


Abstract
Cardiomyocytes derived from human induced pluripotent stem cells (hiPSC-CMs) are a promising cell source for cardiac repair after myocardial infarction (MI) because they offer several advantages such as potential to remuscularize infarcted tissue, integration in the host myocardium, and paracrine therapeutic effects. However, cell delivery issues have limited their potential application in clinical practice, showing poor survival and engraftment after transplantation. In this work, we hypothesized that the combination of hiPSC-CMs with microparticles (MPs) could enhance long-term cell survival and retention in the heart and consequently improve cardiac repair. CMs were obtained by differentiation of hiPSCs by small-molecule manipulation of the Wnt pathway and adhered to biomimetic poly(lactic-co-glycolic acid) MPs covered with collagen and poly(d-lysine). The potential of the system to support cell survival was analyzed in vitro, demonstrating a 1.70-fold and 1.99-fold increase in cell survival after 1 and 4 days, respectively. The efficacy of the system was tested in a mouse MI model. Interestingly, 2 months after administration, transplanted hiPSC-CMs could be detected in the peri-infarct area. These cells not only maintained the cardiac phenotype but also showed in vivo maturation and signs of electrical coupling. Importantly, cardiac function was significantly improved, which could be attributed to a paracrine effect of cells. These findings suggest that MPs represent an excellent platform for cell delivery in the field of cardiac repair, which could also be translated into an enhancement of the potential of cell-based therapies in other medical applications.
Introduction
Myocardial infarction (MI) continues to represent the leading cause of mortality and morbidity worldwide (Reed et al., 2017). Treatments are still palliative despite great medical advances in recent years, with heart transplantation currently constituting the only option providing a definite and reparative solution. However, heart transplantation is severely limited by donor availability and the major complications associated with such an invasive procedure (Tonsho et al., 2014). After a MI, it is estimated that 50 g of the human heart muscle becomes dysfunctional due to the irreversible loss of around 1 billion native cardiac cells (Gepstein, 2002). The endogenous regenerative mechanisms of the heart are insufficient to replace the dead tissue, which remains chronically impaired. Therefore, a large number of patients who suffer from a cardiac ischemic event undergo frequent rehospitalizations, take multiple medications, and progress toward end-stage heart failure (Bahit et al., 2018).
Cardiac cell therapy arose at the turn of the century as a novel strategy aiming to counteract the massive loss of tissue by repopulating the ischemic area with functional cardiac lineage cells. Since then, a large number of cell sources have been explored in preclinical studies, with several progressing to the clinical stage (Telukuntla et al., 2013). Because cardiomyocytes (CMs) are responsible for heart contraction, many efforts have focused on the development of in vitro protocols to generate contractile cardiac muscle cells similar to native heart cells (Boheler et al., 2002; Batalov and Feinberg, 2015). Thus far, embryonic stem cells have been the major source for obtaining CMs (Liu et al., 2018). However, ethical, immunogenic, and availability issues have limited their potential application (Nussbaum et al., 2007). Induced pluripotent stem cells (iPSCs) generated from adult cells (Takahashi and Yamanaka, 2006) and progress in cell reprogramming techniques provide another attractive source of CMs. CMs differentiated from iPSCs (iPSC-CMs) present potential advantages. These cells can be expanded to obtain a large number of cells, are nonimmunogenic (autologous), and do not require the destruction of embryonic tissue (Lalit et al., 2014; Cahill et al., 2017).
Cell therapy research conducted over the last few years has taught us several lessons. Originally, it was believed that transplanted stem cells develop into cardiac cell types. However, when the fate of engrafted cells was investigated, most of the studies reported that only a few cells, if any, actually gave rise to new cells (Nigro et al., 2018). Instead, cells were found to stimulate cardiac functional recovery by the secretion of a wide variety of biologically active molecules that modulate the behavior of nearby cells, which is known as the paracrine effect (Hodgkinson et al., 2016). Moreover, efficacy results that originated from these studies have been controversial due to the low percentage of local engraftment and survival of transplanted cells (Hou et al., 2005; Terrovitis et al., 2010).
In this framework, the association of stem cells and drug delivery systems is now taking cell therapy one step further. Biocompatible implantable/injectable hydrogels have been extensively explored to deliver and retain cells in the target tissue, bringing significant improvements in cardiac function (Saludas et al., 2017). However, these systems have also shown limitations, such as lack of long-term cell engraftment (Chow et al., 2017), administration procedures based on open-chest surgeries (Atluri et al., 2014), or the need for a careful adjustment of the gelation and mechanical properties. An alternative less widely investigated approach to deliver cells could be the use of biodegradable and biocompatible microparticles (MPs) (Saludas et al., 2018). MPs represent an excellent delivery platform, because they allow for minimally invasive administration procedures using catheter technology (Garbayo et al., 2016) and are localizable to the injection site (Leong and Wang, 2015). Among their diverse cell-based applications, MPs may be used as tridimensional scaffolds, as microcapsules, or as key elements to deliver molecules and control the architecture of cell aggregates (Ahrens et al., 2017).
In this study, we developed an effective strategy for cardiac repair by the combination of human iPSC-CMs (hiPSC-CMs) and biomimetic MPs. First, cell-MP complexes were prepared and in vitro cell viability was analyzed. We then studied the potential of the strategy to enhance long-term hiPSC-CM survival and engraftment in a mouse MI model and its effect on cardiac function and adverse ventricular remodeling. Finally, the phenotype, electrical coupling, and maturation of the engrafted cells were characterized. Altogether, the results obtained indicate that the combination of hiPSC-CMs with MPs is a promising approach to stimulate heart repair after a MI.
Materials and Methods
Materials
Poly(lactic-co-glycolic acid) (PLGA) with a monomer ratio (lactic acid/glycolic acid) of 50:50 Resomer RG 503H (molecular mass of 34 kDa) was provided by Boehringer-Ingelheim (Ingelheim, Germany). Polyethylene glycol (PEG; molecular mass of 400 Da), human serum albumin, Sigmacote, fibronectin from bovine plasma, poly(d-lysine) (PDL), cadmium chloride, Fast Red, hydrochloric acid, rabbit anti–connexin-43 antibody (C6219), and rabbit anti–α-sarcomeric actinin (A7811) were provided by Sigma-Aldrich (Barcelona, Spain). Dichloromethane, acetone, and formaldehyde were obtained from Panreac Química S.A. (Barcelona, Spain). Poly(vinyl alcohol) (88% hydrolyzed, with a molecular mass of 125 kDa) was obtained from Polysciences Inc. (Warrington, PA). Collagen I rat protein and phosphate-buffered saline (PBS), pH 7.2, were provided by Gibco-Invitrogen (Carlsbad, CA). The Live/Dead Viability/Cytotoxicity kit was obtained from Molecular Probes (Carlsbad, CA). Mouse anti-human mitochondria (ab92824) and rabbit anti-dystrophin (ab15277) primary antibodies were purchased from Abcam (Cambridge, UK). Alexa Fluor 488 goat anti-mouse (A10680) and Alexa Fluor 594 goat anti-rabbit (A11012) secondary antibodies were supplied by Invitrogen. Anti-cardiac troponin T (MA5-12960), Alamar Blue Cell Viability Reagent, the human episomal iPSC line, Essential 8 Medium, RPMI basal medium, B27 and B27 minus insulin supplements, as well as other cell culture reagents were obtained from Thermo Fisher (Carlsbad, CA). Small molecules CHIR99021 and Wnt-C59 were from Axon Medchem (Groningen, The Netherlands), whereas ROCK inhibitor Y27632 was purchased from Tocris (Bristol, UK) and Growth Factor Reduced Matrigel Matrix was from BD Bioscience (Madrid, Spain).
Differentiation and Isolation of CMs from hiPSCs
CMs were obtained by differentiation of hiPSCs as described in Lian et al. (2013). Briefly, cells were maintained in Essential 8 Medium on 1:180 Growth Factor Reduced Matrigel–coated plates until subconfluence, when hiPSCs were routinely passaged at a 1:15 ratio. For differentiation, cells were plated on 12-well plates as above. When the culture reached 80%–90% confluence, the medium was changed to RPMI supplemented with 1× B27 minus insulin (RPMI minus insulin) plus 8 µM CHIR99021 for 24 hours. Then the medium was replaced with RPMI minus insulin for 48 hours, followed by 5 µM C-59 in RPMI minus insulin for another 48 hours. Next, the medium was shifted again to RPMI minus insulin for 48 hours, before changing to RPMI supplemented with 1× B27 (RPMI-B27). This medium was renewed every other day until the appearance of beating (usually 7–9 days after the start of the differentiation), when a metabolic selection of hiPSC-CMs by culturing for 72 hours in RPMI without glucose supplemented with 1× B27 and 4 mM lactate was applied. Cells were returned to conventional RPMI-B27 for 48 hours before another 72 hours of metabolic selection in the above-mentioned medium. After that, cells were returned to RPMI-B27 for 48 hours and isolated. At this point, hiPSC-CMs formed beating monolayers. The cells were washed three times with PBS plus 0.5 mM EDTA and incubated for 7–15 minutes in warm TrypLE. hiPSC-CMs were then mechanically dissociated with a micropipette tip and counted with trypan blue to exclude dead cells. The required amount of cells was suspended in RPMI-B27 plus 1 µM Y27632 and spun down at 1000 rpm for 10 minutes.
Characterization of hiPSC-CMs
Differentiation purity was assessed by fluorescence-activated cell sorting (FACS), whereas hiPSC-CMs identity was confirmed by functionality (beating), gene expression, and protein expression. For FACS, cells were stained with anti-cardiac troponin T (1:100, MA5-12960) using the Fix & Perm kit and analyzed. RNA extraction was carried out with TRI Reagent, whereas reverse transcription (RT) was performed using the TaKaRa PrimeScript RT Reagent Kit, following the manufacturer’s instructions, with a maximum of 500 ng RNA per sample. Semi-quantitative RT-polymerase chain reaction was performed with primers shown in Supplemental Table 1, using Applied Biosystems PowerUp SYBR Green Master Mix in a QuantStudio 5 system (Thermo Fisher Scientific). Glyceraldehyde-3-phosphate dehydrogenase was selected as a housekeeping gene and the results were analyzed using the 2−ΔΔCt method. For protein expression, cells were labeled with anti–α-sarcomeric actinin (1:200, A7811) and visualized with a Zeiss confocal microscope (Zeiss, Madrid, Spain).
Preparation of MPs
PLGA particles were prepared by the multiple emulsion solvent evaporation method using the Total Recirculation One-Machine System (Formiga et al., 2013; Pascual-Gil et al., 2015). Briefly, the organic phase (O) consisting of 50 mg PLGA 503H dissolved in a mixture of 4 ml acetone/dichloromethane (1:3) was injected into the inner aqueous phase (W1) formed by 5 mg human serum albumin, 5 µl PEG 400, and 200 µl PBS, pH 7.4. The W1/O emulsion was allowed to recirculate through the system for 1 minute and 30 seconds. Then this emulsion was added to the outer aqueous phase (W2) consisting of 20 ml 0.5% poly(vinyl alcohol) and allowed to recirculate for 2 minutes and 30 seconds. Finally, the W1/O/W2 emulsion was stirred at room temperature for 3 hours to allow total solvent evaporation. MPs were washed three times with ultrapure water by consecutive centrifugation at 20,000g at 4°C for 5 minutes and lyophilized for 48 hours (Genesis Freeze Dryer 12 EL; VirTis, Gardiner, NY). Lyophilized MPs were stored at 4°C.
Characterization of MPs
Particle size, size distribution, and ζ potential were determined after lyophilization. Particle size and size distribution were measured by laser diffractometry using a Mastersizer (Malvern Instruments, Malvern, UK). MPs were dispersed in ultrapure water and analyzed under continuous stirring. The average particle size was expressed as the volume mean diameter. Particle surface charge was determined by ζ potential measurement using a Zetasizer Nano ZS (Malvern Instruments), based on the analysis of complete electrophoretic mobility distributions.
Particle Surface Modification
To facilitate the adhesion of hiPSC-CMs to MPs, the particle surface was functionalized by coating with biomimetic molecules. Different mixtures of molecules and concentrations were tested to select the most adequate to favor the adhesion of hiPSC-CMs. The different coatings studied were as follows: 1) a mixture of 30 µg/ml collagen type I and 100 µg/ml PDL, 2) a mixture of 60 µg/ml collagen type I and 100 µg/ml PDL, and 3) a mixture of 30 µg/ml fibronectin and 100 µg/ml PDL. In all cases, particle coating was performed in Sigmacote-coated tubes. MPs were dispersed in acidified PBS (pH 5.7). Then biomimetic molecules were added to the particle solution and the mixtures were incubated at 37°C for 1 hour under rotation. Coated particles were washed with distilled sterile water by consecutive centrifugations (25,000g, 4°C, 10 minutes) and lyophilized for 48 hours. ζ potential was measured to confirm that the particles had been successfully coated.
Adhesion of hiPSC-CMs to MPs
For the adhesion of cells, 0.150 mg coated MPs was dispersed in RPMI-B27 medium prior to the addition of 150,000 CMs. The mixture was plated in a Costar ultra-low cluster flat-bottom sterile polystyrene plate and incubated at 37°C. The evolution of the adhesion of cells to MPs was assessed at 30 minutes and at 1, 1.5, 2, and 4 hours by bright-field microscopy (Nikon TMS, Amsterdam, The Netherlands).
In Vitro Cell Survival
The survival of cells cultured alone or adhered to MPs was evaluated by the Live/Dead and Alamar Blue assays, following the manufacturer’s instructions. For the assays, 150,000 cells alone or 150,000 cells adhered to 0.150 mg MPs were incubated in a Costar ultra-low cluster flat-bottom sterile polystyrene plate at 37°C. After 1 and 4 days of incubation, samples were stained with a Live/Dead staining kit for 30 minutes at 37°C and were then examined using an LSM 800 confocal microscope (Zeiss). The survival rate of cells was quantified by incubation with the Alamar Blue kit at 37°C. After 6 hours of incubation, absorbance was measured at 570 and 600 nm using a Power Wave XS Microplate Spectrophotometer. Survival was calculated following the manufacturer’s instructions. Four to eight replicates were used in each treatment and the test was performed in quadruplicate.
In Vivo Studies Using a Chronic MI Model
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Navarra and were performed according to the requirements of EU legislation.
Permanent myocardial ischemia was induced in male BALB/C (Rag-2) mice aged 8–12 weeks. Briefly, animals were anesthetized with isoflurane and intubated for mechanical ventilation. Prior to surgery, animals received ketoprofen, fentanest, and enrofloxacin. Mice underwent a left thoracotomy through the fourth intercostal space and the left anterior descending coronary artery was permanently occluded. After 15 minutes of artery occlusion, the animals received one of the following treatments: MPs coated with PDL and collagen (0.150 mg, n = 13) or hiPSC-CM-MPs (150,000 hiPSC-CMs adhered to 0.150 mg MPs coated with PDL and collagen, n = 11). Treatments were dispersed in 12 µl injection medium and injected intramyocardially into two areas using a 27G syringe. Finally, animals were closed, administered buprenorphine, and allowed to recover. Buprenorphine was obtained from Schering AG (Berlin, Germany).
Cardiac Function Evaluation
Echocardiography was performed using a Vevo 770 ultrasound system (Visualsonics, Toronto, ON, Canada) at 2 and 60 days after ligation of the left anterior descending coronary artery. Measurements were optimized for small animals and performed as previously described (Benavides-Vallve et al., 2012). The left ventricular ejection fraction (LVEF), fractional area change (FAC), end-systolic volume (ESV), and end-diastolic volume (EDV) were studied. A total of 24 animals with a LVEF below 45% at day 2 after MI were included in the study.
Morphometric and Histologic Studies
Male mice were sacrificed at day 7 (n = 4 for the MP group, n = 3 for the hiPSC-CM-MP group) or at day 60 (n = 9 for the MP group, n = 8 for the hiPSC-CM-MP group) to perform the morphometric and histologic studies. Briefly, mice were anesthetized, injected with 100 µl 0.1 mM cadmium chloride for diastole cardiac arrest, and perfusion-fixed for 15 minutes with zinc formalin under physiologic pressure. The hearts were excised, fixed overnight in zinc formalin at 4°C, cut into three equally sized blocks (apical, midventricular, and basal), dehydrated in 70% ethanol (4°C, overnight), and embedded in paraffin. For histologic analysis, 5-µm serial sections were prepared.
Infarct size and heart wall thickness were determined using Sirius Red–stained sections. For the Sirius Red staining, sections were deparaffinized and immersed for 90 minutes in 0.1% Fast Red, which was diluted in a saturated solution of picric acid. They were then differentiated for 2 minutes in 0.01 N HCl, dehydrated, and mounted in DPX. Infarct size was assessed by quantifying images from 12 serial heart sections, 50 μm apart. Images were analyzed with ImageJ software (version 1.48) and the data were expressed as a percentage of the ischemic area versus the total left ventricle area. For quantifying infarct wall thickness, 12 images from serial heart sections of the infarct zone were analyzed using ImageJ software (version 1.48).
The survival and engraftment of transplanted cells was analyzed and quantified at 7 and 60 days after administration in the ischemic tissue. Due to the human origin of transplanted cells, these cells could be identified in vivo by immunostaining using a mouse anti-human mitochondria antibody (1:1000, ab92824). The number of engrafted cells was calculated by quantifying images from 15 serial heart sections 50 μm apart and extrapolating this number to the whole length of the graft area. Data were expressed as a percentage of the number of engrafted cells versus the number of injected cells. Electrical coupling of transplanted cells and preservation of the cardiac phenotype were studied using rabbit anti–connexin-43 (1:500, C6219) and rabbit anti-dystrophin antibodies (1:100, ab15277), respectively. In addition, Alexa Fluor 488 goat anti-mouse (1:200, A10680) and Alexa Fluor 594 goat anti-rabbit (1:200, A11012) secondary antibodies were used. Fluorescently stained tissue slides were observed with a camera attached to a Zeiss Axio Imager M1 fluorescence microscope.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software (version 5.0; GraphPad Software Inc., San Diego, CA). Differences between both groups for each time point were analyzed by a t test for independent samples. Results are expressed as means ± S.E.M. Statistical significance was determined by P values < 0.05.
Results
Cardiac Differentiation of hiPSCs
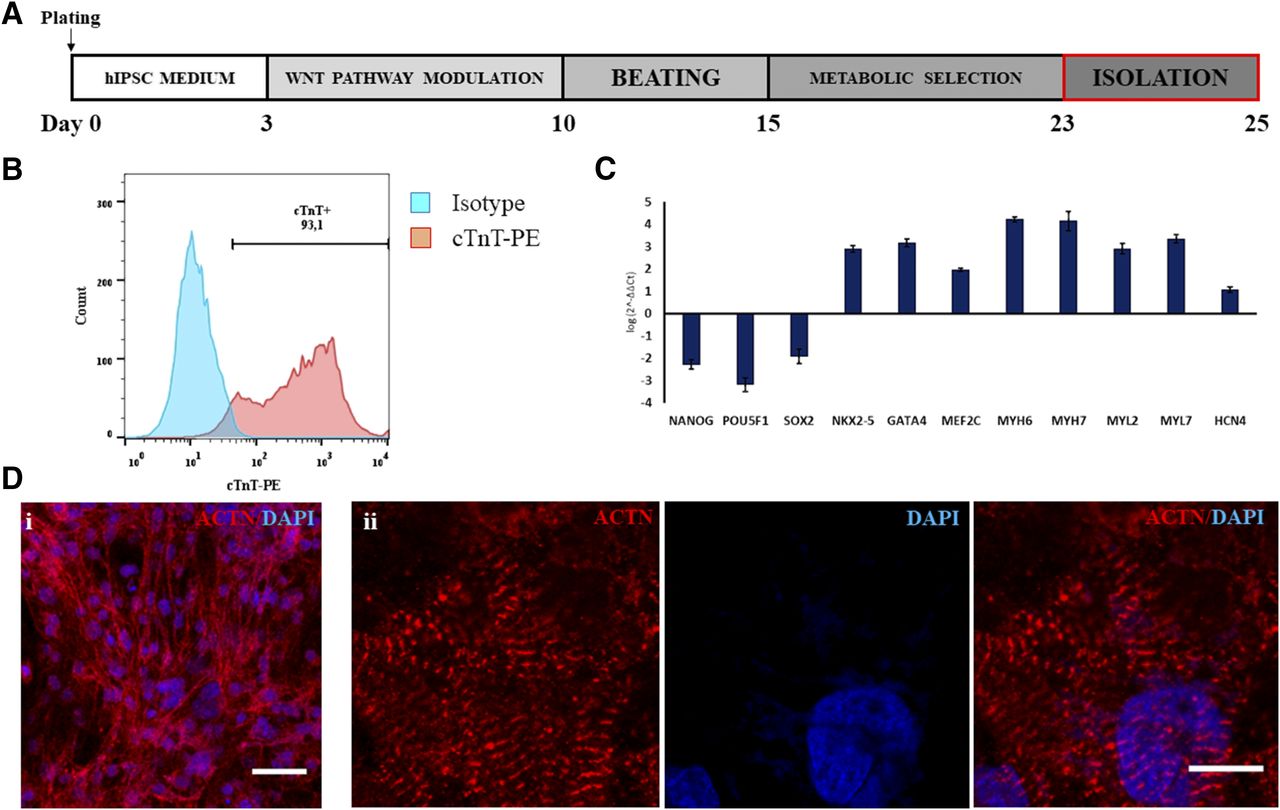
Cardiac differentiation was performed by small-molecule manipulation of the Wnt pathway, followed by enrichment through metabolic selection (Fig. 1A). This resulted in the final obtaining of beating monolayers of hiPSC-CMs (Supplemental Video 1), with all experiments showing a purity above 90% of cardiac troponin T–positive cells by FACS (Fig. 1B). Quantitative RT-polymerase chain reaction analysis (Fig. 1C) showed a robust downregulation of pluripotency-associated genes (NANOG, PU5F1, and SOX2), concomitant with an upregulation of a cardiac-specific gene expression profile (NKX2-5, GATA4, MEF2C, MYH6, MYH7, MYL2, MYL7, and HCN4). hiPSC-CMs were mononucleated, with well defined sarcomeres, as depicted by α-sarcomeric actinin staining (Fig. 1D, i and ii).

Cardiac differentiation of hiPSCs. (A) Diagram of small molecule–based CM differentiation and metabolic selection. (B) Representative FACS staining for cTnT, showing a purity of >90% of hiPSC-CMs. (C) Gene expression profile of hiPSC-CMs, demonstrating proper downregulation of pluripotency genes (NANOG, POU5F1, and SOX2) and upregulation of cardiac transcription factors (NKX2-5, GATA4, and MEF2C) and contractile proteins (MYH6, MYH7). Coexpression of genes for CM subtypes was detected: namely, ventricular (MYL2), atrial (MYL7), and pacemaker (HCN4) subtypes. (D) Immunofluorescence for α-sarcomeric actinin, confirming the high purity of cultures (i), with cells displaying evident sarcomeric striations (ii). Data represent means ± S.D. ACTN, alpha-sarcomeric actinin; cTnT, cardiac troponin T; DAPI, 4′,6-diamidino-2-phenylindole; PE, phycoerythrin. Scale bar, 25 µm (Di); 10 µm (Dii).
Microparticle Characterization
MPs were manufactured using PLGA, a biocompatible and biodegradable U.S. Food and Drug Administration–approved polymer, as a model system (Makadia and Siegel, 2011). PLGA MPs were prepared by the multiple emulsion solvent evaporation method using the Total Recirculation One-Machine System. A homogenous population of MPs with a mean particle size of 10.4 ± 0.8 µm was obtained. Particles presented a negative surface charge with a ζ potential of −20.4 ± 3.3 mV.
Particle Surface Modification
The surface of particles was functionalized with biomimetic molecules to facilitate the adhesion of hiPSC-CMs. Collagen type I and fibronectin are proteins of the cardiac extracellular matrix that bind cell adhesion molecules on the surface of cells, whereas PDL increases the surface charge of MPs to positive values. This switch of ζ potential may attract cells by nonspecific interactions between the negatively charged cell membrane and the positively charged surface of MPs (Delcroix et al., 2011; Garbayo et al., 2011). ζ potential was measured to confirm the successful coating. All coated particles presented positive ζ potential values, with those particles coated with a larger proportion of PDL presenting higher values. In particular, the coating of MPs with 30 µg/ml collagen and 100 µg/ml PDL increased the ζ potential to +20.0 ± 2.2 mV, the coating with 60 µg/ml collagen and 100 µg/ml PDL to +16.3 mV, and the coating with 30 µg/ml fibronectin and 100 µg/ml PDL to +25.0 mV.
Adhesion of hiPSC-CMs to MPs
The evolution of the adhesion of hiPSC-CMs to MPs with different coatings was studied at 30 minutes and at 1, 1.5, 2, and 4 hours by bright-field microscopy. On the one hand, when cells were incubated with MPs covered with 30 µg/ml fibronectin and 100 µg/ml PDL, only a small number of cells were adhered to the particles after 1.5 hours of incubation (Fig. 2A). By contrast, the coating of particles with 30 or 60 µg/ml collagen and 100 µg/ml PDL induced a greater adhesion of hiPSC-CMs. After 1.5 hours of incubation, most of the cells were successfully adhered to the surface of the particles (Fig. 2A). Incubation for longer times did not increase cell adhesion. Similar results were obtained in terms of particle surface charge and cell adhesion properties with both tested concentrations of collagen, which suggests that 30 µg/ml collagen creates a large enough biomimetic surface for the adhesion of 150,000 hiPSC-CMs. From a practical point of view, the coating formed by the lowest concentration of collagen blended with PDL (i.e., 30 µg/ml collagen and 100 µg/ml PDL) was selected for further studies to obtain a less expensive protocol with potential for future clinical translation.

Adhesion of hiPSC-CMs to biomimetic MPs and in vitro cell viability. (A) Bright-field images of the complexes formed by 150,000 hiPSC-CMs and 0.150 mg MPs covered with different mixtures of biomimetic molecules after 1.5 hours of incubation. MPs coated with 30 µg/ml collagen type I and 100 µg/ml PDL showed the largest adhesion of hiPSC-CMs and were selected for further studies. (B) Representative confocal images of hiPSCs cultured alone or in combination with biomimetic MPs for 24 hours. Calcein AM (green) stains for live cells and ethidium homodimer-1 (red) for dead cells. Scale bar, 50 µm.
In Vitro Survival of Cells
The potential of MPs to provide cells with a three-dimensional microenvironment that enhances cell survival and facilitates cell biologic functions was analyzed by the Live/Dead and Alamar Blue assays. As seen in the Live/Dead assay, the number of viable cells after 1 day of incubation was increased when cells were cultured associated with MPs compared with cells alone (Fig. 2B). This higher survival was maintained up to day 4. These results were quantified and confirmed with the Alamar Blue assay. The use of particles as a physical support for cells produced a significant 1.7-fold increase in cell survival over the culture of cells alone after 1 day and a 1.99-fold increase after 4 days. These results demonstrate that the adhesion of cells to the surface of biomimetic MPs stimulates cell viability by providing cells with a three-dimensional support.
Cardiac Functional Analysis
We then tested the efficacy of the complexes to repair the damaged myocardium in a mouse MI model. LVEF, FAC, ESV, and EDV were analyzed by echocardiography at 2 and 60 days after treatment administration. After 2 days, no differences could be found between the MP and hiPSC-CM-MP groups for any of the functional parameters studied. Two months after treatment administration, LVEF was significantly enhanced (P < 0.05) in animals treated with hiPSC-CM-MPs (41.25% ± 2.57%) compared with animals treated with MPs (33.21% ± 1.86%) (Fig. 3A). Similar results were obtained when FAC was studied. This parameter was significantly increased (P < 0.05) in the hiPSC-CM-MP group (30.31% ± 2.28%) compared with the MP group (23.26% ± 1.46%) (Fig. 3B). Furthermore, functional analysis revealed that pathologic hypertrophy of the left ventricle was prevented after 2 months in the combinatorial treatment, as reflected in the ESV and EDV parameters. In this sense, a significant reduction was found in the hiPSC-CM-MP group compared with the MP group in both ESV (MP group: 96.95 ± 8.21 µl; hiPSC-CM-MP group: 63.16 ± 3.09 µl; P < 0.01) and EDV (MP group: 142.70 ± 8.77 µl; hiPSC-CM-MP group: 111.80 ± 4.8 µl; P < 0.05) (Fig. 3, C and D). These findings reveal that the combinatorial use of hiPSC-CMs and MPs as a therapeutic option is able to induce significant cardiac repair, as shown by the improvement in cardiac function.
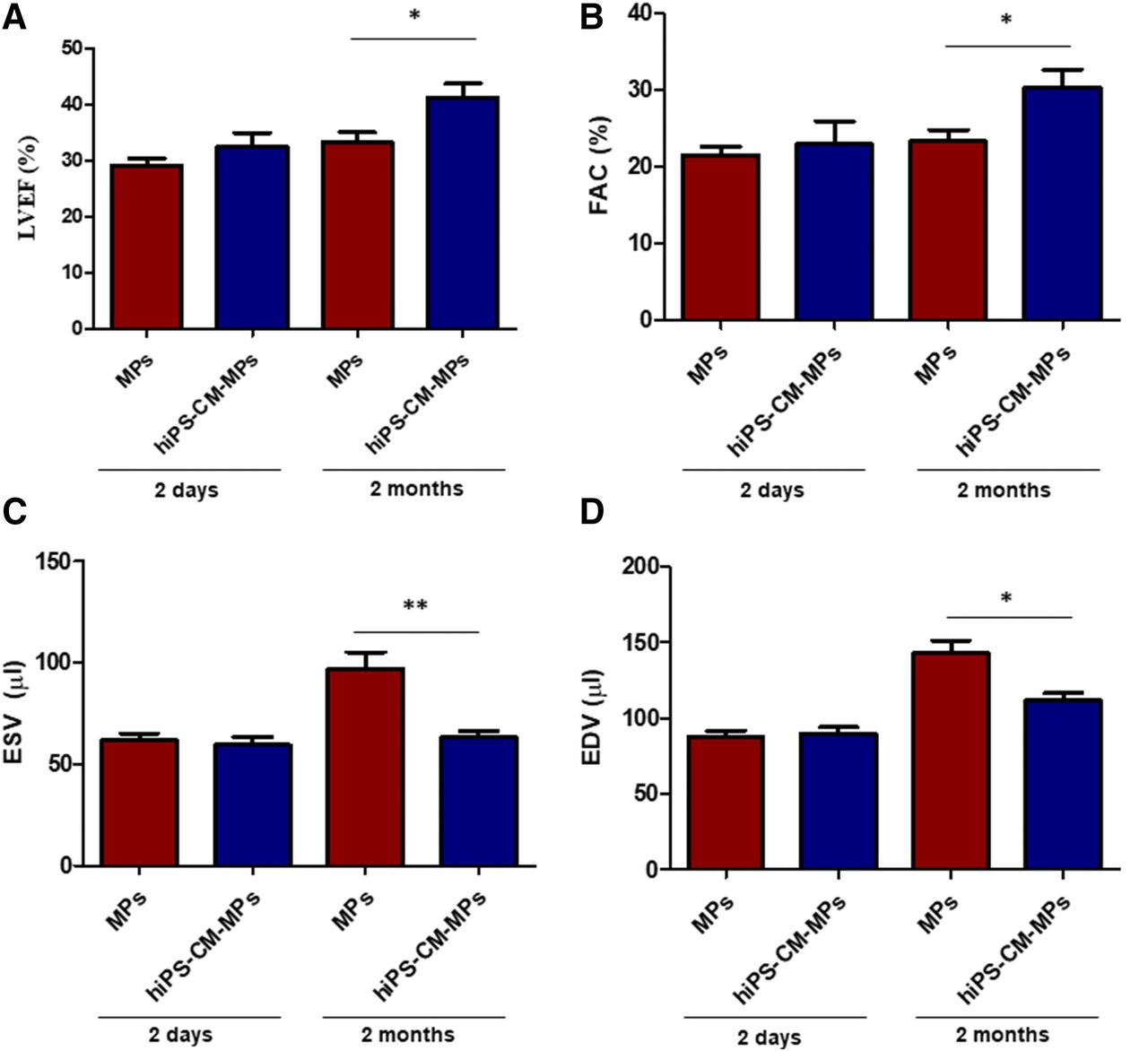
Effects of hiPSC-CM-MPs on cardiac function. (A–D) LVEF (A), FAC (B), ESV (C), and EDV (D) measured by echocardiography at 2 days and at 2 months after treatment in animals injected with MPs alone or with hiPSC-CMs adhered to biomimetic MPs (hiPSC-CM-MPs). Results show that combinatorial treatment of MPs and hiPSC-CMs improves cardiac function after 2 months compared with MPs. Data are expressed as means ± S.E.M. *P < 0.05; **P < 0.01 (t test for independent samples).
Morphometric and Histologic Studies
Left Ventricle Remodeling.
Left ventricle wall thickness and infarct size were analyzed after 2 months of treatment administration. Left ventricle wall thickness was slightly increased in the animals treated with hiPSC-CM-MPs (499.0 ± 41.7 µm) compared with animals treated only with MPs (446.2 ± 58.4 µm) (Fig. 4A). Similarly, a tendency toward a reduction of infarct size was observed in the hiPSC-CM-MP group (19.93% ± 2.91%) compared with the MP group (25.09% ± 4.28%) (Fig. 4B). These findings may suggest that MPs alone or in combination with cells induce a similar prevention of adverse ventricle remodeling.
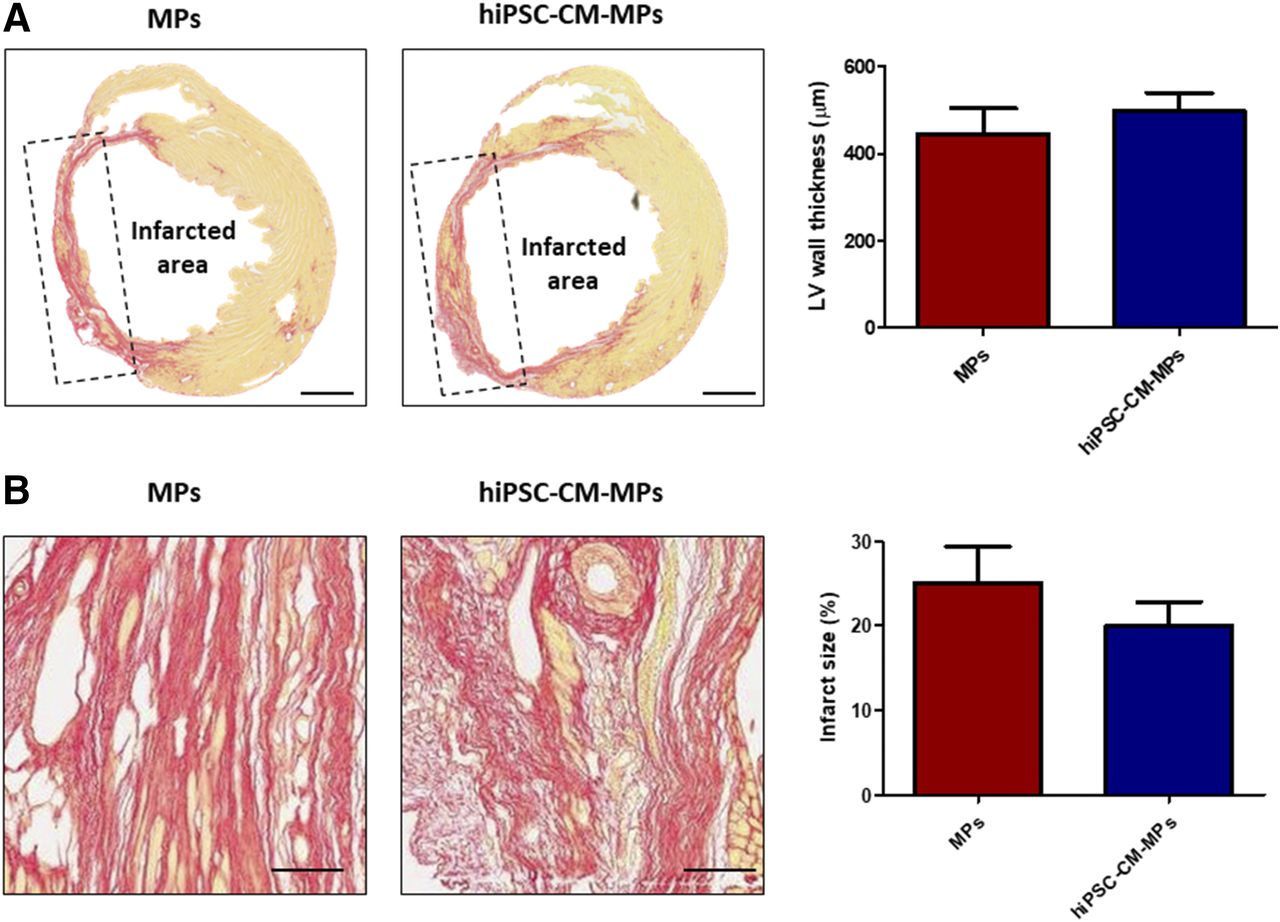
Effects of hiPSC-CM-MPs on ventricle remodeling. (A and B) Representative images and quantification of left ventricle wall thickness (A) and infarct size (B) as measured by Sirius Red staining after 2 months of treatment administration. Both treatment groups presented similar wall thickness of the left ventricle, whereas administration of hiPSC-CM-MPs showed a tendency to reduce infarct size. Data are expressed as means ± S.E.M. LV, left ventricle. Scale bar, 1 mm (A); 100 µm (B).
Fate of Transplanted Cells.
The engraftment and survival of transplanted hiPSC-CMs was analyzed at 1 week and 2 months after administration in the ischemic tissue by immunofluorescence using an anti-human mitochondria antibody, which allows the specific detection of cells of human origin. One week after administration, transplanted cells could be detected at the graft site in the infarct and peri-infarct area. hiPSC-CMs were relatively small, presented a round-shape morphology, and were arbitrarily distributed. Interestingly, when cell survival was analyzed after 2 months, engrafted cells could still be detected at the injection site surrounding the damaged area (<1%) (Fig. 5A). At this time, engrafted cells showed a certain maturation, reflected in their larger and elongated morphology and the appearance of clusters of aligned hiPSC-CMs. In addition, engrafted cells preserved their cardiac phenotype, as reflected in the expression of dystrophin throughout the cytoplasm (Fig. 5B). Finally, we investigated whether engrafted cells formed gap junctions with other cells and therefore if they showed signs of electrical coupling to native cardiac cells. For that, the expression of Cx-43 was analyzed. After 2 months of administration in the cardiac tissue, transplanted hiPSC-CMs showed expression of Cx-43 at the interface between transplanted hiPSC-CMs (Fig. 5C). Altogether, the results indicate that the transplantation of cells in combination with MPs enhances cell survival and engraftment. In addition, engrafted hiPSC-CMs maintain a cardiac muscle phenotype and express cardiac Cx-43 gap-junction protein after 2 months of administration.

Fate of hiPSC-CMs after transplantation in the cardiac tissue. (A) Analysis of cell engraftment and survival at 1 week and 2 months after administration into the hearts of infarcted mice. Representative areas of infarcted myocardium showing hiPSC-CMs (green) engrafted in the tissue at both time points (area delimited by a discontinuous line). (B) Expression of dystrophin (red) was observed in transplanted hiPSC-CMs after 2 months (arrow), suggesting that these cells maintained a cardiac phenotype. (C) Engrafted hiPSC-CMs expressed the gap-junction protein connexin-43 (red) at the interface between CMs, suggesting electrical coupling 2 months after administration (arrows). CX-43, connexin-43; DAPI, 4′,6-diamidino-2-phenylindole; DYS, dystrophin; h-MITO, infarcted myocardium showing hiPSC-CMs. Scale bar, 50 µm.
Discussion
In this article, we have effectively developed a strategy for cardiac repair based on the enhancement of hiPSC-CM therapy potential through combination with biomimetic MPs. Given that the low survival of cells in the cardiac tissue critically limits the clinical application of cell therapy, the main goal of this study was to implement a system to improve long-term cell engraftment. Specifically, in vitro studies revealed that the combination of hiPSCs with biomimetic MPs enhanced cell survival compared with cells alone, indicating the potential of MPs to support cell functions. In consonance, the transplantation of hiPSC-CMs associated with biomimetic MPs in a mouse MI model notably increased cell survival up to 2 months. After this period, engrafted cells showed maturation and expression of Cx-43 gap-junction protein. Importantly, progress in cell delivery shown in this study correlated with a cardiac functional improvement, mainly attributed to a paracrine effect of cells. Overall, these results highlight the potential of MPs to overcome cell delivery issues, which could be translated into a greater stimulation of heart repair.
Numerous preclinical and clinical studies have been conducted aiming to elucidate the cell source that presents the largest potential to stimulate the repair of the necrotic myocardium (Yu et al., 2017). In a previous study, our group demonstrated that the transplantation of adipose-derived stem cells combined with MPs enhances cardiac repair in a rat MI model (Díaz-Herráez et al., 2017). However, when the fate of transplanted cells was analyzed, adipose-derived stem cells did not differentiate to CMs. Lack of differentiation of stem cells has been also reported in other studies (Lin et al., 2010). Transplantation of differentiated CMs could bring some advantages. Injection of cells similar to the native myocardium is related to the mechanical and functional coupling of cells, which reduces the appearance of arrhythmias (Pijnappels et al., 2010). In addition, Tachibana et al. (2017) recently described that hiPSC-CMs salvage the myocardium more effectively than hiPSCs through their differential paracrine secretion. hiPSCs are currently the only realistic CM source. Furthermore, it is possible to apply robust differentiation protocols to generate CMs that can be implemented in regenerative therapies, as demonstrated in this article and by others (Wang et al., 2016; Breckwoldt et al., 2017).
One of the most interesting findings of this study is the demonstration that MPs are powerful carriers to improve cell survival both in vitro and in vivo. Previous studies showed that the number of cells remaining in the cardiac tissue after a few hours is extremely low (Hou et al., 2005; Terrovitis et al., 2010). Interestingly, in a previous study in which hiPSC-CMs were injected in a mouse MI model, 3.8% of cells could be found at 7 days, 0.3% could be found at 14 days, and no cells were found at 1 and 2 months after administration (Iglesias-García et al., 2015). In this study, we were able to localize engrafted cells in the cardiac tissue after 2 months, which greatly encourages the potential of particles to support cell viability. The positive impact that the association with particles has on cell viability and engraftment can be explained by different mechanisms. Although MPs have traditionally been used for protein encapsulation (Pascual-Gil et al., 2015; Garbayo et al., 2016; Suarez et al., 2016) rather than cell delivery, they also constitute suitable scaffolds to provide cells with a biomimetic tridimensional support (Díaz-Herráez et al., 2013). Furthermore, our group proved that MPs similar to those employed in this study are retained in the myocardium for up to 3 months, avoiding mechanical washout of cells from the heart (Formiga et al., 2013). With the same aim of overcoming the low engraftment of cells, other authors have explored the delivery of hiPSC-CMs encapsulated in hydrogels (Wang et al., 2015; Chow et al., 2017). Wang et al. (2015) found an increased survival of hiPSC-CMs in the heart after transplantation in the hydrogel matrix at 2 weeks, but survival rates were not studied at longer times. On the other hand, despite the encouraging results in terms of cardiac repair, Chow et al. (2017) reported the absence of grafted cells after 10 weeks when transplanted in a synthetic PEG hydrogel, a similar endpoint to ours. In view of these results, we suggest that biomimetic MPs could constitute better cell delivery platforms than hydrogels. Further studies using natural hydrogels and hiPSC-CMs would help to compare results with both delivery systems.
Going a step further, our data demonstrate that hiPSC-CM grafts showed a certain degree of maturation in the cardiac tissue, with a larger size and elongated morphology after 2 months. In addition, clusters of cells could be found in an aligned disposition, which could reveal integration within the surrounding myocardium. We have also shown that cells expressed dystrophin after 2 months, which confirms that engrafted cells maintained their cardiac phenotype. In the literature, there are currently only a few studies describing the in vivo engraftment, maturation, and alignment of transplanted cells in the infarcted myocardium in the long term. One example is the work recently carried out by Liu et al. (2018), who found a similar in vivo maturation of CMs derived from embryonic stem cells. Integration in the native myocardium could be suggested by the expression of the gap-junction protein Cx-43 observed in grafted cells. Expression of this protein could be related to the electrical coupling of cells and protection against ventricular arrhythmias (Roell et al., 2007). Apart from electrical coupling, transplanted cells must achieve functional integration to avoid the appearance of arrhythmias. For this, cells must be electromechanically similar to the endogenous adult myocardium (Pijnappels et al., 2010), which supports the transplantation of differentiated CMs instead of stem cells.
Finally, we were able to confirm that the long-term engraftment of hiPSC-CMs resulted in the repair of the damaged cardiac tissue, which is the final objective of this strategy. The enhanced cardiac function observed could result from a dual action of particles and cells, since MPs alone were able to improve cardiac output and stroke volume (data not shown). In line with this, no significant differences were found between the MP and hiPSC-CM-MP groups in infarct size and left ventricle wall thickness. These results could be due to the mechanical reinforcement that particles provide to the ventricle wall, which increases wall thickness, prevents infarct expansion, and therefore enhances the function of the host myocardium. Besides that, cardiac repair is mainly attributed to the delivered cells. Interestingly, the significant prevention in the increase of ESV and EDV after 2 months, reflects that the combinatorial treatment avoided the enlargement of the left ventricle to compensate for the loss of function. The low engraftment of transplanted hiPSC-CMs shows that cells stimulate cardiac repair by the paracrine secretion of growth factors or cytokines. It has been previously described that hiPSC-CMs secrete antiapoptotic, proangiogenic, and cell migration–related factors (Tachibana et al., 2017). These results are in agreement with recent experiments by Tachibana et al. (2017) and Zhu et al. (2018), attributing the functional improvements observed after transplantation of cardiac myocytes or cardiovascular progenitors to the paracrine effect. Moreover, the lack of evident sarcomeric striations 2 months after transplantation further points to the paracrine capacity over contractile support as the driver of the functional benefit.
In summary, the findings of this study confirm that biomimetic particles represent an excellent platform to improve long-term cell engraftment, due to their ability to create a favorable microenvironment that retains cells at the injection site and supports cell functions. Future studies should focus on the mechanisms responsible for the therapeutic response observed, such as characterization of the paracrine secretion of hiPSC-CMs, as well as its impact on the endogenous tissue. Furthermore, specific molecules could be incorporated into MPs to direct transplanted cell function, as shown in other studies (Mahoney and Saltzman, 2001). It should also be considered that in this study, cells of human origin were injected in a mouse MI model. The use of more representative models of human physiology (pig or nonhuman primate) could help to elucidate the potential of hiPSC-CMs combined with MPs to directly remuscularize infarcted tissue. Finally, this platform could be implemented not only in the field of cardiac repair but also in other areas of regenerative medicine.
Authorship Contributions
Participated in research design: Saludas, Garbayo, Mazo, Pelacho, Raya, Prósper, Blanco-Prieto.
Conducted experiments: Saludas, Garbayo, Mazo, Pelacho, Abizanda, Iglesias-Garcia.
Performed data analysis: Saludas, Garbayo, Mazo, Pelacho, Abizanda.
Wrote or contributed to the writing of the manuscript: Saludas, Garbayo, Mazo, Pelacho, Prósper, Blanco-Prieto.
Footnotes
- Received December 12, 2018.
- Accepted January 29, 2019.
L.S. and E.G. contributed equally to this work.
This work was supported by the Spanish Ministry of Economy and Competitiveness (SAF2013-42528-R, SAF2015-69706-R, SAF2017-83734-R and Cardiomesh), funds from the ISCIII and FEDER (Tercel RD16/0011/0005, RD16/0011/0024, PI16/00129, CPII15/00017 and ERANET II-Nanoreheart), “Asociación de Amigos de la Universidad de Navarra” and “la Caixa” Banking Foundation.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- CM
- cardiomyocyte
- EDV
- end-diastolic volume
- ESV
- end-systolic volume
- FAC
- fractional area change
- FACS
- fluorescence-activated cell sorting
- hiPSC
- human induced pluripotent stem cell
- hiPSC-CM
- cardiomyocyte derived from human induced pluripotent stem cell
- LVEF
- left ventricular ejection fraction
- MI
- myocardial infarction
- MP
- microparticle
- PBS
- phosphate-buffered saline
- PDL
- poly(d-lysine)
- PEG
- polyethylene glycol
- PLGA
- poly(lactic-co-glycolic) acid
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}