatively charged liposomes (PC/PG/Chol). Also, PEGylated liposomes are seen to interact less with the cells, compared with the non-PEGylated liposome types.
Potential to Load CVs with Drugs by Applying the DRV Technique.
To date, the DRV method had never been evaluated for the loading of CVs or extracellular vesicles with aqueous soluble compounds, so we compared it with three other commonly applied methods: incubation, sonication, and the freeze-thawing method. Calcein-loaded CVB16F10 prepared by the DRV method were found to have between 2 and 2.8 times higher calcein content (7% loading efficiency) compared with the CVs loaded with the other techniques (Fig. 2A). The lowest loading (2.49%) was achieved by incubation, while sonication and freeze-thawing methods resulted in similar loading (3.28% and 3.45%, respectively), 30% higher compared with incubation.

(A) Calcein encapsulation (expressed as calcein/lipid [mole/mole] Final/C/L Initial) in CVB16F10 using different loading methods. (B) Initial calcein latency values (%) in PBS (pH 7.40) of CVB16F10 loaded with different methods. (C) Integrity of CVB16F10 during incubation in PBS (37°C, 24 hours). (D and E) Mean hydrodynamic diameter and polydispersity index of CVB16F10 immediately after preparation and after 3 days. Each value is the mean value from four different preparations, and the bars represent the S.D. value of each mean. One-way ANOVA P values for significant differences are shown on each graph.
Calcein-Latency Method for Evaluation of CV Integrity.
Initial calcein latency values were measured immediately upon dilution of the CVs (from B16F10 cells) in buffer. The values ranged between 14% and 52% (Fig. 2B), irrespective of the loading method used, which were very low compared with the usual values reported for liposomes composed of phospholipids and Chol (usually >85%) (Kokkona et al., 2000; Markoutsa et al., 2011, 2014), indicating poor vesicle integrity. Nevertheless, the CVs prepared by DRV method have the highest initial calcein latency values compared with those prepared by other methods (3 to 3.3 times lower latency values).
The release of CV-encapsulated calcein from vesicles was observed during 24 hours of incubation in buffer, as seen in Fig. 2C. Calcein was gradually released from all CVs within the time period monitored. Concerning the size and polydispersity of the vesicles produced by the four different methods, we observed that all had similar mean diameters (between 135 and 153 nm) and PDIs (from 0.22 to 0.29) (Fig. 2, D and E). For all CVs, the size and PDI were stable after 3 days of storage at 4°C.
The conclusion from the above studies is that the DRV method produces CVs with substantially increased amounts of vesicle-associated calcein, a higher fraction of which is retained into the aqueous compartment of the vesicle upon dilution compared with the vesicles produced by other methods evaluated. Nevertheless, all CV types released their calcein content after 24 hours of incubation in buffer.
The integrity of CVs prepared by DRV method from other cells (HEK-293 and hCMEC/D3) was additionally studied to confirm that the previous findings were not specific for CVB16F10. The initial calcein latency values of CVHEK-293 were similar to those of CVB16F10. The corresponding values of CVhCMEC/D3 were much lower (approximately half) (Table 2). No significant difference was observed between the different CVs considering their lipid/protein (w/w) ratios (Table 2).
Initial calcein latency (%) in PBS and FCS, and lipid/protein (w/w) ratios of CVs from different cells (mean (n = 5 preparations) ± S.D.)
The kinetics of calcein release from all CV types during 24-hour incubation in buffer and FCS are seen in Fig. 3. In all cases (Fig. 3, A–C) the calcein release was higher when CVs were incubated in buffer compared with FCS, which is not the case for liposomes (Fig. 3D). Additionally, all CVs tested gradually released most of their calcein content during the 24-hour incubation period, confirming that this was not the case for CVB16F10 only. Furthermore, the substantial difference between the CVs and liposomes in terms of their integrity was obvious; the initial calcein latency values for liposomes were between 86% and 94%. PEG-LIP demonstrated significantly higher stability during the 24-hour incubation period studied (retaining >65% of the encapsulated calcein). The non-PEGylated LIP gradually lost a significant amount of calcein during incubation in FCS, but even non-PEGylated liposomes are very stable in buffer.

Time course of calcein latency from CVs of various origin and liposomes (for comparison) during their incubation in PBS buffer and FSC (80% v/v) for 24 hours (37°C) (mean values ± S.D.; n = 4). CV (or LIP) lipid concentration was 0.5 mg/ml. (A) CVHEK-293; (B) CVB16F10 and PEG- CVsB16. (C) CVhCMEC/D3 and PEG- CVhCMEC/D3. (D) PC/PG/Chol liposomes and PEG-LIP (PC/PG/Chol/PEG). Two-way ANOVA P values are reported on graphs for the effect of PEG.
When CVs were engineered to increase their Chol content and to incorporate PEG in their membrane (as described in the Materials and Methods section) their integrity, especially in FCS, was significantly increased (P < 0.05), as demonstrated for PEG-CVB16F10 (Fig. 3B) and PEG-CVhCMEC/D3 (Fig. 3C). Additionally their initial calcein latency values (Table 2) were increased. The physicochemical properties of the vesicles are shown in Table 3.
Physicochemical properties of the vesicles used in calcein release, cell interaction (in vitro), and in vivo studies
Mean diameter, polydispersity index (PDI), and ζ-potential of CVs from HEK-293 cells (CVHEK-293), B16F10 cells (CVB16F10), and hCMEC/D3 cells (CVhCMEC) as well as pegylated CVB16F10 (PEG-CVB16F10) and pegylated CVhCMEC/D3 (PEG-CVhCMEC/D3). Liposomes (LIP), pegylated liposomes (PEG-LIP) (PC/PG/Chol/PEG) and targeted liposomes (t-LIP) properties are also reported.
Potential of CVs Derived from Brain Endothelial Cells to Target the Brain.
CVs from the three different cell types used were studied for their interaction with the hCMEC/D3 cellular model of the blood-brain barrier (BBB). Cell-viability experiments verified that the CVs did not exhibit cytotoxic effects toward hCMEC D3 cells (Fig. 4A) under the conditions applied in the studies. The cytotoxicity of some CV types after 24 hours of incubation with B16F10 and hCMEC/D3 cells was additionally evaluated, and the results showed that cell viability was always slightly lower when the cells were incubated with CVB16F10 compared with CVhCMEC/D3. However CVB16F10 had similar viability values as PEG-LIP, which was also studied for comparison (see Supplemental Fig. 1).

(A) Effect of CVs on viability of hCMEC/D3 cells after incubation of various concentrations for 4 hours. LIP (PC/PG/Chol) are also tested as controls. (B) Uptake of various CV types and liposomes by hCMEC/D3 cells after 4 hours of incubation of 200 nmol lipid/106 cells. (C) Effect of serum concentration of media on the interaction between CVs and hCMEC/D3 cells. (D) Uptake of CVhCMEC/D3 by hCMEC/D3, B16F10, and HEK-293 cells. The CVs were produced by cells grown in EndoGro or RPMI medium. Statistical significance of individual differences is presented as asterisks on the top of the bars (or on connecting lines). Significant one-way (B) or two-way (C and D) ANOVA P values are shown in the graphs.
The uptake of CV-associated FITC into the brain endothelial cells was calculated after CV/cell incubation as a measure of the uptake of the CVs by the cells. Negative-charged liposomes (LIP) and also ligand-targeted liposomes (t-LIP) decorated with OX-26 monoclonal antibody against the transferrin receptor were also tested for comparison. The physicochemical properties of the vesicles used in this study are presented in Table 3. As seen in Fig. 4B, the uptake of CVhCMEC/D3 by the brain endothelial cells was several times higher compared with that of all the other CVs tested, as well as the conventional liposomes. In fact, compared with nontargeted LIP, all CVs demonstrated substantially higher uptake by hCMEC/D3 cells.
Surprisingly, the uptake of CVhCMEC/D3 by the brain cells was also significantly higher compared with the uptake of the ligand-targeted liposomes (Fig. 4B). It is important to point out that it was proven that the use of trypsin (as a cell-detaching method) during CVhCMEC/D3 preparation did not decrease the hCMEC/D3 cells uptake compared with CVs prepared without using trypsin (see Supplemental Fig. 2).
As seen in Fig. 4C the uptake of CVhCMEC/D3 is slightly decreased when the experiment is performed in the presence of 20% (v/v) of serum (compared with 10%), while the corresponding differences for the other two CVs are not significant. When hCMEC/D3 cells are cultured in EndoGro medium they express specific proteins on their membranes, which contribute to the formation of “tight” cell monolayers (Schrade et al., 2012).
We sought to investigate whether the CVs produced by the same cells grown in a different medium, such as RPMI, would demonstrate modulated cell interactions. Indeed, the uptake of CVhCMEC/D3 from cells grown in RPMI by hCMEC/D3 cells is approximately 2 times lower compared with the uptake of CVhCMEC/D3 produced from cells grown in EndoGro (Fig. 4D). A similar result was seen for the uptake of the same CVs by B16F10 cells (at a lower magnitude), while their uptake by the HEK-293 cells was substantially modulated but in the opposite direction: CVs from cells grown in RPMI were taken up by HEK-293 cells 2.5 times more compared with CVs from cells grown in EndoGro. Thereby, it is indicated that the cell-culturing conditions have a substantial effect on the interactions of the cell-derived CVs with different cells.
The CVhCMEC/D3 cell-uptake results were confirmed by flow cytometry (Fig. 5A) and confocal microscopy studies (Fig. 5B). Indeed, interaction of CVhCMEC/D3 produced by cells grown in EndoGro toward hCMEC/D3 cells was higher compared with CVs produced by cells grown in RPMI (Fig. 5A). Furthermore, the interaction of CVB16F10 with hCMEC/D3 cells was 3.0–4.7 times lower compared with the CVhCMEC/D3, in accordance with the results of the cell-uptake experiment (Fig. 4). Confocal microscopy confirmed that CVs (dually labeled with FITC as aqueous phase marker and RHO as membrane marker) are endocytosed in the cell cytoplasm (Fig. 5B). Confocal microscopy micrographs demonstrating quantitatively lower interactions (compared with Fig. 5B) between B16F10 cells and CVhCMEC/D3 or CVB16F10 can be seen in Supplemental Fig. 3.

(A) Flow cytometry interactions of various CVs (indicated in graphs) with hCMEC/D3 cells shown in corresponding morphologic plots. (B) Representative laser-confocal micrograph (and magnification of indicated part) of hCMEC/D3 cells after interaction with CVhCMEC/D3. Cells for CV preparation were cultured in EndoGro. CVs were labeled with FITC (green, aqueous phase label) and RHO (red, membrane label); Hoeckst 33342 (blue) shows the nucleus. (C) Transport of vesicle-associated FITC across hCMEC/D3 cell monolayers (% of total). Samples (200 nmol of each vesicle type) were added on transwell-mounted monolayers, and transport was calculated by measuring FITC FI. Each result is the mean from six different experiments. Statistical significance of individual differences is presented as asterisks (connecting lines indicate comparison). Two-way ANOVA P values (for comparison of all cases (all), and comparison between CVs and t-LIP) are shown.
TEM micrographs of CVs from hCMEC/D3 cells grown in the two different media (RPMI and EndoGro) prove that they have no morphologic differences (Supplemental Fig. 4).
To explore the magnitude of the membrane composition differences between CVhCMEC/D3 produced by cells grown in the different media, proteomic analysis was performed. A total of 1460 confident protein groups were identified on CVs, and the statistical comparison of quantitative (LFQ) values for each protein in RPMI versus EndoGro culturing conditions gave a list of 171 proteins that were significantly altered, as shown by a volcano plot in Fig. 6 (see also Supplemental Table 1 for the full protein list). These were analyzed by the protein subcellular annotation available at subcellbarcode.org (Orre et al., 2019), which revealed 28 proteins annotated to be localized as secretory and subcategorized into plasma membrane, endoplasmic reticulum, mitochondrial, lysosomal, peroxisomal, and so on (Supplemental Table 2).

Volcano plot with the 171 proteins identified to be significantly altered between the two CV groups evaluated. The surface proteins that may be interesting to further evaluate are highlighted.
CVhCMEC/D3 were evaluated for their transport across the hCMEC/D3 cell monolayer model of the BBB. Plain negative-charge liposomes (PC/PG/Chol) (LIP) as well as ligand-targeted liposomes (t-LIP) were also evaluated for comparison. The TEER of the control monolayer (without any sample) was measured during monolayer formation and was found to gradually increase from 36 Ω cm2 (at day 3) to 52.1 ± 2.4 Ω cm2 and finally to 59.6 ± 4.4Ω cm2 (after simvastatin treatment), and LY permeability was 1.04 × 10−3 ± 8.56 × 10−5 cm/min, all values being in good agreement with previously reported ones (Poller et al., 2008; Markoutsa et al., 2014). No significant differences were found in the TEER and LY permeability values between all the monolayers used and the control (Table 4), proving that monolayer permeability was not affected by any of the samples.
Transendothelial resistance (TEER), LY-permeability values (measured on the monolayers where the corresponding vesicle samples were placed), and FITC permeability values of the vesicle-associated FITC for the vesicle types tested
Each value is the mean of six monolayer experiments from two different samples.
Concerning vesicle-associated FITC translocation across the monolayer, transport of CVhCMEC/D3 for the first 45 minutes was much higher compared with that of t-LIP, but after that the transport rate of CVhCMEC/D3 was markedly decreased (Fig. 5C). By contrast, t-LIP transport was initiated at a substantially lower rate and gradually increased; finally the FITC amounts transported were similar to those from CVhCMEC/D3. As expected, the nontargeted liposomes were transported at a significantly lower degree compared with both t-LIP and CVhCMEC/D3. The permeability values are presented in Table 4. The values for LIP and t-LIP are in agreement with previously reported permeability values (Markoutsa et al., 2011, 2014).
In Vivo Studies.
In vivo live animal imaging and ex vivo imaging of explanted organs after injection of DiR-labeled CVhCMEC/D3 (in FVB mice) and CVB16F10 (in syngeneic C57BL/6 mice) were used to explore CV pharmacokinetics and potential for brain targeting. Because the integrity of CVs was found to be low (as presented earlier), PEG-CVs were also studied. Physicochemical properties of vesicles used for in vivo studies are shown in Table 3.
As seen in Fig. 7, engineered CVs (for Chol and PEG incorporation) demonstrate lower distribution in the liver and localize in the brain at significantly higher amounts compared with the corresponding non-PEG vesicles. In fact the nonengineered CVs (depicted as CTR in the graphs) rapidly accumulated in the liver and lung area as seen from the 15-minute postinjection images (Fig. 7, A and D). The very fast accumulation of the DiR signal in liver and lungs indicates that CVs have low integrity and thus are most probably rapidly opsonized and cleared from circulation, having limited chances to distribute to other tissues. For both types of CVs (those from B16F10 and hCMEC/D3 cells) CV engineering does not significantly modify the CV-associated signal in lungs; however, PEG-CVB16F10 have a substantially lower signal in the spleen as compared with the corresponding control CVs. On the other hand, the DiR signal in the spleen is not significantly altered when the CVhCMEC/D3 are engineered.

In vivo live animal and ex vivo imaging studies. In all cases CVs as well as corresponding PEG-CVs were tested under identical conditions to evaluate the effect of CV engineering on their in vivo fate. Representative images of mice after injection of 200 μg lipid/mouse, 15 minutes and 4 hours after injection (A and D). Ex vivo imaging of extracted organs 4 hours after injection (B and E). Graphs showing the comparative DiR signals of extracted organs (brain, liver, lung, and spleen) from mice injected with nonpegylated CVs (control, CTR) versus PEG-CVs (PEG) (C and F). A–C correspond to CVB16F10 injected in syngeneic C57BL/6 mice; D–F correspond to CVhCMEC/D3 injected in FVB mice. Each value is the mean from at least six animals.
In Fig. 8A the brain signal/dose ratios measured 4 hours after injection for the four different CV types are compared. As shown, the normalized DiR brain signal of PEG-CVhCMEC/D3 is highest, followed by that of CVhCMEC/D3; the brain signals of CVs from B16F10 cells are lower, even those of the engineered CVs (although the PEG-CVB16F10 brain signal is 3 times higher than the corresponding signal acquired by the nonengineered CVB16F10). In fact, the different brain targeting potential between CVs derived from B16F10 and hCMEC/D3 cells (especially as they were studied in different animal models) is better realized by comparison of the brain/liver + spleen (DiR signal) ratios (Fig. 8B), which is a more accurate measure of brain-targeting capability. These ex vivo results are in good correlation with the in vitro hCMEC/D3 uptake results (Fig. 4B).

(A) Comparison of ex vivo DiR signals (photon/s) normalized to the total DiR signal injected (dose) in brain of mice 4 hours after injection of various types of CVs. FVB mice (for CVhCMEC/D3) and C57BL/6 mice (for CVB16F10) were used. (B) Comparison of the 4-hour postinjection brain/liver + spleen (B/L+S)*100 DiR signal ratios of the same CVs as in (A). (C) Comparison of DiR-signals (photon/s) normalized to the total DiR-signal injected (dose) in brain of FVB mice 4 hours after injection of CVs derived from hCMEC/D3 cells grown in EndoGro (ENDO) and in RPMI. All values are mean values from at least four or six animals.
In another in vivo study, two sets of CVs were evaluated: CVs from cells grown in RPMI (CVRPMI) and CVs from cell grown in EndoGro (CVEndo). The kinetics of CV-associated DiR for both CV types was disappointing: a very rapid uptake of the CVs by liver, lungs, and spleen was evident as soon as 15 minutes after injection (Supplemental Fig. 5). Thereby, the potential of the CVs to target the brain could not be verified in vivo. The ex vivo bio fluorescence values, measured 4 hours after injection, proved the high accumulation of the CVs in liver, lungs, and spleen (Supplemental Fig. 5C) and also showed that both CV types evaluated (those derived from cells grown in RPMI and those from EndoGro grown cells) seemed to have slight difference in regards to their biodistribution.
The decay of biofluorescence signals with time is demonstrated in the ex vivo imaging of organs from two mice that received CVRPMI and were harvested 4 and 24 hours after injection, respectively (Supplemental Fig. 5A). The decay in signals of all organs was obvious, with the exception of the spleen where the signal was increased at 24 hours compared with 4 hours. The very fast accumulation of the DiR signal in the liver and lungs indicates that these natural CVs have low integrity (as indeed proven by the calcein latency study). As a consequence, they are probably rapidly opsonized and cleared from circulation. However, when the ex vivo brain signal measured 4 hours after injection of the two CV types was compared after being normalized to the total DiR signal injected (dose), it is obvious that the CVEndo was localized at higher amounts in the brain compared with CVRPMI (Fig. 8C).
Discussion
The current study provides new insights in the CVs for drug delivery field. First of all, concerning the role of parental cells on CV potential to facilitate delivery into cells, it was clearly demonstrated that homologous origin CVs do not always display increased interaction with parent cells; the CVs from HEK-293 cells were taken up more by B16F10 cells compared with the CVs of homologous origin (Fig. 1D).
Regarding the potential to load CVs with drugs by the DRV technique, it was proven that this well-known in liposome field method can be applied for loading hydrophilic compounds into CVs. CV loading achieved by the DRV method was more than double compared with the commonly used methods of sonication, incubation, or freeze-thaw cycles (Fig. 2A) (Antimisiaris et al., 2018). Interestingly, the loading method was also observed to influence the initial calcein latency of CVs; the DRV method was found to be superior not only because larger amounts of calcein were loaded but also because a higher fraction of the load was retained in vesicles (Fig. 2, B and C).
Nevertheless, the CV-loading values conferred by the DRV method were lower compared with those reported for liposomes (between 9% and 30%) (Fatouros et al., 2001; Mourtas et al., 2015). The later result is connected with the low initial calcein latency values of CVs (Table 2). Low initial calcein latency may be attributed to the rapid “reorganization” of vesicles immediately upon dilution, as demonstrated for liposomes composed of plain dimyristoyl-phosphatidylcholine (Kokkona et al., 2000), for “elastic vesicles” with no Chol in their membrane (Ntimenou et al., 2012), and when liposomes are diluted in membrane-disrupting media (Mourtas et al., 2008). The fact that some CV types were found to have higher mean diameters compared with the membrane pores they were extruded through (Table 1) agrees with the possibility of high elasticity due to (possible) low cholesterol content (Ntimenou et al., 2012). Initial calcein latency values of CVs from hCMEC/D3 cells were substantially lower compared with those from B16F10 and HEK-293(Table 2), indicating that CV integrity is determined not only by the method used for preparation/loading but also by the parent cell type.
Another possible explanation for the low initial calcein latency of CVs is that a high percent of “liposome-associated” calcein is “adsorbed” on vesicle surface rather than “entrapped” in vesicles, which is relevant with the high protein content of CVs.
The utility of calcein latency method for evaluation of CV integrity was confirmed. Calcein latency is routinely used as a method to monitor liposome integrity during incubation in various media and to predict blood circulation time. It has the advantage of measuring vesicle-encapsulated calcein leakage in real time without separation from encapsulated dye. The integrity of CVs during incubation in buffer and FCS, as monitored by calcein latency, was found to be lower compared with liposomes (Fig. 3), while the liposome integrity was in agreement with previous reports (Kokkona et al., 2000; Mourtas et al., 2008). Because we did not perform lipidomic analysis of the current CVs, we cannot be sure about the reason for their low integrity. Increased fractions of Chol in exosomes compared with cellular membranes had been reported previously (Llorente et al., 2013), and the higher rigidity of extracellular vesicles compared with cells was attributed to higher sphingomyelin, disaturated lipid, and cholesterol contents (Huang et al., 2013, Ridder et al., 2014). However, little is known about CV rigidity/integrity. Zhang et al. (2017) found it necessary to enrich with cholesterol to load doxorubicin into CVs; the same was required for drug loading into platelet-derived CVs (Ying et al., 2018).
Furthermore, although there are some studies about in vivo therapeutic effects of CV-associated drugs, not much is known about their in vivo kinetics. Jang et al. (2013) performed numerous experiments with doxorubicin-loaded exosomes and CVs after injection into mice; a high accumulation of vesicle-associated dye in the liver, lungs, and spleen was found 12 hours after injection. However, nothing was mentioned about the circulation time of vesicles or their tissue distribution at shorter periods.
Despite our limited knowledge about CV in vivo kinetics, several studies have demonstrated rapid accumulation of exosomes in liver, spleen, and lungs. Smyth et al. (2015) studied the kinetics of several exosomes types and found that the vast majority of injected unmodified exosomes are cleared by the reticuloendothelial system (RES) before reaching tumors. Elsewhere, pegylation significantly increased the circulation half-life of exosomes (Kooijmans et al., 2016).
When the current CVs were engineered for Chol and PEG incorporation in their membranes, they demonstrated increased integrity according to the calcein integrity study (Fig. 3). Based on the poor integrity of the nonengineered CVs, we would predict a short blood-circulation period and a rapid accumulation in the RES after in vivo injection; accordingly, slower accumulation in liver and higher distribution in other organs would be expected for engineered CV types. Indeed, in vivo and ex vivo studies in which engineered and nonengineered CVs were compared (Figs. 7 and 8A) confirm the calcein-integrity–based predictions, proving the value of this in vitro test. Furthermore, our results agree with those of the previous studies.
In regards to the potential of brain-endothelial-cell–derived CVs to target the brain, the high uptake of CVhCMEC/D3 by the BBB model compared with other CV types evaluated—as verified by FACS and confocal microscopy (Figs. 4 and 5) as well as their rapid/high transport across thee monolayer BBB model (Fig. 5C)—proves that they have high potential for brain targeting. In fact, this is the first study to prove the higher BBB targeting potential of CVs compared with OX-26 liposomes. Because recognition of targeting ligands on nanoparticles may be blocked after adsorption of serum proteins on their surface (Salvati et al., 2013), it was proposed that vesicle/cell interaction studies in presence of increased protein concentrations may provide insights about the former possibility (Markoutsa et al., 2014).
The interaction of CVhCMEC/D3 with BBB cells was slightly decreased when the cell-medium protein concentration was increased (Fig. 4), suggesting a possibility for such “blocking” effects. Coating of the CVs surface with PEG may—in addition to providing increased integrity—help in this direction. The results of the in vivo studies (Figs. 7 and 8) clearly demonstrate the higher brain-targeting potential of CVhCMEC/D3 compared with CVB16F10 in accordance to previous reports (Weskler et al., 2005; Yang et al., 2015; Chen et al., 2016). The difference between the two CV types with respect to their brain-targeting potential is better realized when the brain/liver + spleen (DiR signal) ratios (Fig. 8B) are compared. Furthermore, although the unmodified CVhCMEC/D3 did not demonstrate exceptionally high brain-targeting potential (most probably due to low circulation time and rapid uptake by liver and spleen), engineering methods (similar to those used in liposome technology) could increase the integrity of natural CVs in order for the latter to exhibit their high organotropism after systemic administration.
Comparison of the 4-hour postinjection brain/liver + spleen DiR signal ratios of the current CVhCMEC/D3 (4.90 ± 0.83) and especially the PEG-CVhCMEC/D3 (12.1 ± 1.6) with the reported ratios of BBB-targeted liposomes (with one and two ligands, which range between 0.3 and 1.6 at 8 hours after injection and 0.6–1.5 at 24 hours after injection) (Markoutsa et al., 2014; Papadia et al., 2017) reveal the enhanced potential of the current CVs as brain-targeting carriers for theragnostic agents.
The finding that culturing conditions (media) of parent cells may have such a significant effect on the targeting capability of the CVs, as demonstrated herein in vitro (Figs. 4 and 5) and in vivo (Fig. 8C), is very interesting and may be used as a methodology to identify exosomal surface proteins (or protein combinations), which are important for targeting, thus facilitating the development of artificial exosome mimetics. Previously hCMEC/D3 exosomes were characterized for their signature profiles, and 1179 proteins were identified (Weskler et al., 2005), many of them similar to the 1460 protein groups currently identified in the CVs from the same cells.
Some of the surface proteins (highlighted in the volcano plot of Fig. 6), such as PHB2, HSPA8, PDIA4, ITGA2, NT5E, EPHA2, HLA-C, PHB, HLA-B, AIMP1, and TLN1, may be interesting for further exploitation to assess whether the observed differences could explain the deregulation in CV interaction with hCMEC/D3 cells. Interesting groups are the proteins that share their involvement in cellular adherence (heat shock cognate 71 kDa protein, integrin alpha-2, ephrin type-A receptor 2, and talin-1), which might influence the vesicle–cellular junction interaction. Additionally, proteins PHB and PHB2, although mitochondrial, might confer to the vesicle biology.
Acknowledgments
The authors are thankful to Dr. Mary Kollia and the Laboratory of Electron Microscopy and Microanalysis (L.E.M.M.), Faculty of Natural Sciences, University of Patras, for the TEM studies. The help provided in CV experiments by Konstantina Pefani Antimisiari, MPharm, University of Patras, and in the proteomic analysis study by G. Stamatakis, Fleming Institute, is highly acknowledged. Financial support was provided to A.M. by the Stavros Niarchos Foundation within the framework of the project ARCHERS (“Advancing Young Researchers’ Human Capital in Cutting Edge Technologies in the Preservation of Cultural Heritage and the Tackling of Societal Challenges”).
Authorship Contributions
Participated in research design: Marazioti, Stathopoulos, Panayotou, Antimisiaris.
Conducted experiments: Marazioti, Papadia, Kannavou, Spella, Basta, de Lastic, Rodi, Samiotaki.
Contributed new reagents or analytic tools: Panayotou, Samiotaki, Mouzaki, Stathopoulos.
Performed data analysis: Marazioti, Papadia, Samiotaki, Antimisiaris.
Wrote or contributed to the writing of the manuscript: Marazioti, Samiotaki, Mouzaki, Antimisiaris.
Footnotes
- Received January 31, 2019.
- Accepted April 30, 2019.
Financial support was provided by the Stavros Niarchos Foundation [ARCHERS] (to A.M.).
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ACN
- acetonitrile
- B16F10
- C57BL/6 mouse B16F10 skin melanoma cells
- BBB
- blood-brain barrier
- Chol
- cholesterol
- CVs
- cellular vesicles
- DiR
- 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide
- DRV
- dried reconstituted vesicles
- FCS
- fetal calf serum
- FI
- fluorescence intensity
- FITC
- fluorescein-isothiocyanate-dextran-4000
- FVB
- Friend leukemia virus B
- hCMEC/D3
- immortalized human cerebral microvascular endothelial cells
- HEK-293
- human embryonic kidney 293 cells
- LFQ
- label-free quantification
- LIP
- liposomes
- LY
- lucifer yellow-CH dilithium salt
- MS
- mass spectrometry
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OX-26
- mouse anti-rat CD71 IgG2a
- PC
- 1,2-distearoyl-sn-glycerol-3-phosphatidylcholine
- PDI
- polydispersity index
- PEG
- 1,2-distearoyl-sn-glycerol-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000]
- PEG-MAL
- 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000]
- PG
- 1,2-distearoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt)
- RES
- reticuloendothelial system
- RHO
- lissamine rhodamine B phosphatidylethanolamine
- TEER
- transendothelial electrical resistance
- TEM
- transmission electron microscopy
- t-LIP
- targeted liposomes
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}