


Abstract
Cocaine abuse is a public health concern with seizures and death being one consequence of overdose. In the present study, dopamine D3/D2 receptor agonists dose dependently and completely prevented the convulsant and lethal effects of cocaine. The D3-preferring agonists R-(+)-trans-3,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano[4,3-b]-1,4-oxazin-9-ol) [(+)-PD 128,907], (+)-7-hydroxy-dipropylaminotetralin, and the mixed D3/D2 agonists quinpirole and quinelorane were all effective against cocaine toxicity in mice. The anticonvulsant effects of these compounds occurred at doses below those that produced motor impairment as assessed in the inverted screen test. Protection against the convulsant effects of the selective dopamine uptake inhibitor 1-[2-[bis(4-fluorophenyl)methoxy] ethyl]-4-[3-phenyl-propyl]piperazine (GBR 12909) was also conferred by (+)-PD 128,907. The possible selectivity of the effects of (+)-PD 128,907 (3 mg/kg) for these dopaminergic compounds was demonstrated by its general lack of protective efficacy against a host of convulsants acting through other neural mechanisms [pentylenetetrazol, (+)-bicuculline, and picrotoxin, 4-aminopyridine, and t-butylbiclyclophosphoorothionate, N-methyl-d-aspartate, kainate, pilocarpine, nicotine, strychnine, aminophylline, threshold electric shock, and 6-Hz electrical stimulation]. Direct and correlational evidence suggests that these effects were mediated by D3 receptors. Protection was stereospecific and reversible by an antagonist of D3 receptors [3-{4[1-(4-{2[4-(3-diethyamino-propoxy)-phenyl]-benzoimidazol-l-yl}-butyl)-1H-benzoimidazol-2-yl]-phenoxy}-propyl)-diethyl-amine; PD 58491] but not D2 receptors [3[[4-(4-chlorophenyl)-4hydroxypipeidin-1-yl]methyl-1H-indole; L-741,626]. Anticonvulsant potencies were positively associated with potencies in a functional assay of D3 but not D2 receptor function. Together, these findings suggest that the prevention of cocaine convulsions and lethality by (+)-PD 128,907 may be due to D3 receptor-mediated events.
Dependence upon cocaine and other psychomotor stimulants is associated with medical problems and death (Derlet and Albertson, 1989; Colliver et al., 1992; Benowitz, 1993; SAMHSA, 1997). It has been estimated that cocaine abuse accounts for about 25% of all emergency medical department drug-related episodes (SAMHSA, 1997). Up to 12% of patients presenting to emergency departments with cocaine intoxication require anticonvulsant therapy (Derlet and Albertson, 1989; Dhuna et al., 1991). Although anticonvulsant therapies are generally effective in controlling cocaine-related seizures, some seizures and status epilepticus from cocaine can also be resistant to standard therapies (e.g., benzodiazepines and barbiturates) and can be fatal (Dhuna et al., 1991). Rodent models have confirmed the existence of anticonvulsant-resistant cocaine seizures (Witkin and Tortella, 1991; Gasior et al., 1999; Witkin et al., 1999. Although the mechanisms responsible for these seizures have begun to be delineated, a full understanding of these processes has yet to be described.
Difficulties in understanding the mechanisms underlying cocaine dependence and toxicity derives from the multiplicity of toxic effects and the diversity of the physiological actions of cocaine with which these effects may be associated. Although the precise regulatory role of dopamine D3 receptors remains debated, investigations over the past several years have suggested a potentially pivotal role for this mesolimbic receptor in the actions of cocaine. Both anatomical and pharmacological evidence has implicated D3 receptors in the subjective and reinforcing effects of cocaine (Caine and Koob, 1993; Acri et al., 1995; Spealman, 1996; Caine et al., 1997; Levant, 1997; Pilla et al., 1999). The possibility that D3 receptors are involved in the toxic effects of cocaine is suggested from observations of up-regulation in densities of D3 receptors in striatum and substantia nigra of cocaine overdose victims (Staley and Mash, 1996) and more directly from our preliminary observations that the dopamine D3/D2 agonist, (+)-PD 128,907 produced dose-dependent and complete protection against the convulsant effects of cocaine in mice under conditions where a host of clinically used anticonvulsant agents (Witkin and Tortella, 1991; Gasior et al., 1999; Witkin et al., 1999) were relatively ineffective. The present sequence of experiments was designed to examine the potential contribution of D3 receptors to the anticonvulsant effects of (+)-PD 128,907 and to explore whether (+)-PD 128,907 had anticonvulsant effects against other convulsant agents. In the present report, effects of a series of structurally distinct dopamineD3/D2 agonists against cocaine convulsions are described. The lethal effects of cocaine were also evaluated. Additional direct and correlative pharmacological evidence is presented that ultimately points to a role for D3 receptors in the mediation of this anticonvulsant activity.
Materials and Methods
Animals. The following mice were used: experimentally naive male Swiss-Webster mice weighing between 28 and 37 g (Taconic Farms, Germantown, NY); adult male NSA (CF1 mice) (Harlan, Indianapolis, IN) weighing between 20 and 30 g; and 3–4-week-old DBA/2 mice of either sex (Taconic Farms), weighing between 8 and 12 g. Mice were group housed in a temperature-controlled vivarium and were used only once in these experiments. All animals were acclimated to their home cages and to the light/dark cycle (12 h) for at least 5 days before testing. Food and water were continuously available in their living cages. Animals used in the present study were maintained in facilities fully accredited by the American Association for the Accreditation of Laboratory Animal Care. All experimentation was conducted in accordance with the guidelines issued by the Institutional Care and Use Committees and included in the Guide for Care and Use of Laboratory Animals (National Research Council 1996).
Cocaine and Other Chemical-Induced Seizures. Convulsions were engendered with cocaine and other chemical or electrical stimuli (see below). Swiss-Webster mice were injected with a test compound or vehicle s.c. 30 min before cocaine (75 mg/kg i.p.) or other chemical convulsants (Table 1), and mice were immediately placed in individual Plexiglas containers (14 × 25 × 36 cm in height) for observation. Cocaine-induced convulsions were defined as loss of the righting response for at least 5 s, and the occurrence of clonic limb movements; tonus and death were rarely observed. For the lethality experiments, higher doses of cocaine were studied in separate groups of mice. The dose of the chemical and electrical convulsants (below) was selected based upon literature values and pilot experiments to match the effects of cocaine on the percentage of animals exhibiting convulsions. Although all of the convulsant stimuli resulted in clonic seizures, some but not all produced tonic seizures and death. The effects of the dopamine agonists were evaluated against each of the specific seizure types measured. For convulsants other than cocaine, vehicle or (+)-PD 128,907 (3 mg/kg) was administered, and the animals were tested 30 min postinjection as with cocaine. This dose of (+)-PD 128,907 was used based upon its nearly complete attenuation of cocaine seizures. Higher doses of (+)-PD 128,907 were also tested against many of the convulsants.
Effects of (+)-PD 128,907 on the toxicity induced by other agents
Different convulsants produced distinct seizure types (clonic, tonic, limbic). (+)-PD 128,907 s.c. was administered 30 min before application of the convulsant stimulus.
Electroshock-Induced Clonic-Tonic Seizures [Threshold Electroshock (TES) Test]. CF1 mice were used. TES seizures were induced via corneal stimulation (60 Hz, 0.2 s, 15 mA; White et al., 1995) using an apparatus similar to that originally described by Woodbury and Davenport (1952). At the time of drug administration and again before stimulation, a drop of 0.5% tetracaine solution was applied to the eyes of all animals. The presence or absence of hind-limb tonus was recorded in each animal. Vehicle or (+)-PD 128,907 was administered to groups of five mice each, and the animals were tested 30 min postinjection.
Electroshock-Induced Limbic Seizures (6-Hz Test). Limbic seizures were induced via corneal stimulation (6 Hz, 32 mA, 0.2-ms rectangular pulse width, 3-s duration). The stimuli were delivered using a Grass model S48 stimulator with a 2500-ohm resistance. The 6-Hz-induced seizure was characterized by immobility, forelimb clonus, twitching of the vibrissae, and Straub-tail (Barton and Shannon, 2003) and lasted approximately 10 to 15 s, after which the animals rather abruptly resume normal locomotion and exploratory behavior. The presence or complete absence of a limbic seizure was recorded for each mouse. Vehicle or (+)-PD 128,907 was administered to groups of five mice each, and the animals were tested 30 min postinjection.
Sound-Induced (Audiogenic) Seizures. DBA/2 mice were injected with vehicle or a dose of (+)-PD 128,907. Thirty minutes later, the mice were placed individually in clear tubes (8.5 cm in diameter, 16 cm in length) located within an acoustic chamber (model SR-Lab; San Diego Instruments, San Diego, CA). After an adaptation period (1–5 min), a broadband sound stimulus of 120 dB was presented for 60 s. In untreated mice, the sound stimulus typically produces a seizure response progressing from hyperactivity to clonic and then tonic seizures; seizures typically last approximately 45 s or less. The presence or absence of tonus was recorded in each animal.
Inverted Screen Test. This test was used to evaluate doses of the test compounds that produce ataxic and/or sedative effects (Ginski and Witkin, 1994). Swiss-Webster mice (n = 8/group) were pretreated with either vehicle or test compound as described above and returned to their home cage for the appropriate pretreatment interval. They were then individually placed on a 14 × 14-cm wire mesh screen (0.8-cm screen mesh) elevated 38 cm above the ground. After slowly inverting the screen, the mice were tested during a 2-min trial for their ability to climb to the top. Mice not climbing to the top (all four paws on upper surface) were counted as a failure.
Compounds. Compounds were synthesized in-house (PD designated compounds) or obtained from commercial sources (Sigma/RBI, Natick MA; Sigma-Aldrich, St. Louis, MO). SCH 39166 was a gift from Vicki Coffin (Schering Plough, Kenilworth, NJ). Drug solutions were prepared in sterile water with sonication as needed except for L741,626 which was suspended in dimethyl sulfoxide/water.
Data Analyses. ED50 values (with 95% confidence limits) for the protective effects of the test drugs or for their effects on motor performance and ID50 values for blocking the anticonvulsant effects of (+)-PD 128,907 were calculated from dose-effect curves (Litchfield and Wilcoxon, 1949). Protective indices were calculated from the respective anticonvulsant and inverted-screen test dose-effect functions (ED50 for motor impairment/ED50 for anticonvulsant effect; Löscher and Nolting, 1991). Comparison of effects of individual doses to vehicle control was made through Fisher's exact probability test. Comparison of dose-effect curves was accomplished through relative potency analysis using analysis of variance to determine the dose of the comparison compound needed to produce a comparable effect relative to the compound standard. Thus, a relative potency of 1.0 means that the two compounds were equally potent; statistical differences were assigned when the 95% confidence limits of the relative potency estimate did not contain the value of 1.0. Effects for all statistical evaluations were considered significant when the probability of the results occurring by chance was less than 0.05.
Results
Cocaine (75 mg/kg i.p.) produced clonic seizures in male Swiss-Webster mice as reported previously (Witkin and Tortella, 1991) with an incidence of about 80%. Pretreatment with (+)-PD-128,907 or (+)-7-OH-DPAT resulted in dose-dependent and full protection against the convulsant effects of cocaine (Fig. 1). The ED50 values and associated 95% confidence limits (in milligrams per kilogram) were 7-OH-DPAT = 0.04 (0.01–0.11) and (+)-PD 128,907 = 0.15 (0.06–0.36). The protective effects of (+)-7-OH-DPAT and (+)-PD 128,907 were stereoselective with no protection observed by their inactive, levorotatory isomers (Fig. 1, squares). The D3/D2 agonists quinelorane and quinpirole also blocked cocaine-induced convulsions (Fig. 1). The ED50 (95% confidence limits) values (milligrams per kilogram) were quinelorane = 0.01 (0.002–0.06) and quinpirole = 0.06 (0.02–0.14). In contrast to the D3/D2 agonists, the nonselective dopamine receptor agonist apomorphine was not fully efficacious against cocaine-induced clonic seizures, producing a maximum inhibition to 37.5% at 0.3 and 1 mg/kg; higher doses (3 and 10 mg/kg) were without effect (not shown). The ED50 (95% confidence limits) for apomorphine was 0.32 (0.08–1.2) mg/kg.

Dopamine D3/D2 receptor agonists dose dependently protected against the convulsant effects of cocaine in mice. Compounds were administered s.c., 30 min before cocaine (75 mg/kg i.p.) Top, active (circles) but not the inactive enantiomers (squares) of PD 128,907 and 7-OH-DPAT blocked cocaine convulsions. Bottom, D3/D2 receptor agonists quinelorane and quinpirole also blocked cocaine-induced convulsions The shaded region represents the mean ± S.E.M. effect of cocaine alone (75 mg/kg i.p.). Each data point represents the effect in a group of at least 8–16 mice. Asterisk (*) indicates significant protection (Fisher's exact probability test; p < 0.05).
The highest dose of each of the agonists was evaluated in the inverted screen test. The anticonvulsant effects of the D3/D2 agonists occurred at doses well below those that produced motor impairment. The lack of motor impairment at these doses resulted in protective indices of >20 (PD 128,907), >17 (quinpirole), >100 (quinelorane), and >75 (7-OH-DPAT).
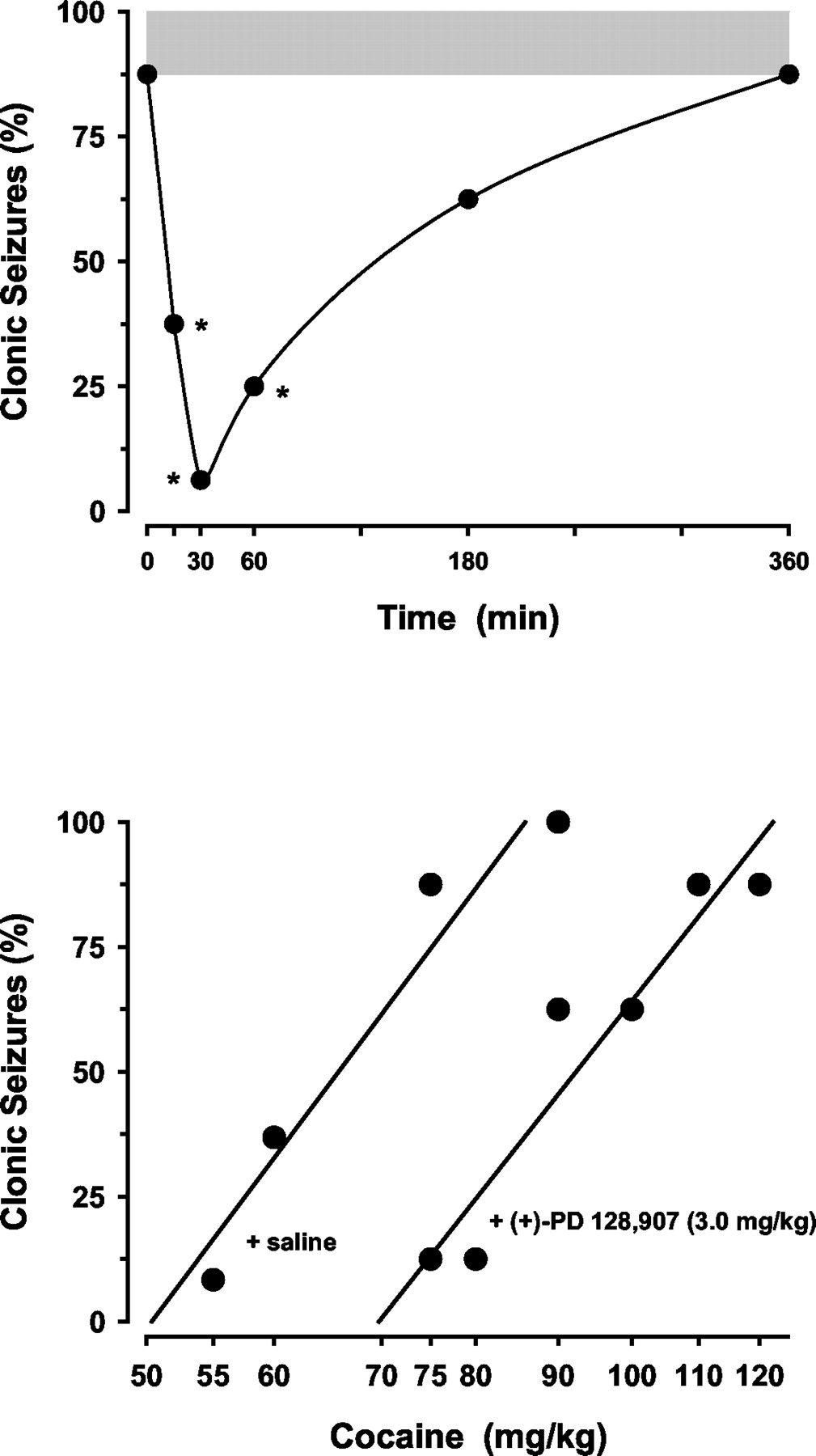
The anticonvulsant protection conferred by (+)-PD 128,907 was time-dependent (Fig. 2). Protective effects of (+)-PD 128,907 evident at pretreatment times from 15 to 60 min, were lost at 180 min. The t1/2 value for this effect was 134.8 min (95% CL, 81.3–223.6). (+)-PD 128,907 (3 mg/kg) shifted the dose-effect curve to the right for the convulsant effects of cocaine (Fig. 2). ED50 values (in milligram per kilogram) for cocaine alone and in the presence of (+)-PD 128,907 were 64.3 (59.4–69.7) and 86.7 mg/kg (78.4–95.8). A relative potency estimate of 1.35 (1.19–1.54) indicated a significant shift of the cocaine curve in the presence of (+)-PD 128,907.

Top, time course for the protective effects of (+)-PD 128,907 against cocaine-induced convulsions. (+)-PD 128,907 was administered at different times before cocaine (75 mg/kg i.p.). Bottom, (+)-PD 128,907 (3 mg/kg) significantly shifted the dose-effect curve to the right for the convulsant effects of cocaine. Other details as in Fig. 1.
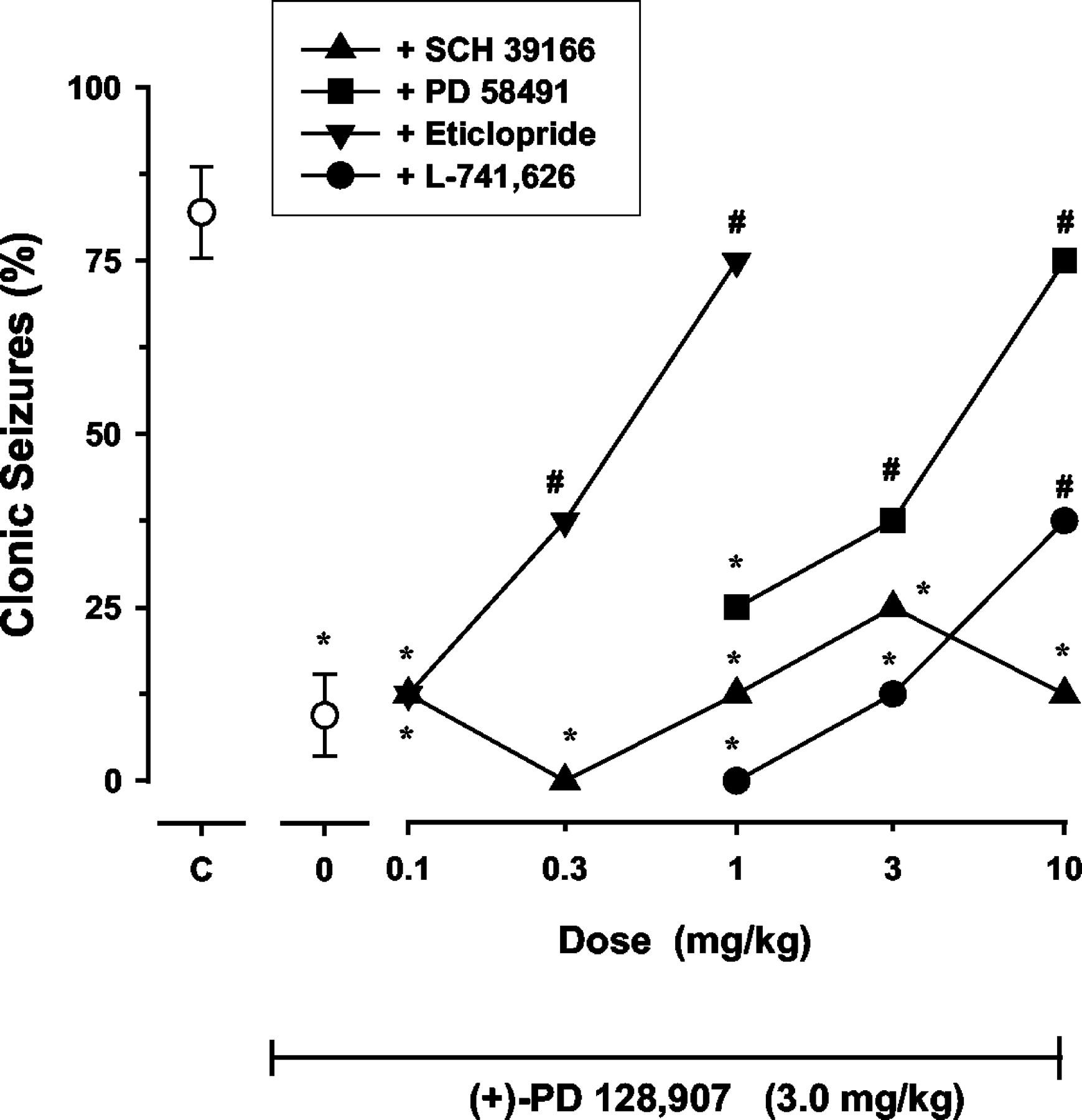
A specific role for D3 receptors in the anticonvulsant effects of (+)-PD 128,907 was evaluated with antagonists (Fig. 3). The D3-selective antagonist PD 58491 dose dependently prevented the anticonvulsant effect of (+)-PD 128,907 with complete blockade occurring at 10 mg/kg (Fig. 3). The ID50 for PD 58491 was 3.7 mg/kg (95% CL, 1.4–9.7). Eticlopride more potently blocked the effects of (+)-PD 128,907 with an ID50 of 0.42 mg/kg (95% CL, 0.20–0.88). The D2 receptor selective antagonist L-741,626 produced only a modest blockade when given up to a dose of 10 mg/kg; higher doses were not tested due to poor solubility. The selective D1 receptor blocker SCH 39166 was inactive against the protective effects of (+)-PD 128,907.

D3 antagonist PD 58491 and the D3/D2 antagonist eticlopride blocked the anticonvulsant effects of (+)-PD 128,907. The more selective D2 receptor antagonist L-741,626 had effects against (+)-PD 128,907 only at the highest dose tested. A selective D1 receptor antagonist, SCH 39166, did not block the anticonvulsant effects of (+)-PD 128,907. The unconnected point above C represents the mean ± S.E.M. effect of cocaine alone (75 mg/kg i.p.). Each data point represents the effect in a group of at least 8 to 16 mice. Asterisk (*) indicates significant protection against cocaine; # represents effects significantly different from cocaine + PD 128,907 (unconnected point above 0) (Fisher's exact probability test; p < 0.05).
Additional evidence for a role for D3 receptors in the convulsant effects of cocaine was gleaned from correlation analysis with data from functional assays in cell lines expressing either D3 or D2s receptors (Sautel et al., 1995). There was a positive association between anticonvulsant potencies of dopamine agonists and their potencies to increase mitogenesis as measured by incorporation of [3H]thymidine in CHO cells expressing human D3 receptors (r = 0.94, p < 0.05) (Fig. 4). In contrast, there was no significant correlation between anticonvulsant potencies and [3H]thymidine incorporation into CHO cells expression human D2S receptors (r = 0.26, p > 0.05)(Fig. 4). The relationship between in vivo potency as anticonvulsants and affinity for D3 or D2 receptors was also evaluated; r = 0.87, p = 0.058 for D3 receptors and r = –0.47, p > 0.05 for D2 receptors (receptor affinities from the literature as noted in paragraph 1 of Discussion.

A, positive association of anticonvulsant potencies of dopamine agonists with potencies to increase mitogenesis as measured by incorporation of [3H]thymidine in CHO cells expressing human D3 receptors. B, lack of association between anticonvulsant potencies and [3H]thymidine incorporation into CHO cells expressing human D2S receptors. Mitogenesis data are from Sautel et al. (1995) in which compounds were incubated with cells expressing D3 or D2S receptors, and the degree of [3H]thymidine incorporation was assessed.
Lethal effects of cocaine were also blocked by pretreatment with (+)-PD 128,907 (Fig. 5). The ED50 for this effect of (+)-PD 128,907 was 0.35 mg/kg (0.12–0.97). The minus enantiomer of PD 128,907 was devoid of significant effects. In the presence of a fixed dose of (+)-PD 128,907, the lethal effects of cocaine were shifted significantly to the right (Fig. 5). ED50 values (in milligrams per kilogram) for cocaine alone and in the presence of (+)-PD 128,907 were 98.3 (92.6–104.2) and 150.8 (133.3–170.6), respectively. A relative potency estimate of 1.54 (1.33–1.75) indicated a significant shift of the cocaine dose-effect curve.

Top, (+)-PD 128,907 protected against the lethal effects of cocaine. The unconnected point above C represents the mean ± S.E.M. lethal effect of cocaine alone (110 mg/kg i.p.). Asterisk (*) indicates significant protectant efficacy against death (Fisher's exact probability test; p < 0.05). The ED50 for the lethality protectant effects of (+)-PD 128,907 was 0.35 mg/kg (0.12–0.97). The minus enantiomer of PD 128,907 was devoid of significant protective effects. Bottom, (+)-PD 128,907 significantly shifted the dose-effect function for cocaine to the right.
The selective dopamine transport inhibitor GBR 12909, like cocaine, produced dose-dependent increases in clonic convulsions with 75% of mice seizing at 300 mg/kg i.p. (Table 1). (+)-PD 128,907 (3 mg/kg) significantly reduced the number of mice exhibiting convulsions to 13% at 3 mg/kg.
To compare the effects of (+)-PD 128,907 on cocaine seizures, we used a series of chemical and electrical convulsants. Although the dose of these agents was chosen to be generally comparable with the seizure incidence produced by cocaine, this matching was not always exact. Moreover, other convulsants produced a high percentage of lethality that was not comparable with cocaine at the dose studied for convulsions per se and some convulsants produced tonic seizures in addition to clonus that was the primary seizure type observed with cocaine (Table 1). In contrast to the protection against cocaine and GBR 12909, (+)-PD 128,907 was generally ineffective or less potent against a host of chemical convulsants acting through other neural mechanisms (Table 1). These include γ-aminobutyric acidA receptor ligands with various mechanisms of action [pentylenetetrazol, (+)-bicuculline, and picrotoxin, and t-butylbiclyclophosphoorothionate], excitatory amino acid receptor ligands (N-methyl-d-aspartate and kainate), a muscarinic cholinergic agonist (pilocarpine), a nicotinic cholinergic agonist (nicotine), a glycine receptor agonist (strychnine), a K+ channel blocker (4-aminopyridine), and an adenosine receptor antagonist (aminophylline). A higher dose of (+)-PD 128,907 was able to prevent the tonic convulsions induced by bicuculline and the lethality produced by bicuculline and picrotoxin. Clonic seizures induced by these compounds were not significantly affected by 3 mg/kg (+)-PD 128,907 that almost completely prevented both seizures and lethality produced by cocaine and GBR 12909. (+)-PD 128,907 also did not significantly decrease seizures induced by electrical stimuli (Table 1). However, audiogenic seizures were significantly reduced by (+)-PD 128,907 (Table 1).
Discussion
The present findings provide the first experimental evidence that D3 dopamine receptors may have control over the convulsant and lethal effects of acutely administered cocaine. In the present experiment, (+)-PD 128,907 and other D3/D2 agonists prevented the convulsant and lethal effects of cocaine in a rodent model of cocaine seizures that is relatively unresponsive to conventional anticonvulsants (Witkin and Tortella, 1991; Witkin et al., 1999). However, these D3/D2 agonists are not the best tools for defining D3 receptor-mediated events. Affinities of the agonists for D3 receptors (Ki) and selectivities over D2 receptors have been reported as follows: (+)-PD 128,907: D3 = 1.8 and 2.3 nM, D2/D3 = 216 and 18 by Sautel et al. (1995) and Pugsley et al. (1995), respectively; (–)-PD 128,907: D3 >10,000 nM by D. Dijkstra (unpublished data); (+)-7-OH-DPAT: D3 = 0.78 nM and D2/D3 = 78 by Levesque et al. (1992); quinpirole: 5.1 and 39 nM; D2/D3 = 113 and 36 by Sokoloff et al. (1990, 1992), respectively; quinelorane: D3 = 3.6 nM and D2/D3 = 95 by Sokoloff et al. (1992); apomorphine: D3 = 20 nM and D2/D3 = 1.2 by Sokoloff et al. (1992); and PD 58491: D3 = 19.5 nM; D2/D3 = 121 by Whetzel et al. (1997).
(+)-PD 128,907 is not selective for D3 receptors especially at higher doses in vivo. For example, the decrease in dopamine efflux induced by (+)-PD 128,907 occurs only at high doses in mice devoid of D3 receptors, suggesting that (+)-PD 128,907 likely acts through D2 receptors in vivo at higher doses (Zapata et al., 2001). Nonetheless, evidence from the present series of experiments support a conclusion that D3 receptors are involved in the protection conferred by this compound. Evidence linking D3 receptors to cocaine convulsions comes from several observations: 1) the anticonvulsant effects of (+)-PD 128,907 and other D3/D2 agonists were not likely due to sedative or motor-impairing effects because the compounds prevented convulsions at doses with at least a 20-fold separation relative to doses that produce motor impairment; 2) prevention by (+)-PD 128,907 of the toxicities of another indirect dopamine agonist but generally not of convulsants operating through nondopaminergic mechanisms; 3) stereoselectivity of the protection conferred by (+)-PD 128,907; 4) blockade of (+)-PD 128,907-induced protection by a selective D3 receptor antagonist; 5) the rank order of potencies of compounds to block the anticonvulsant effects of (+)-PD 128,907 followed rank order affinities for D3 but not for D2 receptors; and 6) there was a positive association between the potencies of compounds to protect against convulsions and their functional effects at D3 but not at D2 receptors. However, although a number of pieces of evidence have been brought to bear on the question of D3 receptor involvement, the general conclusion must be viewed in light of the relatively small number of agonists and antagonists tested along with their limitations of selectivity and ability to modulate the specific receptor targets in vivo to which they are directed.
The anticonvulsant effects of (+)-PD 128,907 displayed pharmacological specificity against compounds with a mechanism of action at the dopamine transporter (cocaine and GBR 12909). (+)-PD 128,907 was generally ineffective against a host of chemical convulsants acting through other neural mechanisms. These include γ-aminobutyric acidA receptor ligands with various mechanisms of action [pentylenetetrazol, (+)-bicuculline, picrotoxin, and t-butylbiclyclophosphoorothionate], excitatory amino acid receptor ligands (N-methyl-d-aspartate and kainate), a muscarinic cholinergic agonist (pilocarpine), a nicotinic cholinergic agonist (nicotine), a glycine receptor agonist (strychnine), a K+ channel antagonist (4-aminopyridine), and an adenosine receptor antagonist (aminophylline). (+)-PD 128,907 was also without effect on the seizures engendered by two forms of electrical stimulation but was effective against sound-induced seizures in DBA/2 mice. The efficacy of (+)-PD 128,907 against audiogenic seizures is not surprising when viewed against the liberal manner in which this model detects anticonvulsant agents including apomorphine (cf., Chapman et al., 1984). However, the issue of pharmacological specificity must be tempered by the observation that doses of (+)-PD 128,907 higher than those that were effective in blocking toxicities produced by cocaine did attenuate the convulsant or lethal effects of bicuculline and pilocarpine Furthermore, the other convulsants tested often produced lethality, whereas convulsant doses of cocaine and GBR 12909 generally did not produce as high an incidence of death. Nonetheless, the generally more potent and efficacious effects of (+)-PD 128,907 against cocaine and GBR 12909 versus a range of other chemical convulsants, helps narrow the mechanism of the anticonvulsant effects of this compound as discussed below. These data are also consistent with a specific anticonvulsant mechanism distinct from a decrease in general neuronal or motoric excitability.
That the protection conferred against cocaine toxicity involved dopaminergically driven toxic effects was given further support from experiments with GBR 12909. Although cocaine inhibits monoamine transport of serotonin, norepinephrine, as well as dopamine, GBR 12909 is selective in its inhibition of dopamine transport (van der Zee et al., 1980). The convulsant effects of GBR 12909 were clonic like those of cocaine and were also prevented by a dose of (+)-PD 128,907 that was effective against the convulsant effects of cocaine. The mechanism by which (+)-PD 128,907 exerts anticonvulsant effects against cocaine and GBR12909 and the lethality induced by cocaine is likely due to its effects on D3 receptors. The effects of PD 128,907 and 7-OH-DPAT were stereoselective. Although these data are consistent with a D3 receptor target, the loss of affinities of the minus isomers for D2 receptors makes the use of stereoisomer effects nondiscriminatory for D3 over D2 receptors. There was a positive association between the anticonvulsant potencies of the D3/D2 agonists and the functional effects of activation of D3 but not D2 receptors. Blockade of the protective effects of (+)-PD 128,907 by PD 58491 further substantiated the claim that D3 receptors play a role in the anticonvulsant effects of (+)-PD 128,907. PD 58491 is a D3 antagonist with a 100-fold selectivity over D2 receptors (Ki, D3 = 19.5 nM; Ki, D2 = 2362 nM; D4 > 3000 nM) (Whetzel et al., 1997). The anticonvulsant effect of (+)-PD 128,907 were also prevented by pretreatment with (–)-eticlopride with equivalent and high affinity for both D3 and D2 receptors (Ki = about 0.1 nM; Tang et al., 1994). L-741,626 is a preferential antagonist of D2 receptors (Ki,D3 = 100 nM; D2 = 2.4 nM; Kulagowski et al., 1996). This compound only partially prevented the anticonvulsant effects of (+)-PD 128,907 when given up to doses that pushed the limits of compound solubility. Potencies of the compounds to block the anticonvulsant effects of (+)-PD 128,907 were in accord with their affinities to bind D3 receptors (eticlopride > PD 58491 > L-741,626). The preferential D1 receptor antagonist SCH 39166 did not significant modify the effects of (+)-PD 128,907 consistent with the low affinity of (+)-PD 128,907 for D1 receptors (Pugsley et al., 1995).
Although a number of pieces of evidence linking D3 receptors to the anticocaine toxicity of (+)-PD 128,907 have been brought forward, skepticism regarding D3 receptor involvement cannot be fully dispelled for at least two reasons. First, the available agonist tools lack high selectivity for the target. For example, Zapata et al. (2001) found that dopamine efflux in ventral striatum of mice was decreased by (+)-PD 128,907. However, when the dose of (+)-PD 128,907 was 1 mg/kg and higher, these neurochemical changes also were observed in mice devoid of D3 receptors, indicating that these higher doses of (+)-PD 128,907 decrease dopamine efflux via D2 autoreceptors. A second reason for uncertainty with regard to an involvement of D3 receptors is the obscurity that surrounds the physiological function and relevance of this receptor subtype. Although an autoreceptor function for D3 receptors has been debated, evidence for an autoreceptor function for D3 receptors have been presented with these receptors being implicated in regulating the synthesis and release of dopamine (Aretha et al., 1995; Gobert et al., 1995; Levant, 1997). In the present case, D3 receptor agonists would function to reduce the extracellular dopamine concentrations as has been reported from in vivo microdialysis studies (Pugsley et al., 1995; Zapata et al., 2001) and electrochemical experiments (Joseph et al., 2002). Reduction in dopamine overflow via D3 receptor-mediated control would in turn reduce the intensity of the pharmacological stimulus for convulsions and lethality as reflected in potency shifts in the dose-effect functions in the presence of (+)-PD 128,907.
The detailed mechanisms responsible for the anticonvulsant and antilethality-inducing effects of (+)-PD 128,907 and other D3/D2 receptor agonists can only be speculated upon at present. One possibility may be through the ability of (+)-PD 128,907 to facilitate the uptake of synaptic dopamine and thereby reduce the extracellular levels of dopamine engendered under these extreme toxic conditions. In the strain of mouse used in the present experiments, (+)-PD 128,907 was shown to facilitate dopamine transport in vivo (Zapata and Shippenberg, 2002). However, it is not at all clear from such a proposed mechanism how the kinetics of cocaine and GBR at the dopamine transporter would play into the enhanced uptake of dopamine that may be driven by D3 receptors.
The present study documents the anticonvulsant and antilethal effects of (+)-PD 128,907 against cocaine. Despite any firm understanding of mechanism, convergent data are presented in this report that suggest that D3 receptors mediate the effects of (+)-PD 128,907 as an anticocaine toxicity agent. These findings have a number of implications. First, they suggest the possible utility of a D3 receptor agonist in the treatment of cocaine overdose toxicity, the convulsions from which can be unresponsive to standard anticonvulsant agents in the emergency room (cf., Dhuna et al., 1991; Benowitz, 1993) as well as in animal models (Witkin and Tortella, 1991; Witkin et al., 1999). Second, the present findings are suggestive of the drug interaction safety of D3 receptor agonists with cocaine, an issue that arises with the proposed utility of these compounds for cocaine abuse therapeutics (cf., Pilla et al., 1999). Finally, these data add to the body of literature that documents pharmacological effects that may be linked to D3 receptors in vivo. The later point is not trivial because pharmacological effects attributed to D3 receptors have not always been supported by additional information (cf., Depoortere, 1999; Chaperon et al., 2003). Too, the functional significance of D3 receptors for physiology and behavior under normal and pathophysiological conditions (cf., Joyce, 2001) is still little known despite the identification of D3 receptors through cloning more than 10 years ago.
Acknowledgments
We are grateful to Drs. Jonathan L. Katz and Roy A. Wise for comments provided on an earlier version of the manuscript and to Dr. Kjell Svensson for helpful discussion and comments on the current version.
Footnotes
-
DOI: 10.1124/jpet.103.059980.
-
A.Z. is a visiting fellow in the National Institutes of Health granted from Fogarty International Center (Bethesda, MD). M.G. was a visiting fellow in the National Institutes of Health granted from Fogarty International Center.
-
ABBREVIATIONS: (+)-PD 128,907, R-(+)-trans-3,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano[4,3-b]-1,4-oxazin-9-ol; TES, threshold electroshock; SCH 39166, (–)-trans-6,7,7a,8,9,13b-hexahydro-3-chloro-2-hydroxy-N-methyl-5H-benzo[d]naptho-{2–1-b}azepine; 7-OH-DPAT, (+)-7-hydroxy-dipropylaminotetralin; CL, confidence limits; PD 58491, (3-{4[1-(4-{2[4-(3-diethyamino-propoxy)-phenyl]-benzoimidazol-l-yl}-butyl)-1H-benzoimidazol-2-yl]-phenoxy}-propyl)-diethyl-amine; L-741,626, 3[[4-(4-chlorophenyl)-4hydroxypipeidin-1-yl]methyl-1H-indole; CHO, Chinese hamster ovary; GBR 12909, 1-[2-[bis(4-fluorophenyl)methoxy] ethyl]-4-[3-phenyl-propyl]piperazine.
-
↵1 Current address: National Institute of Neurological Disorders and Stroke, Epilepsy Research Section, National Institutes of Health, Bethesda, MD 20824.
- Received September 12, 2003.
- Accepted November 25, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}