


Abstract
Convulsions associated with cocaine abuse can be life threatening and resistant to standard emergency treatment. Cocaine (75 mg/kg, i.p.) produced clonic convulsions in ∼90% of male, Swiss-Webster mice. A variety of clinically used antiepileptic agents did not significantly protect against cocaine convulsions (e.g., diazepam and phenobarbital). Anticonvulsants in clinical practice that did significantly protect against convulsion did so only at doses with significant sedative/ataxic effects (e.g., clonazepam and valproic acid). In contrast, functional N-methyl-d-aspartate (NMDA) antagonists all produced dose-dependent and significant protection against the convulsant effects of cocaine. Anticonvulsant efficacy was achieved by blockade of both competitive and noncompetitive modulatory sites on the NMDA receptor complex. Thus, competitive antagonists, ion-channel blockers, polyamine antagonists, and functional blockers of the strychnine-insensitive glycine modulatory site all prevented cocaine seizures. The role of NMDA receptors in the control of cocaine-induced convulsions was further strengthened by the positive correlation between the potencies of noncompetititve antagonists or competitive antagonists to block convulsions and their respective affinities for their specific binding sites on the NMDA receptor complex. Although some NMDA blockers produced profound behavioral side effects at efficacious doses (e.g., noncompetitive antagonists), others (e.g., some low-affinity channel blockers, some competitive antagonists, and glycine antagonists) demonstrated significant and favorable separation between their anticonvulsant and side effect profiles. The present results provide the most extensive evidence to date identifying NMDA receptor blockade as a potential strategy for the discovery of agents for clinical use in averting toxic sequelae from cocaine overdose. Given the literature suggesting a role for these drugs in other areas of drug abuse treatments, NMDA receptor antagonists sit in a unique position as potential therapeutic candidates.
The incidence of cocaine abuse continues to be high, with estimates of 1.7 million regular users of cocaine in the United States (National Institutes on Drug Abuse, 1996). Cocaine dependence is a public health concern, especially with the ready availability of drug forms with greater addiction potential and toxicity (e.g., crack). Although physiological targets (cf. Isner and Chokshi, 1991 and Catravas and Waters, 1981) and receptor targets (cf. Witkin et al., 1993a and Ritz and George, 1997) have been suggested as possible mechanisms associated with cocaine toxicity, definitive identification of the mechanisms associated with the convulsant and lethal effects of cocaine remains elusive.
Cocaine abuse has medical consequences of morbidity and mortality. There was an estimated 150,000 cocaine-related emergency department incidents in 1995, which accounted for 27% of all emergency department drug-related episodes. These toxicity data represent an increasing trend since 1990 (Substance Abuse and Mental Health Services Administration, 1997). Benzodiazepines and phenobarbital are drugs of choice for the emergency treatment of seizures and/or status epilepticus resulting from cocaine intoxication. Unfortunately, status epilepticus following cocaine poisoning is often resistant to standard therapy and can be fatal (Dhuna et al., 1991). Preclinical models have likewise demonstrated that convulsions induced by cocaine are relatively insensitive to standard anticonvulsant therapies (Witkin and Tortella, 1991; Gasior et al., 1997). Witkin and Tortella (1991)reported a rodent model of cocaine overdose in which convulsions induced in mice by bolus injection were resistant to the standard anticonvulsants, diazepam and phenobarbital. In this model, theN-methyl-d-aspartate (NMDA) receptor ion channel blockers, dizocilpine and phencyclidine, and the competitive blockers, CPP [(±)-2-carboxypiperazine-4yl-propyl-1-phosphonic acid] and NPC 12626 [(±)-2-amino-4,5-(1,2-cyclohexyl)-7-phosphonoheptanoic acid], conferred dose-dependent protection against cocaine-induced convulsions.
The present study was initiated to extend the observations of Witkin and Tortella (1991) to a broader range of standard clinically used anticonvulsant/antiepileptic agents, and to a broader range of compounds that confer functional blockade of the NMDA receptor. The NMDA receptor blockers were selected from several classes affecting different binding domains on the NMDA receptor/ionophore complex: noncompetitive blockers of the ion pore, competitive antagonists, functional antagonists of the strychnine-insensitive glycine site, and antagonists of the polyamine site. In addition to providing efficacy information on the anticonvulsant effects of these compounds, data were also collected on their sedative/ataxic effects. These later data permitted the calculation of a protective index (PI), a ratio of the potency of the drug to produce side effects (e.g., ataxia) to its anticonvulsant potency.
Materials and Methods
Animals.
Experimentally naive, male Swiss-Webster mice (Taconic Farms, Germantown, NY) between 10 and 12 weeks old were housed six per cage in a temperature-controlled vivarium. The facilities in which the animals were maintained are fully accredited by the American Association for the Accreditation of Laboratory Animal Care, and the studies described beneath were conducted in accordance with the Guide for Care and Use of Laboratory Animals provided by the Nationals Institutes of Health and adopted by National Institutes on Drug Abuse. All animals were acclimated to their home cages and to the light/dark cycle for at least 5 days before testing. Water and food were continuously available for the mice in their home cages. Experiments were conducted during the light phase of a 12-h light/dark cycle.
Behavioral Toxicity.
Immediately before administration of drugs, mice were first tested on the inverted screen test. The inverted screen test was used to assess one form of behavioral toxicity induced by the test compounds. In this test, compounds with sedative and/or ataxic properties produce dose-dependent increases in screen failures, whereas other classes of drugs (e.g., psychomotor stimulants) do not (Ginski and Witkin, 1994). Mice (at least 8 per group) were pretreated with either vehicle or test compound and returned to their home cage for the appropriate pretreatment interval (see Drugs below). They were then individually placed on a 14 × 14-cm wire mesh screen (0.8-cm screen mesh) elevated 38 cm above the ground. After slowly inverting the screen, the mice were tested during a 2-min trial for their ability to climb to the top. Mice not climbing to the top (all four paws on upper surface) were counted as a failure. Results were expressed as a TD50 value. Each TD50 value, calculated from a dose-response curve, represents the dose of a drug (in milligrams per kilogram) producing screen failure in 50% of the mice tested. After the screen test, cocaine was administered and anticonvulsant efficacy was assessed as described below.
Anticonvulsant Efficacy.
After the screen test, a convulsant dose of cocaine (75 mg/kg, i.p.) was administered and the mice were immediately placed in individual Plexiglas containers (14 × 25 × 36 cm high) for observation. Mice in these experiments were used only once to evaluate the anticonvulsant efficacy of drugs. The dose of cocaine was chosen to be close to its ED85 to ED95 values as determined from the literature (Witkin and Tortella, 1991). The presence or absence of convulsions was monitored for 30 min following cocaine. Cocaine-induced convulsions were defined as loss of the righting response for at least 5 s and the occurrence of clonic limb movements; tonus and death were rarely observed. Locomotor depression with loss of righting response often preceded clonic episodes in cocaine-challenged mice. Once seizures developed in cocaine-treated mice, loss of the righting response often persisted over the 30-min observation period.
Data Analysis.
The quantal data for both the anticonvulsant and behavioral toxicity tests were evaluated according to the methods described by Litchfield and Wilcoxon (1949) and ED50 and TD50 values with 95% CLs were derived from this analysis. Specific comparisons between control and drug treatments were made with Fisher’s exact probability test. To make the statistical comparisons as conservative as possible, each dose was compared with a control group of only 16 mice that received cocaine alone during the determination of those effects. Statistical probabilities of <0.05 were considered to be significant.
Drugs.
The following compounds were dissolved in distilled water or 0.9% NaCl: 1-amino-1-cyclopropanecarboxylic acid (ACPC; Aldrich Chemical Company, Milwaukee, WI), 5-aminocarbonyl-10,11-dihydro-5h-dibenzo[a, d]cyclohepten-5,10-imine (ADCI; Neurogen Corporation, Branford, CT), CPP [Research Biochemicals International (RBI), Natick, MA], dextrorphan d-tartrate (RBI), R(+)-3-amino-1-hydroxypyrrolid-2-one (HA-966; RBI), (−)-cocaine HCl (Sigma Chemical Co., St. Louis, MO), phencyclidine (PCP) HCl [National Institute on Drug Abuse (NIDA), Rockville, MD], 1-[1-(2-thienyl)-cyclohexyl]piperidine (TCP) HCl (NIDA), 5-methyl-10,11-dihydro-5H-dibenzo[a, d]cyclophepten-5,10-imine hydrogen maleate (dizocilpine hydrogen maleate; (+)-MK-801; RBI), (−)-MK-801 hydrogen maleate (RBI), ketamine HCl (Sigma),N-allylnormetazocine (SKF 10,047; Smith-Kline and French, Philadelphia, PA), LY 235959 [(−)-(phosphonomethyl)-decahydroisoquinoline-3-carboxylic acid; Lilly Research Laboratories, Indianapolis, IN], (±)-(phosphonomethyl)-decahydroisoquinoline-3-carboxylic acid (LY 274614; Lilly), (±)-6-(1(2)H-tetrazol-5-yl)methyldecahydroisoquinoline-3-carboxylic acid (LY 233536; Lilly), cis-4-phosphonomethyl-2-piperidine carboxylic acid (CGS 19755; Ciba-Geigy Corp., Summit, NJ), 2R,4R,5S2-amino-4,5-(1,2-cyclohexyl)-7-phosphonoheptanoic acid (NPC 17742; Nova Pharmaceutical Corp., Baltimore, MD), NPC 12626 (Nova), phenobarbital (Ruger Chemical Company, Inc., New York), memantine (Merz & Co., Frankfurt, Germany), ifenprodil (Synthelabo Recherche, Bagneux, France), eliprodil (SL82.0715–10; Synthelabo), and valproate Na (Sigma).
Diazepam (Hoffmann-La Roche, Nutley, NJ), primidone (Sigma), clonazepam (Sigma), phenytoin (RBI), and trimethadione (Abbott Laboratories, Chicago, IL) were suspended in propylene glycol and prepared in final concentration (20% v/v) by addition of water. 1,2,3,4,-tetrahydo-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX; Novo Nordisk, Måløv, Denmark) and ethosuximide (Sigma) were dissolved in distilled water with 1 drop of Tween 80 per 5 ml. Carbamazepine (RBI) was suspended in 40% hydroxypropyl-β-cyclodextrin (RBI). The compounds requiring suspending agents just described were mildly heated and sonicated before injection. 7-Chlorokynurenic acid (7-CKA; RBI) was dissolved in distilled water with the minimal NaOH required for solution with mild heat and sonication.
Routes of administration and pretreatment times were based upon biological activity existing in the literature and on pilot experiments. 7-CKA was given i.p., 10 min before cocaine; phenytoin was given s.c., 120 min before cocaine. All other drugs were administered s.c., 30 min before cocaine. Drugs were injected in a volume of 0.01 ml/g with the exception of higher concentrations of some of the standard anticonvulsant agents, which were administered in double volume due to solubility problems.
Results
Under control conditions, only 5.2 ± 3.2% (mean ± S.E.M.) of the control mice treated with drug vehicles failed the inverted screen test. Cocaine (75 mg/kg, i.p.) produced clonic convulsions in 87.2 ± 5.1% of the mice tested. Although different vehicles were used for a number of the compounds tested, the vehicles per se did not significantly alter these control values.
A host of classical anticonvulsant/antiepileptic agents were studied (Fig. 1). Most of these compounds only produced significant protection against convulsions at the highest doses tested. Others did not produce significant protection (primidone, carbamazepine, phenobarbital, and phenytoin). Phenytoin was notable in exacerbating the toxicity of cocaine. Status epilepticus, not observed with cocaine alone, was engendered in 1/10 (30 mg/kg), 3/10 (56 mg/kg), and 2/10 (100 mg/kg) mice pretreated with phenytoin. The drugs ranged widely in potency from clonazepam to ethosuximide (Table1). In contrast to their general lack of anticonvulsant efficacy, all of these antiepileptic agents dose dependently increased ataxia as measured by the inverted screen test (Fig. 1B). Primidone was not studied in higher doses than 100 mg/kg due to solubility limitations. The TD50 values derived from the screen test were close to or lower than their respective ED50 values as anticonvulsants, rendering the PIs (TD50/ED50) less than or approximately equal to unity (Table 1).
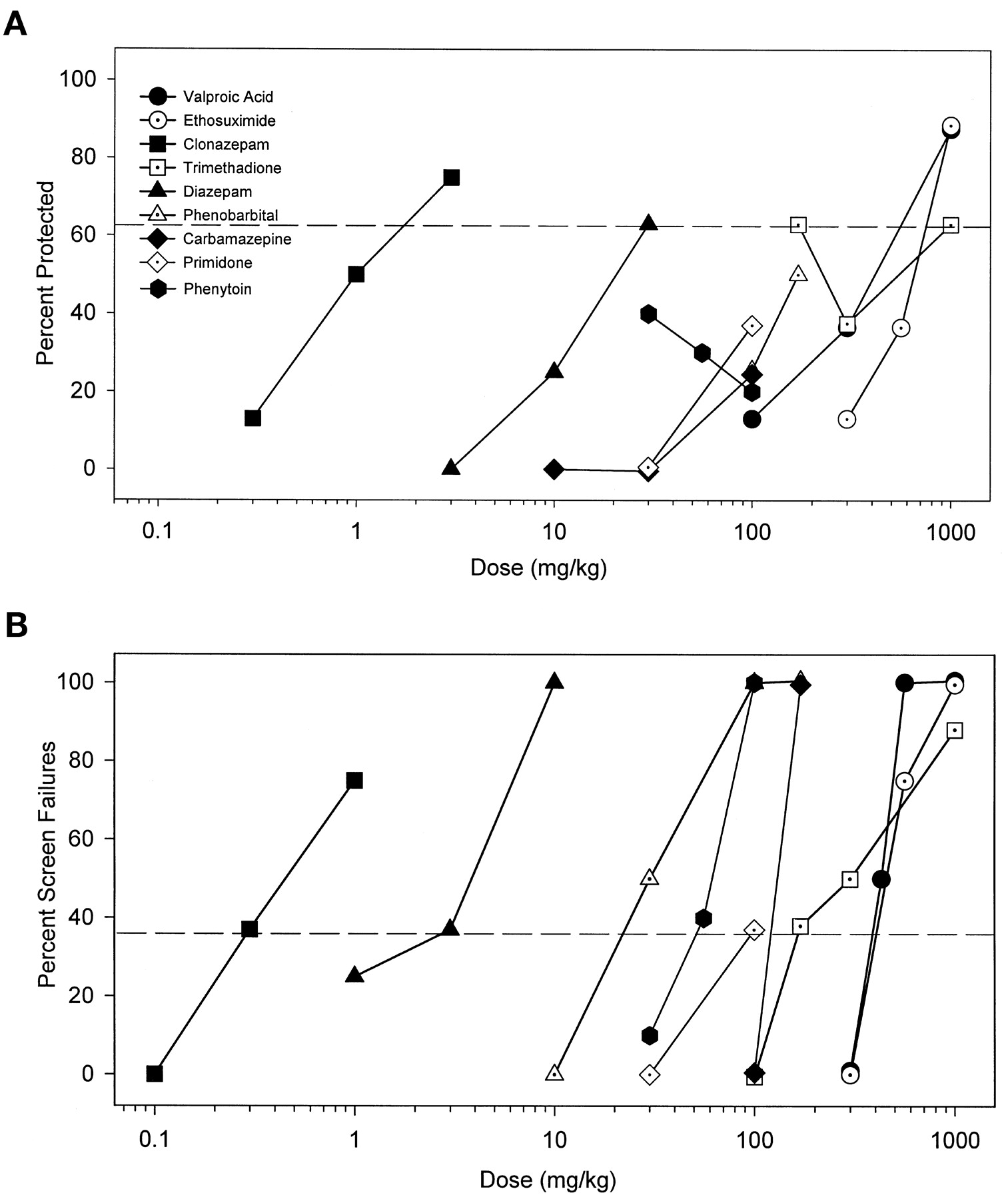
A, classical anticonvulsant drugs do not demonstrate marked efficacy for protecting mice from the convulsant effects of cocaine. Drugs were administered before a challenge dose of cocaine that produced convulsions in 87.2 ± 5.1% of mice tested (75 mg/kg, i.p.) and were then observed for 30 min for convulsions. Points above the dashed line represent significant anticonvulsant protection as determined by Fischer’s exact probability test (p < .05). B, the classical anticonvulsants tested all produced significant behavioral toxicity when given alone as measured in the inverted screen test. Only 5.2 ± 3.2% of control mice failed the inverted screen test. Points above the dashed line represent significant effects on the inverted screen test as determined by the Fischer’s exact probability test (p < .05).
Potencies (ED50 in mg/kg) of standard anticonvulsant drugs to prevent convulsant effects of cocaine (75 mg/kg, i.p.) and potencies (TD50 in mg/kg) of these drugs to produce behavioral side effects on the inverted screen test
All of the noncompetitive NMDA antagonists dose dependently protected against cocaine-induced convulsions (Fig.2A) and also dose dependently increased the percentage of screen failures (Fig. 2B). A wide range of potencies was observed for these effects (Table 2). Although the PI values were close to unity for many of the noncompetitive ligands, the PI values for dizocilpine and its optical isomer, (−)-MK-801, were <1. At the opposite extreme, dextrorphan displayed a slightly better separation between efficacy and ataxia. ADCI displayed the largest separation between potencies for anticonvulsant efficacy and ataxia with a PI value of 18.5. The potencies of these NMDA receptor ion-channel blockers to protect against cocaine convulsions was positively associated with their affinities for the ion channel ([3H]dizocilpine binding) (r = 0.80, p < .05; Fig.3A). When the low-affinity ligand ADCI (K i = 11,300 nM; Monn et al., 1990) was excluded from the analysis, the correlation coefficient was increased (r = 0.90, p < .01). Potencies of the noncompetitive antagonists to produce failures on the inverted screen test were also positively associated with potencies to inhibit [3H]dizocilpine binding) (r = 0.92, p < .01; Fig. 3B).

A, dose-dependent protection against convulsant effects of cocaine by noncompetitive NMDA receptor antagonists. B, dose-dependent increases in sedation/ataxia by noncompetitive NMDA receptor antagonists given alone as assessed in the inverted screen test. Other details as in Fig. 1.
Potencies (ED50 in mg/kg) of noncompetitive NMDA antagonists to prevent convulsant effects of cocaine (75 mg/kg, i.p.) and potencies (TD50 in mg/kg) of these drugs to produce behavioral side effects on the inverted screen test

A, Potencies of noncompetitive NMDA receptor ion-channel blockers to protect against cocaine convulsions was positively associated with their affinities for the ion channel. B, potencies of noncompetitive antagonists to produce failures on the inverted screen test were also positively associated with potencies to inhibit [3H]dizocilpine binding. Binding data are fromWong et al., 1988.
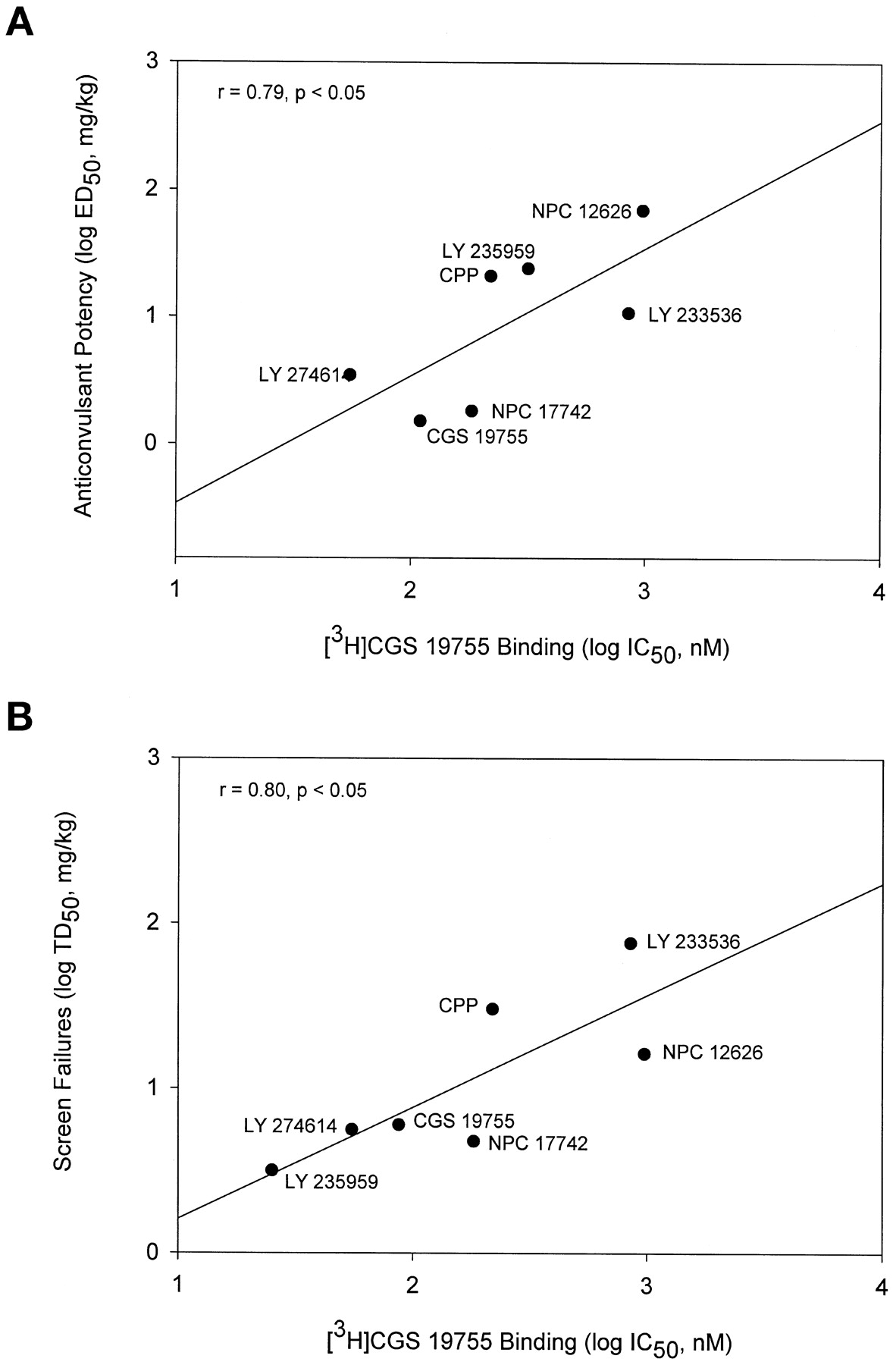
Competitive NMDA antagonists also protected against convulsions in a dose-dependent manner (Fig. 4A) with a range of potencies (Table 3). These compounds also increased the percentage of mice failing the inverted screen test (Fig. 4B). The highest doses of these drugs produced 100% screen failures with the exception of LY 233536. All of the competitive antagonists had PI values >1 except for NPC 12626 (Table 3). LY 233536 was the most impressive compound in this regard, with a PI value of 7. The potencies of the competitive ligands to inhibit cocaine convulsions was positively correlated with their affinities for the glutamate binding site of the NMDA receptor as measured by displacement of [3H]CGS 19755 binding (r = 0.79, p < .05; Fig. 5A). Likewise, the potencies of these compounds to produce failures on the inverted screen test was also positively associated with their affinities for the glutamate binding site (r = 0.80,p < .05; Fig. 5B).

A, dose-dependent protection against convulsant effects of cocaine by competitive NMDA receptor antagonists. B, dose-dependent increases in sedation/ataxia by competitive NMDA receptor antagonists as assessed in the inverted screen test. Other details as in Fig. 1.
Potencies (ED50 in mg/kg) of competitive NMDA antagonists to prevent convulsant effects of cocaine (75 mg/kg, i.p.) and potencies (TD50 in mg/kg) of these drugs to produce behavioral side effects on the inverted screen test

A, potencies of competitive NMDA receptor antagonists to inhibit cocaine convulsions was positively correlated with their affinities for the glutamate binding site of NMDA receptor. B, potencies of competitive NMDA receptor antagonists to produce failures on the inverted screen test were also positively associated with their affinities for the glutamate binding site. Binding data are from Murphy et al., 1988; Ornstein et al., 1992; Ferkany et al., 1993.
Compounds interacting with the strychnine-insensitive binding site of the NMDA receptor complex were also efficacious in protecting against cocaine-induced convulsions (Fig. 6A) but did not significantly engender ataxia in the screen test (Fig. 6B). PI values for these compounds were all estimated to be greater than unity (Table 4). The polyamine antagonists, ifenprodil and eliprodil, also dose dependently blocked cocaine convulsions without affecting behavior on the screen test (Table 4); PI values were therefore also greater than unity. The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) antagonist, NBQX, potently blocked the convulsant effects of cocaine and did not affect performance on the screen test up to a dose of 100 mg/kg; the PI value for NBQX was estimated to be >36 (Table 4).
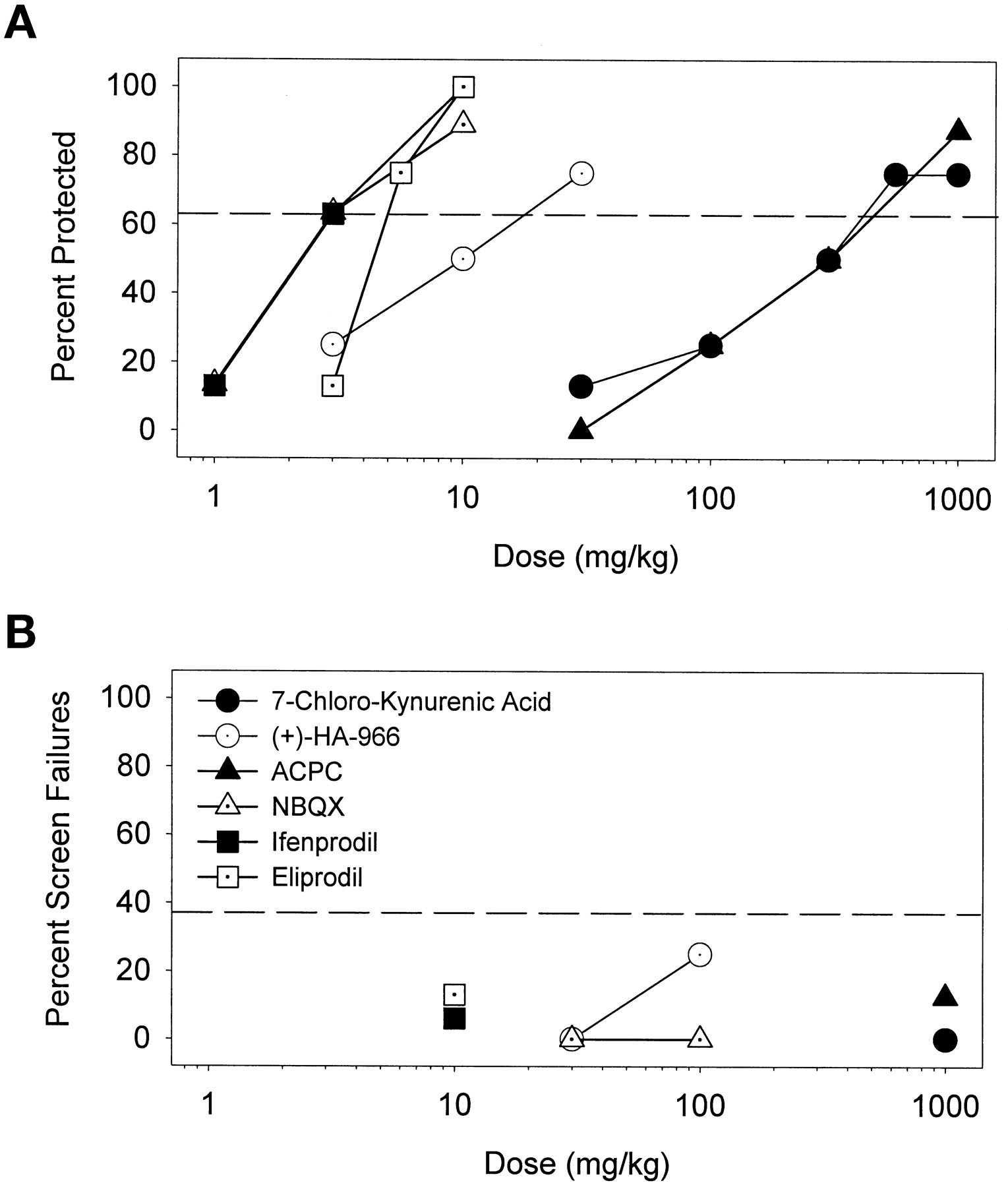
A, protective effects of partial agonists (HA-966 and ACPC) as well as an antagonist (7-CKA) of the strychnine-insensitive glycine site on the NMDA receptor complex, the AMPA antagonist (NBQX), and polyamine antagonists (ifenprodil and eliprodil) against convulsant effects of cocaine. B, these drugs were devoid of significant effects on behavior of mice on the inverted screen test at doses that were effective anticonvulsants. Other details as in Fig. 1.
Potencies (ED50 in mg/kg) of some polyamine antagonists, glycine receptor ligands, and an AMPA antagonist to prevent convulsant effects of cocaine (75 mg/kg, i.p.) and potencies (TD50 in mg/kg) of these drugs to produce behavioral side effects on the inverted screen test
Discussion
The present findings document that classical anticonvulsants have limited efficacies and narrow therapeutic windows against cocaine-induced convulsions, as previously reported for diazepam and phenobarbital (Witkin and Tortella, 1991). The present results extend earlier findings to include a host of anticonvulsant/antiepileptic drugs with a range of therapeutic uses and mechanisms of action (MacDonald and Meldrum, 1989). In contrast to these clinically used anticonvulsants, functional blockers of the NMDA receptor produce full and dose-dependent protection against cocaine convulsions as reported previously for dizocilpine and a few other NMDA antagonists (Derlet and Albertson, 1990; Rockhold et al., 1991; Witkin and Tortella, 1991;Witkin and Acri, 1995; Matsumoto et al., 1997). The positive correlation between affinities of compounds for the NMDA receptor and their potencies to protect against cocaine convulsions suggests an important role for NMDA receptor blockade in their anticonvulsant efficacy against cocaine.
The present results provide the most extensive evidence to date identifying NMDA receptor blockade as a potential strategy for the discovery of agents that could find clinical use in averting toxic sequelae from cocaine overdose. In addition to the blockade of cocaine convulsions by NMDA receptor antagonists, noncompetitive NMDA blockers also prevent the sensitization that develops to the convulsant effects of repeated cocaine exposure (kindling) (Karler et al., 1989; Itzhak and Stein, 1992). NMDA antagonists are also capable of blocking the development of tolerance, dependence, and sensitization that has been observed with the repeated administration of a number of drugs (cf.Witkin, 1995 and Popik and Danysz, 1997). Thus, functional NMDA receptor antagonists would appear to be in a unique position as potential therapeutic candidates to treat cocaine abuse and overdose.
Although we have shown here and elsewhere that cocaine-induced seizures are resistant to standard emergency medical treatments such as diazepam and phenobarbital (Witkin and Tortella, 1991; Gasior et al., 1997), under other conditions these compounds are capable of blocking convulsions engendered by cocaine (Derlet and Albertson, 1990). For example, whereas convulsions induced by 75 mg/kg cocaine are relatively unresponsive to diazepam or phenobarbital, as shown in the present experiment, convulsions induced by 60 mg/kg cocaine were fully blocked by these drugs in mice (Witkin and Tortella, 1991). The general unresponsiveness of cocaine convulsions to standard anticonvulsant drugs is further illustrated by the findings that carbamazepine and ethosuximide did not significantly prevent cocaine convulsions that were sensitive to diazepam or phenobarbital; the broad-spectrum anticonvulsant, valproate, only incompletely attenuated convulsions (Derlet and Albertson, 1990). Although positive allosteric modulators of the γ-aminobutyric acid type A receptor (diazepam and phenobarbital) have limited efficacy against cocaine convulsions, a novel class of positive allosteric modulator, the neuroactive steroids, have been reported to be fully efficacious and demonstrate favorable PI values (Gasior et al., 1997).
Because some drugs that block the NMDA receptor ion channel also bind to ς receptors, the role of ς receptors in toxic effects of cocaine should be addressed. Although the anticonvulsant effects of (+)-SKF 10,047 have been attributed to its effects on ς receptors (Ritz and George, 1997), evidence suggests that the NMDA antagonist actions of this and related compounds are critical. First, a positive association was found between anticonvulsant potencies and binding to the NMDA receptor ion channel but not to sigma receptors r = 0.40, N.S.; binding data for ς receptors defined by [3H]SKF 10,047 binding by Wong et al., 1988). Second, selective ς receptor ligands do not fully prevent cocaine convulsions (Witkin et al., 1993b), whereas (+)-SKF 10,047 and other NMDA antagonists completely block convulsions. Conversely, it has been argued that the ς/NMDA receptor ligand dextromethorphan (Tortella et al., 1994), should block the convulsant effects of cocaine because the related compound dextrorphan is effective (Rockhold et al., 1991). However, the high-affinity binding of dextromethorphan to ς receptors combined with its marginal efficacy against cocaine (Witkin and Tortella., 1991 and our unpublished observations), suggests that the affinity of this compound for ς receptors and not its effects on NMDA receptors is likely the primary mechanism responsible for its effects against cocaine convulsions. Taken together with the findings that NMDA antagonists with different sites of action on the receptor complex are all capable of preventing cocaine-induced convulsions, these arguments should help establish NMDA receptor blockade as a distinct and independent mechanism responsible for drug efficacy against convulsant effects of cocaine.
Although demonstrating efficacy, some NMDA antagonists display side effects like those produced by the psychotomimetic, dissociative anesthetic PCP (cf. Witkin, 1995). Indeed, the noncompetitive NMDA antagonists displayed poor separation between anticonvulsant potency and doses producing ataxia and hypermotility (see also Ginski and Witkin, 1994). Likewise, the possibility of PCP-like side effects has been a concern also in the development of compounds that modulate other sites on the NMDA receptor complex. In contrast to the ion-channel blockers, competitive NMDA receptor antagonists generally blocked the convulsant effects of cocaine at lower doses than those producing failures on the inverted screen test (PI values greater than unity). The positive association between the potencies of the ion channel blockers and the competitive antagonists to produce screen failures and their affinities for their ligand binding sites on the NMDA receptor support the idea that ligand binding initiates both the anticonvulsant effects and the sedative/ataxic side effect profile of these drugs.
Of the competitive NMDA receptor antagonists, LY 233536, with a PI of 7.04, showed the greatest separation between efficacious doses and those producing side effects. This compound has previously been reported to display a less sedating profile from other competitive antagonists (Ginski and Witkin, 1994). Despite the fact that most of the competitive NMDA blockers showed marginally favorable PIs, this class of compounds can produce PCP-like motor side effects (cf. Witkin, 1995) and can fully substitute for the discriminative stimulus effects of dizocilpine in mice (Geter-Douglass and Witkin, 1997). Side effects of the competitive antagonists d-CPP-ene and CGS 19755 were observed in clinical trials and included hallucinations, ataxia, and sedation (cf. Kornhuber and Weller, 1997).
ADCI and memantine are noncompetitive NMDA receptor channel blockers with fast on/off kinetics, an action that has been implicated in their generally favorable side effect profiles (cf., Rogawski, 1993; Parsons et al., 1995; Bubser et al., 1997). Regional differences in the binding of these compounds in brain from that of high-affinity channel blockers like dizocilpine may also contribute to their pharmacological profile (cf. Porter and Greenamyre, 1995). There is general agreement that low-affinity blockers of the NMDA receptor ion channel like ADCI do not fully replicate the discriminative stimulus effects of dizocilpine or PCP (Grant et al., 1996). However, somewhat higher affinity compounds including memantine have been shown to fully substitute (Sanger et al., 1992; Grant et al., 1996). In contrast to ADCI, memantine also demonstrated no separation between anticonvulsant potency and behavioral side effect potency in the present study. Nonetheless, memantine has been in clinical use for about 15 years for Parkinson’s disease and dementia; any psychotomimetic effects of this drug appear to be mitigated by dose escalation.
In contrast to other NMDA antagonists, glycine site antagonists do not appear to produce PCP-like behavioral effects in preclinical studies (cf. Koek and Colpaert, 1990; Witkin, 1995; Balster et al., 1995;Witkin et al., 1997). A consequence of the non-PCP-like behavioral profile of the glycine site ligands was that protective indicies against cocaine convulsions were all greater than unity. In previous studies (Witkin and Tortella, 1991), functional antagonists of the glycine site were effective against cocaine convulsions that were also fully and potently blocked by diazepam (60 mg/kg versus 75 mg/kg studied here). In the present study, this class of NMDA blockers was effective as an anticonvulsant against diazepam-resistant cocaine convulsions. Antagonists of the strychnine-insensitive glycine site, in addition, prevent the lethal effects of cocaine and attenuate lethality when administered after seizure induction (Matsumoto et al., 1997). That the blockade of NMDA receptors is the transduction mechanism responsible for the anticonvulsant effects of these drugs gains additional support from data in which the glycine site partial agonist,d-cycloserine, reversed the anticonvulsant effects of the quinoxalinedione glycine antagonists (Matsumoto et al., 1997).
Two polyamine antagonists blocked the convulsions induced by cocaine at doses that did not affect screen failures. The cocaine-blocking effects of ifenprodil have been reported previously (Witkin and Acri, 1995), an observation extended in this report to include the structural analog, eliprodil. Although sedation/ataxia was not seen at anticonvulsant doses of these drugs, other sedative-like effects have been detected on behavioral measures (e.g., locomotor activity; Ginski and Witkin, 1994). The generally favorable side effect profiles of these polyamines has been suggested to be due to their selective binding with the NR1A/NR2Bheteromeric receptor (Avenet et al., 1997). This argument is based on the finding that, in contrast to the polyamines, dizocilpine, memantine, and PCP antagonize NMDA induced currents with equivalent potency in NR1A/NR2Aconfigured receptors as in the 2B variants. However, the fact that the glycine antagonist 7-CKA produced high-potency blockade of the NR1A/NR2A receptor heteromer, and is, like the polyamines generally devoid of PCP-like effects (cf. Witkin, 1995 and Ginski and Witkin, 1994), indicates that this assay generates false negatives. Results of the present study also revealed that the AMPA antagonist NBQX was an effective blocker of cocaine convulsions, with an exceptionally high PI. Matsumoto et al. (1997) did not find significant protection against cocaine convulsions with NBQX, an inconsistency that cannot be accounted for at present.
In conclusion, pharmacological evidence implicates NMDA receptors in the convulsive effects of cocaine. Some functional blockers of the NMDA receptor (e.g., glycine receptor blockers) may be viable candidates for drug development in this therapeutic area, as well as for the clinical management of aspects of cocaine dependence.
Acknowledgments
The conscientious support and oversight of Jesse T. Ungard was indispensable to the conduct of this research and in various phases of manuscript preparation. We are grateful to the pharmaceutical companies that provided us with generous supplies of compounds (seeMaterials and Methods). We thank Dr. Beth Geter-Douglass for contributing data on behavioral side effects of two of the compounds.
Footnotes
-
Send reprint requests to: Jeffrey M. Witkin, Ph.D., Drug Development Group, NIDA, Addiction Research Center, 5500 Nathan Shock Dr., Baltimore, Maryland 21224. E-mail:jwitkin{at}intra.nida.nih.gov
-
↵1 Parts of this research were reported in abstract form (Tortella et al., 1992).
-
↵2 A visiting fellow in the National Institutes of Health granted from Fogarty International Center, Bethesda, MD. Permanent affiliation: Department of Pharmacology, Medical University School, Lublin, Poland.
-
↵3 A summer research fellow supported by Marion Merrell Dow Pharmaceuticals Inc. Currently at Yale University, New Haven, Connecticut.
- Abbreviations:
- ACPC
- 1-amino-1-cyclopropanecarboxylic acid
- ADCI
- 5-aminocarbonyl-10,11-dihydro-5h-dibenzo[a, d]cyclohepten-5,10-imine
- CGS 19755
- cis-4-phosphonomethyl-2-piperidine carboxylic acid
- 7-CKA
- 7-chlorokynurenic acid
- CPP
- (±)-2-carboxypiperazine-4yl-propyl-1-phosphonic acid
- HA-966
- R(+)-3-amino-1-hydroxypyrrolid-2-one
- LY 233536
- (±)-6-(1(2)H-tetrazol-5-yl)methyldecahydroisoquinoline-3-carboxylic acid
- LY 274614
- (±)-(phosphonomethyl)-decahydroisoquinoline-3-carboxylic acid
- MK-801
- 5-methyl-10,11-dihydro-5H-dibenzo[a, d]cyclophepten-5,10-imine hydrogen maleate
- NBQX
- 1,2,3,4,-tetrahydo-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide
- NMDA
- N-methyl-d-aspartate
- NPC 12626
- (±)-2-amino-4,5-(1,2-cyclohexyl)-7-phosphonoheptanoic acid
- NPC 17742
- 2R,4R,5S2-amino-4,5-(1,2-cyclohexyl)-7-phosphonoheptanoic acid
- PCP
- phencyclidine
- PI
- protective index
- SKF 10
- 047,N-allylnormetazocine
- TCP
- 1-[1-(2-thienyl)-cyclohexyl]piperidine
- TD
- toxic dose
- LY 235959
- (−)-(phosphonomethyl)-decahydroisoquinoline-3-carboxylic acid
- Received August 14, 1998.
- Accepted December 21, 1998.
- U.S. Government




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}