


Abstract
Epilepsy continues to be a significant clinical problem as current medications neither adequately control seizures nor are free of untoward side-effects. Modulation of the neuroactive steroid site on the γ-aminobutyric acid (GABA)A receptor complex may be an important new direction for pharmaceutical interventions in epilepsy. In this study we evaluated the protective actions of four neuroactive steroids, 3α-hydroxy-5α-pregnan-20-one, the 3β-methylated analog, ganaxolone (3α-hydroxy-3β-methyl-5α-pregnan-20-one), 3α-hydroxy-5β-pregnan-20-one and Co 2–1068 (3β-(4acetylphenyl)ethynyl-3α,21-dihydroxy-5β-20-one-21-hemisuccinate), against several standard convulsive tests in male, Swiss-Webster mice. Consistent with their GABAergic actions, the neuroactive steroids as well as diazepam and phenobarbital dose-dependently protected against clonic convulsions induced by pentylenetetrazol; the N-methyl-d-aspartate receptor antagonist, dizocilpine, was ineffective. In contrast to diazepam and phenobarbital, however, all of the neuroactive steroids and dizocilpine produced full protection against cocaine-induced convulsions. Some of the neuroactive steroids, as well as dizocilpine, were efficacious against the seizures and lethality induced by N-methyl-d-aspartate. Pregnenolone, a steroid devoid of GABAergic activity, was not effective in any of the convulsant models. Although all of the compounds produced motor toxicity in high doses as measured by the inverted-screen test, the neuroactive steroids demonstrated an equivalent or improved separation between anticonvulsant potency and motoric impairment. Inactive doses of the neuroactive steroids markedly enhanced the anticonvulsant effects of diazepam against pentylenetetrazol without significantly increasing motor toxicity. This adjunct treatment resulted in protective indices ranging from 60 to 360 compared to 12 for diazepam alone. The distinct profile of anticonvulsant activity of the neuroactive steroids may be related to their combined actions on γ-aminobutyric acid, N-methyl-d-aspartate receptors, or voltage-operated Ca++ channels. These results help to define the neuroactive steroids as a novel class of antiepileptic agents and suggest their potential in clinical practice.
Epilepsy, one of the most common neurological disorders, affects approximately 1.65 million individuals in the United States and 50 million people worldwide (Rogawski and Porter, 1990). In about two-thirds of all patients afflicted, seizures are well controlled with currently available antiepileptic drugs, while in the remainder seizures are refractory to treatment (Sander, 1993; McNamara, 1996). Moreover, many of the existent antiepileptic agents produce a host of undesirable side-effects including drowsiness, mental dullness, nausea, ataxia, paresthesia, hematologic changes, hirsutism, weight gain, hypertrophy of gums and congenital malformations (Plaa and Willmore, 1995). For these reasons, new antiepileptic drugs are needed to improve seizure control and side-effect profile (Stables et al., 1995).
At the neuronal level, seizure activity often occurs when glutamatergic excitatory neurotransmission overrides GABAA-mediated inhibition (Bradford, 1995). Therefore, glutamatergic and GABAergic systems are rational targets for antiepileptic drug development. Pharmacological manipulation leading to increased levels of GABA (by inhibition of GABA degradation or reuptake) and/or positive allosteric modulation of the GABAA receptor complex are among the approaches that have been used to facilitate inhibitory GABAergic neurotransmission.
The GABAA receptor complex is an ionotropic receptor of pentametric structure containing combinations of α, β, γ, δ or ρ subunits, which form an intrinsic Cl− ion channel (Macdonald and Olsen, 1994;Lüddens et al., 1995). Subunit composition determines the extent and manner in which drugs modulate Cl− ion conductance. Benzodiazepines and barbiturates are positive GABAA receptor modulators that act via different recognition sites (Skolnick and Paul, 1988; Macdonald and Olsen, 1994). Benzodiazepines (such as DZP or clonazepam) and barbiturates (such as PB) are antiepileptics used for the emergency treatment of status epilepticus and to control various types of epilepsy (McNamara, 1996). However, the development of tolerance and dependence are but some of the side-effects accompanying long-term therapy with these compounds that complicate their clinical utility (McNamara, 1996). Therefore, drug discovery efforts are still active in this area. That GABAergic neurotransmission may be also modulated by neuroactive steroids (Harrison and Simmonds, 1984) has suggested the neurosteroid site as a target for improved antiepileptic agents.
Data accumulated since Selye’s experiment uncovering anticonvulsant properties of progesterone against PTZ-induced seizures (Selye, 1942) have suggested that neuroactive steroids constitute a potential new direction for pharmaceutical interventions in epilepsy (Belelliet al., 1990). Neuroactive steroids are devoid of genomic action; rather, they bind to a site on the GABAAreceptor complex distinct from the binding site for benzodiazepines and barbiturates and thereby potentiate GABA-induced Cl− influx (Morrow et al., 1987,1990; Gee, 1988; Lan et al., 1990; Puia et al., 1990). Neuroactive steroids may be endogenously-derived metabolites of progesterone or desoxycorticosterone or produced synthetically in the laboratory (Simmonds, 1991; Paul and Purdy, 1992; Gee et al., 1995). Endogenous neurosteroids like 3α-hydroxy-5α-pregnan-20-one and 5α-pregnan-3α,21-diol-20-one, derived from progesterone and desoxycorticosterone, respectively, are among the most potent positive-allosteric modulators of the GABAA receptor complex (Puia et al., 1990; Lambert et al., 1995). Neuroactive steroids inhibit the development of seizures in kindled rats (Holmes et al., 1984; Carter et al., 1997) and penicillin-induced epileptic foci in the cerebral cortex of cats (Landgren et al., 1987). They also show protective effects in PTZ, bicuculline, picrotoxin, nicotine and strychnine convulsive tests but are ineffective, at nonataxic doses, against maximal electroshock-induced tonic hindlimb extension (Belelli et al., 1990; Luntz-Leybman et al., 1990; Kokate et al., 1994; Wieland et al., 1995).
It is likely that naturally occurring neurosteroids serve as endogenous anticonvulsants at physiologically-relevant concentrations through homeostatic regulation of neuronal excitability (Belelli et al., 1990). Thus, although neuroactive steroids are devoid of hormonal action per se, their anticonvulsant activity is related to cyclic hormonal fluctuation (Finn and Gee, 1993, 1994). For example, low levels of the progesterone metabolites correlate with high seizure susceptibility in women with catamenial epilepsy (Rosciszewskaet al., 1986). Furthermore, lower seizure thresholds for a number of chemical convulsants were measured in female rats in estrus (low level of progesterone) than in diestrus (high level of progesterone) (Finn and Gee, 1994). Correspondingly, a higher threshold for chemically driven seizures was observed in intact female rats than in males or ovariectomized females (Schwartz-Giblin et al., 1989; Kokka et al., 1992; Wilson, 1992).
In this study we characterized the protective actions and safety indices of four neuroactive steroids, 3α-hydroxy-5α-pregnan-20-one (allopregnanolone; 3α,5α-P), 3α-hydroxy-5β-pregnan-20-one (pregnanolone; 3α,5β-P), 3α-hydroxy-3β-methyl-5α-pregnan-20-one (a 3β methyl analog of 3α,5α-P; CCD 1042, ganaxolone) and 3β-(4acetylphenyl)ethynyl-3α,21-dihydroxy-5β-20-one-21-hemisuccinate (Co 2–1068), against several, standard chemical convulsive tests in mice (PTZ, NMDA and cocaine). For comparative purposes, we evaluated DZP, PB and MK-801 in the same tests. These experiments represent the first attempt to characterize the efficacy of neuroactive steroids against cocaine- and NMDA-induced seizures in which DZP and PB are not generally effective (Leander et al., 1988; Witkin and Tortella, 1991). Potencies in the anticonvulsant tests were compared to potencies to produce motor and behavioral side-effects. Moreover, as polytherapy is routinely employed for treatment of refractory epilepsy (Lammers et al., 1995) and because new antiepileptic drugs are often used as adjuvants to classical therapies (Patsalos and Duncan, 1994; Meinardi, 1995), combined treatments were examined for their anticonvulsant and side-effect profiles. The results of the present study document the unique anticonvulsant profile of neuroactive steroids. In addition, the marked enhancement of the anticonvulsant potency of DZP by neuroactive steroids, without significantly increasing motoric side-effects, further substantiates the idea that neuroactive steroids may be of clinical utility.
Methods
Subjects.
Experimentally-naive, male Swiss Webster mice (Taconic Farms, Germantown, NY) between 10 and 12 wk old were housed six per cage in an environmentally controlled room. All animals were acclimated to their home cages and to the light-dark cycle for at least 5 days before testing. Water was continuously available for the mice in their living cages. Experiments were conducted during the light phase of a 12-hr light/dark cycle.
Motor toxicity.
Immediately before administration of convulsant agents, mice were first tested on the inverted screen test. The inverted screen test was used to assess one form of behavioral toxicity induced by the test compounds. This test was an adaptation (Ginski and Witkin, 1994) of that initially described by Coughenouret al. (1977). In this test, compounds with sedative and/or ataxic properties produce dose-dependent increases in screen test failures whereas other classes of drugs (e.g., psychomotor stimulants) do not (Ginski and Witkin, 1994). Mice (at least eight per group) were pretreated with either vehicle or test compound and returned to their home cage for the appropriate pretreatment interval. They were then individually placed on a 14 × 14 cm wire mesh screen (0.8 cm screen mesh) elevated 38 cm above the ground. After slowly inverting the screen, the mice were tested during a 2-min trial for their ability to climb to the top. Mice not climbing to the top (all four paws on upper surface) were counted as a failure. Results were expressed as a TD50 value. Each TD50 value, calculated from a dose-response curve, represents the dose of a drug (in mg/kg) predicted to produce screen failure in 50% of the mice tested. After the screen test, the anticonvulsant tests, as described below, were conducted.
Anticonvulsant testing.
After the screen test, a convulsant dose of cocaine (75 mg/kg), NMDA (0.4 ml of a 20 mg/ml solution/mouse that equals approximately 200 mg/kg) or PTZ (70 mg/kg) was administered and the mice were immediately placed in individual Plexiglas containers (14 × 25 × 36 cm high) for observation. Mice in these experiments were used only once to evaluate the anticonvulsant efficacy of drugs. Doses of the convulsants were chosen to be close to their ED85 - ED95 values as determined during pilot experiments and from the literature (Leanderet al., 1988; Witkin and Tortella, 1991). The presence or absence of convulsions was recorded either for 15 min (PTZ) or 30 min (cocaine, NMDA) following injection. Additionally, acute toxicity of NMDA, reflected by number of deaths within 60 min after its injection was also evaluated.
PTZ-induced seizures were defined as repetitive, rapid clonic convulsions of fore- and hindlimbs lasting continuously at least 5 sec with accompanying loss of righting response. Locomotor depression without loss of righting responses often preceded clonic episodes in PTZ-challenged mice. Between seizure bouts, mice displayed an intact righting response. Cocaine-induced convulsions were defined as loss of the righting response for at least 5 sec and the occurrence of clonic limb movements; tonus and death were rarely observed. Locomotor depression with loss of the righting response often preceded clonic episodes in cocaine-challenged mice. Once seizures developed in cocaine-treated mice, loss of the righting response often persisted over the observation period. NMDA-induced seizures were defined as repetitive, rapid clonic convulsions of limbs lasting continuously at least 5 sec with accompanying loss of righting response. In NMDA-treated mice, tonus often occurred and was usually associated with death. Short durations of locomotor agitation and scratching often preceded seizures produced by NMDA (Leander et al., 1988). Between clonic seizure episodes, mice usually showed loss of righting response.
Locomotor activity.
To further characterize behavioral effects of neuroactive steroids, DZP and PB in doses selected for combined treatments, ambulatory locomotor activity was assessed. Immediately after injections with drugs or vehicle, mice were individually placed into Digiscan activity monitors with a surface area of 40 × 40 cm (Omnitech Electronics, Columbus, OH) for the first time. The activity monitors were equipped with photoelectric detectors spaced 1.8 cm apart along the perimeter that detected motion at a height of up to 2.5 cm above the floor. The activity of the mice was recorded over a 60-min period.
Drugs and administration regimen.
The following neuroactive steroids were used: 3α,5α-P, 3α,5β-P, ganaxolone and Co 2–1068. Pregnenolone, a parent drug for 3α,5α-P that is devoid of neuronal effects, was used as a negative control for neurosteroidal actions. All steroids were synthesized at CoCensys. Steroids were dissolved in 40% (w/v) hydoxypropyl-β-cyclodextrin [Research Biochemicals International (RBI) Natick, MA] with mild heat and sonication. PB sodium (Ruger Chemical Co., Inc., New York, NY), DZP (Hoffmann-La Roche, Nutley, NJ) and (+)-MK-801 (RBI) were studied for comparison. PB, MK-801, pentylenetetrazol (Sigma Chemical Co., St. Louis, MO), and (-)-Cocaine HCl (Sigma) were dissolved in 0.9% NaCl, whereas DZP was suspended in 20% propylene glycol (Sigma) with mild heat. NMDA (RBI) was dissolved in distilled water. All drugs, except for NMDA, were injected in a volume of 0.01 ml/g. NMDA was administered in a volume of 0.4 ml/mouse (20 mg/ml solution) = approximately 200 mg/kg. The steroids were given s.c., 15 min before testing. PB, DZP and MK-801 were administered s.c., 30 min before testing. The convulsants were given by the i.p. route after the appropriate pretreatments.
Two experiments on drug interactions were performed. First, the degree to which a fixed dose of the neuroactive steroids or PB shifted the dose-effect curve for DZP was determined. The selection of a dose of the neuroactive steroids and of PB was based on effects of the individual drugs alone. The doses selected were ineffective against PTZ-induced seizures and did not significantly alter motor behavior in the inverted screen test. In a second drug-interaction experiment, increasing doses of the neuroactive steroids or PB were given in combination with a fixed, ineffective dose of DZP (0.1 mg/kg). Pretreatment times and routes of administration for drug combination experiments were the same as for their individual administrations as listed above.
Data analysis.
The protective potencies of the drugs against the convulsant agents was reflected by respective ED50 values (in mg/kg). A drug, at its ED50 value, was predicted to protect 50% of mice against convulsant-induced seizures or NMDA-induced lethality. ED50 and TD50 values (with 95% confidence limits) of drugs alone or in combination were calculated from dose-effect curves according to the method described byLitchfield and Wilcoxon (1949). For specific comparisons between treatments, Fisher’s exact probability test was used. Although separate control groups (convulsant + vehicle) were run for each dose-effect function, these data were pooled into a common control group for statistical analysis since there were no significant differences across the individual control groups. For drug combinations, slopes (with 95% confidence limits) of regression lines derived from the log(dose)-response function of a drug alone or in combination were calculated and compared statistically for parallelism. Calculations and statistical analysis were performed using the software package accompanying Tallarida and Murray (1987). In drug combinations, relative potencies of the drugs were calculated by dividing the TD50 or ED50 value of a drug alone by the corresponding TD50 or ED50 value of the drug + an adjuvant.
Locomotor activity was expressed as the mean ± S.E.M. of total activity counts within 60 min for each treatment group, consisting of eight mice each. The results from the locomotor activity tests were compared statistically by using Student’s t test for unpaired groups. Effects with statistical probabilities of error of greater than 0.05 were considered to be nonsignificant.
A PI for a drug alone or in combination was calculated by dividing the respective TD50 value for the drug alone or drug combination by the corresponding anticonvulsant ED50 value. A protective index is a quantitative measure of the margin between doses producing behavioral toxicity and doses producing anticonvulsant protection (Löscher and Nolting, 1991).
Results
Motor toxicity.
The neuroactive steroids dose-dependently increased the percentage of mice falling off the screen (fig.1, top panel). Pregnenolone, which lacks neuronal activity, was inactive up to 30 mg/kg. DZP, PB and MK-801 also dose-dependently increased the percentage of mice exhibiting motor toxicity (fig. 1. bottom panel). The following rank order of potency was observed: MK-801 > DZP > ganaxolone = 3α,5α-P = 3α,5β-P > Co 2–1068 > PB (see table1).
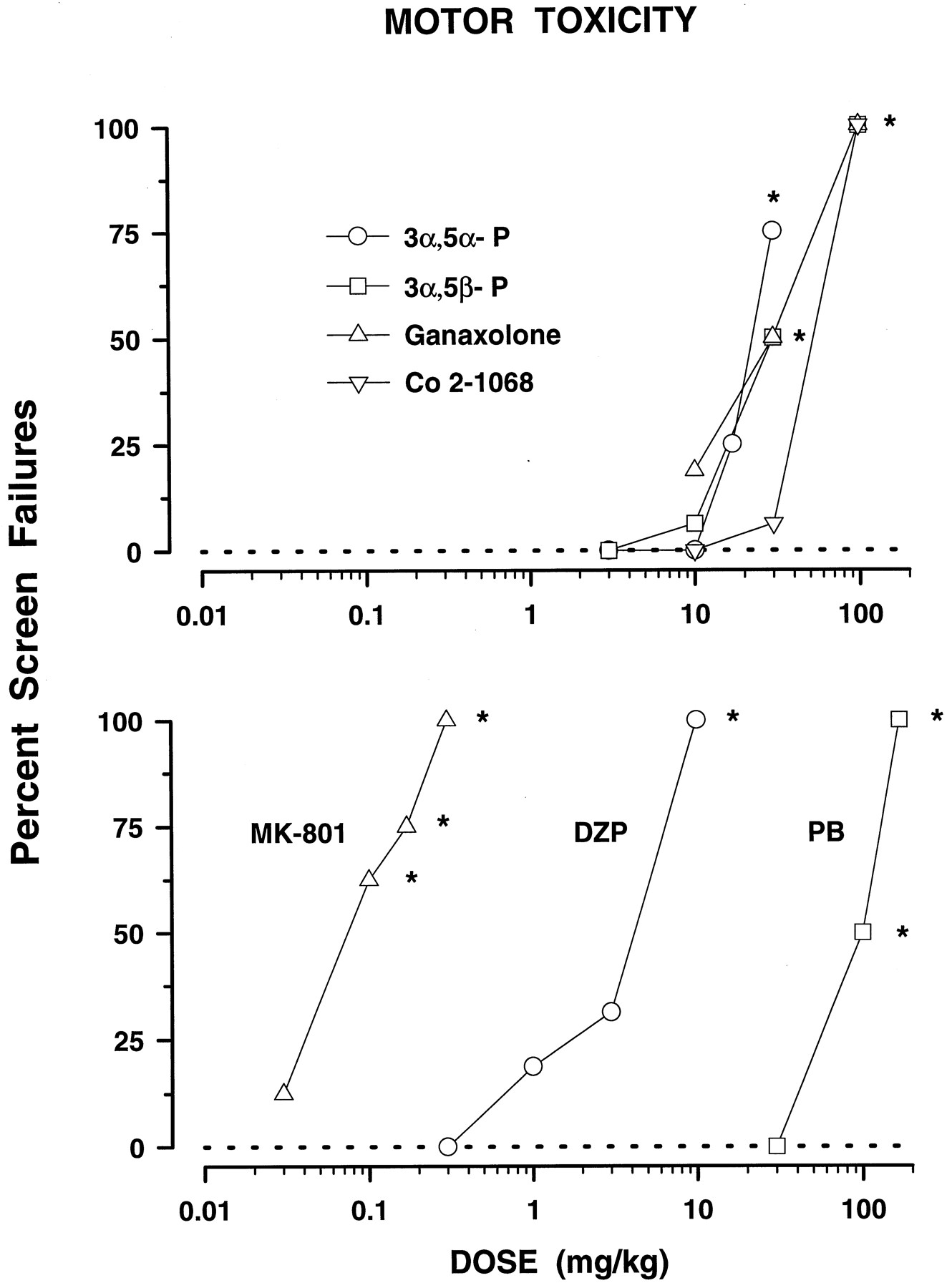
Effects of neuroactive steroids (top panel) and DZP, PB and MK-801 (bottom panel) against the inverted screen test in mice. Each data point reflects the percentage of mice falling off the screen (n ≥ 8 mice per one data point). All vehicle-treated mice correctly performed the test (dashed line at 0). TD50 values with 95% confidence limits, calculated from these dose-response curves, are shown in table 1. *P < .05; compared to vehicle-treated mice (Fisher’s exact probability test).
Motor toxicity (TD50) and anticonvulsant profile (ED50) of the neurosteroids in comparison with pregnenolone, DZP, PB and MK-801
Anticonvulsant effects.
All four neuroactive steroids fully and dose-dependently blocked the convulsant effects of cocaine (fig.2, top panel). ED50values and associated 95% CLs are shown in table 1. Pregnenolone, in the dose range 10 to 30 mg/kg, failed to show any protective action against cocaine-induced seizures. Complete and dose-dependent protection was also observed after pretreatment with MK-801. In contrast, DZP and PB offered poor anticonvulsant efficacy against cocaine, both drugs achieving only 50% effect (fig. 2, bottom panel and table 1) and only at the highest doses that had marked behavior toxicity (fig. 1, bottom panel). The rank order of protective potency against cocaine-induced seizures was MK-801 > 3α,5β-P = 3α,5α-P = ganaxolone = Co 2–1068 > DZP > PB.

Effects of neuroactive steroids (top panel) and DZP, PB and MK-801 (bottom panel) against convulsions induced by 75 mg/kg cocaine in mice. Each data point represents the percentage of mice protected against seizures (n ≥ 8 mice per one data point). Cocaine alone produced convulsions in 85.51% (S.E.M. ± 1.21) of mice challenged. The dashed line at 14.49 shows the percentage of control mice not seizing after cocaine challenge. Respective ED50 values are given in table 1. *P < .05; compared to cocaine alone (Fisher’s exact probability test).
Two of the neuroactive steroids, 3α,5β-P and Co 2–1068, dose-dependently protected against NMDA-induced convulsions with 3α,5β-P offering full protection in the dose of 100 mg/kg (fig. 3, top panel, table 1) which also was toxic in the inverted screen test. In contrast, Co 2–1068 protected 50 and 75% of mice (P < .05vs. (NMDA + vehicle)-treated control group) at doses that did not impair motor performance on the inverted screen test (10–30 mg/kg). Ganaxolone and 3α,5α-P (3–30 mg/kg) and pregnenolone (10–30 mg/kg) were completely ineffective against NMDA-produced seizures. MK-801 produced full and dose-dependent protection against NMDA. In contrast, neither DZP nor PB were fully efficacious up to high doses (fig. 3, bottom panel and table 1). The rank order of potency for this test was MK-801 > Co 2–1068 > DZP = 3α,5β-P followed by 3α,5α-P, ganaxolone and PB which had ED50 values greater than 30, 30 and 170 mg/kg, respectively (table 1).

Anticonvulsive properties of neuroactive steroids (top panel) and DZP, PB and MK-801 (bottom panel) against NMDA-induced seizures in mice. Each data point represents the percentage of mice protected against seizures (n ≥ 8 mice per one data point). NMDA was administered in the dose of 200 mg/kg, in which it produced seizures in 94.17% (S.E.M. ± 3.63) of control animals. The dashed line at 5.83 shows the percentage of control mice not seizing after NMDA challenge. See table 1 for ED50 values for each group. *P < .05; compared to NMDA alone (Fisher’s exact probability test).
Although 3α,5α-P did not block NMDA-induced seizures, it was the only neuroactive steroid producing full, dose-dependent protection (3–30 mg/kg) against the lethal effects of NMDA (fig.4, top panel). Similarly, ganaxolone did not protect against NMDA-induced seizures but diminished acute NMDA-produced lethality. Both 3α,5β-P and Co 2–1068 were effective against the seizures and the lethality produced by NMDA. The ED50 of 3α,5β-P could not be calculated because the drug significantly decreased lethality without producing a clear dose-effect relationship (fig. 4). Neither seizures nor lethality decreased after pretreatment with pregnenolone in doses of 10 and 30 mg/kg.

Protective effects of neuroactive steroids (top panel) and DZP, PB and MK-801 (bottom panel) against NMDA-induced lethality in mice. Each data point (n ≥ 8 mice per one data point) represents the percentage of mice protected against lethal effects of NMDA (200 mg/kg), which when given alone produced lethality in 88.33% (S.E.M. ± 6.09) of the animals. The dashed line at 11.67 shows the percentage of mice surviving after NMDA challenge. Respective ED50 values may be found in table 1. *P < .05; compared to NMDA alone (Fisher’s exact probability test).
As with 3α,5α-P, PB was not effective against NMDA-induced seizures, but fully and dose-dependently reduced the lethal effects of NMDA (fig. 4, bottom panel). MK-801 produced full and dose-dependent protection against NMDA-induced lethality with same potency as it did against seizures (table 1). In contrast to MK-801, DZP was less effective against seizures than lethality produced by NMDA (ED50 values were 16.99 compared to 4.01 mg/kg, respectively). The rank order of potency in this test was MK-801 > DZP = 3α,5α-P = Co 2–1068 > ganaxolone > PB (table 1).
Full protection against PTZ-induced seizures was achieved by three of four neuroactive steroids, while 87.5% protection was produced by 3α,5α-P in doses of 3 and 10 mg/kg (fig.5, top panel). Pregnenolone (30 mg/kg) failed to block PTZ seizures. Potencies of the neuroactive steroids ranged from 2.27 to 6.17 (table 1). DZP and PB, in doses devoid of behavioral side-effects (fig. 1), fully and dose-dependently suppressed seizures produced by PTZ (fig. 5, bottom panel). DZP was the most potent drug studied against PTZ with an ED50 of 0.26 mg/kg. MK-801, at the highest dose tested, 0.3 mg/kg, failed to protect mice from PTZ-induced clonic convulsions (table 1). Higher doses of MK-801 were not tested due to the maximal behavioral toxicity achieved at 0.3 mg/kg (fig. 1). The rank order of potency for this test was DZP > neuroactive steroids = PB (table 1).

Anticonvulsant effect of neuroactive steroids (top panel) and DZP, PB and MK-801 (bottom panel) against PTZ-induced seizures in mice. Each data point represents the percentage of mice protected against seizures (n ≥ 8 mice per one data point). PTZ, administered in the dose of 70 mg/kg, produced seizures in 84.11% (S.E.M. ± 1.92) of animals. The dashed line at 15.89 shows the percentage of mice not seizing after PTZ alone. See table 1 for ED50 values of these groups. *P < .05; compared to PTZ alone (Fisher’s exact probability test).
Protective indices.
As a measure of the separation in potencies between anticonvulsant effects and motor impairment, protective indices were calculated (table 2). The neuroactive steroids displayed PI values against cocaine that ranged from 6.4- to 14.6-fold more than that of MK-801. PI values for DZP or PB could not be calculated because the drugs failed to show full protection against cocaine-induced seizures. Against NMDA-induced convulsions, the PI value for Co 2–1068 (7.0) was substantially greater than the PI for 3α,5β-P and MK-801 (PI = 1.5 and 1.3, respectively) and DZP. Against the lethal effects of NMDA, PI values for Co 2–1068 and 3α,5α-P (7.8 and 5.3, respectively) were better than for the other compounds for which PIs could be calculated (PI values ranging from 0.8 to 1.9 for DZP, MK-801, ganaxolone and PB). In the case of PTZ-induced seizures, PI values of neuroactive steroids, DZP and PB were similar and ranged from 7.2 to 14.
Protective indices (PI) of the neurosteroids, DZP, PB and MK-801
Drug combinations.
Doses of 1.7 mg/kg of 3α,5α-P and 3 mg/kg of 3α,5β-P, ganaxolone, Co 2–1068 and PB were selected for combined experiments with DZP because the drugs in these doses were ineffective in the inverted screen test (fig. 1) and against PTZ-induced seizures (fig. 5). Coadministration of the neuroactive steroids or PB did not affect the overall dose-response curve for DZP + vehicle on motor toxicity. The TD50 values of the drug combinations ranged from 1.50 (DZP + 3α,5β-P) to 2.88 (DZP + PB) mg/kg and none of these values differed significantly from the TD50 value of the (DZP + vehicle)-treated group (3.13 mg/kg). The relative potency of DZP to produce motor toxicity ranged from 1.1 (DZP + PB) to 2.1 (DZP + 3α,5β-P) respectively (table3).
Motor toxicity (TD50), anticonvulsant potency (ED50) against PTZ-induced seizures and protective indices (PI) of DZP alone and in combination with the neurosteroids and PB
In contrast, the neuroactive steroids and PB significantly and markedly potentiated the anticonvulsant potency of DZP against PTZ-induced seizures. Potentiation was reflected in the statistically significant decrease in the protective ED50 value of DZP (table 3). Protective potencies of the drug combinations ranged from 7.4-fold (DZP + 3α,5α-P) to 49.1-fold (DZP + ganaxolone) greater than DZP alone and a shift to the left in the dose-effect curve of DZP (fig. 6). However, the dose-effect functions for DZP and DZP + drug combinations were not parallel. The slopes of regression lines derived from dose-response functions (slope ± 95% CL) of DZP in combination with 3α,5α-P (S = 31.95 ± 21.44), 3α,5β-P (S = 31.25 ± 45.84), ganaxolone (S = 20.91 ± 19.93), Co 2–1068 (S = 25.00 ± 91.70) and PB (S = 39.99 ± 22.52) were significantly different from the slope of the regression line derived from the dose-response function in the (DZP + vehicle)-treated group (S = 87.62 ± 65.99).

Effects of neurosteroids and PB on the protective potency of DZP. Each graph shows dose-response functions for DZP + vehicle (solid symbols) and DZP + an adjuvant (open symbols) against PTZ. Computed linear regressions (solid lines) were drawn for each dose-response function (see “Results” for statistical comparison of slopes of each regression line). Neurosteroids and PB were administered in ineffective doses against PTZ (see table 3 for ED50 values of DZP evaluated in these combinations). Each data point represents the percentage of mice protected against PTZ-induced convulsions (n ≥ 8 mice per data point).
The potentiation of the anticonvulsant potency of DZP occurred in a dose range of DZP that did not significantly enhance the behavioral toxicity of DZP (table 3). This resulted in substantial improvement of the therapeutic window of DZP that ranged from 58.6 to 359 for the combination with 3α,5α-P and Co 2–1068, respectively (table 3). In contrast, the PI value of the (DZP + vehicle) treatment was 12.0.
DZP in the dose of 0.1 mg/kg did not display either motor toxicity (fig. 1) or protective effects against PTZ-induced convulsions (fig. 5) and was evaluated for its ability to modify the potency of the neuroactive steroids. DZP (0.1 mg/kg) failed to alter the anticonvulsant potency of 3α,5α-P against PTZ (table4). In contrast, DZP significantly potentiated the protective potencies of 3α,5β-P, ganaxolone, Co 2–1068 and PB (table 4). This was reflected in significant decreases in the ED50 values of drugs combined with DZP that were between 3.6-fold (ganaxolone + DZP) and 8.3-fold (PB + DZP) lower than respective ED50 values of drugs alone (table 4). Pretreatment with DZP produced significant, leftward shifts in the dose-response curves of 3α,5β-P, ganaxolone, Co 2–1068 and PB (fig. 7). The shift was parallel because slopes of dose-response curves for 3α,5β-P (S = 62.65 ± 70.56), ganaxolone (S = 49.96 ± 16.86), Co 2–1068 (S = 45.93 ± 225.30) and PB (S = 74.56 ± 208.56) in combination with DZP did not differ from that of 3α,5β-P (S = 87.82 ± 153.76), ganaxolone (S = 120.48 ± 487.12), Co 2–1068 (S = 88.01 ± 245.40) and PB (S = 66.25 ± 119.98) alone.
Anticonvulsant potency of neurosteroids and PB alone (vehicle-treated group) and in combination with DZP (0.1 mg/kg) against PTZ-induced seizures

Effects of DZP on the protective potency of neurosteroids and PB against PTZ-induced seizures. DZP was administered in the dose 0.1 mg/kg, which alone was ineffective against PTZ-induced seizures (fig. 5). Each graph shows dose-response functions of 3α,5α-P (top left), 3α,5β-P (top right), ganaxolone (middle left), Co 2–1068 (middle right) and PB (bottom) alone (solid symbols) or in combination with DZP (open symbols). Slopes of regression lines derived from dose-response curves may be found in “Results”. Each data point on a graph represents the percentage of mice protected against PTZ-induced convulsions (n ≥ 8 mice per data point). See table 4 for ED50 values and their statistical comparison.
Locomotor activity.
The effects of drugs studied in the drug combination experiments were assessed on ambulatory locomotor activity of mice. All four neuroactive steroids at doses which significantly potentiated the protective potency of DZP (table 3), were devoid of significant effects on locomotor activity over a 60 min recording period. Total activity counts of 4950.4 ± 1117.9 (mean ± S.E.M.) were recorded in the vehicle-treated group. In groups treated with 3α,5α-P (1.7 mg/kg), 3α,5β-P (3 mg/kg), ganaxolone (3 mg/kg) or Co 2–1068 (3 mg/kg), total activity counts of 7029.3 ± 1282.4, 4632.0 ± 603.0, 4888.0 ± 508.5 and 4929.9 ± 948.2 were registered, respectively. Similarly, DZP (0.1 mg/kg) had no significant effect on locomotor activity (7319.3 ± 680.1). In contrast, PB (3 mg/kg) significantly increased (P < .05vs. vehicle-treated group) spontaneous locomotor activity (7886.4 ± 696.3) in mice.
Discussion
Evaluation of the anticonvulsant effects of four neuroactive steroids demonstrated their anticonvulsant efficacy and revealed both similarities and differences in their pharmacological profiles to those of benzodiazepine, barbiturate and glutamate antagonist-type anticonvulsants. The neuroactive steroids displayed margins of safety or protective indices (PI = TD50/ED50) comparable to or better than those of standard anticonvulsant agents (Löscher and Nolting, 1991). Further, all of the neuroactive steroids significantly potentiated the anticonvulsant effects of the benzodiazepine, DZP. The combined treatment of neuroactive steroids and DZP resulted in exceptional margins of safety. These results demonstrate a more extensive profile of antiepileptic effects of the neuroactive steroids than previously realized. Further, although quantitative variation in the anticonvulsant properties of neuroactive steroid structures has been demonstrated (cf. Kokate et al., 1994), our findings document qualitatively distinct anticonvulsant effects among structural analogs. Finally, the negative findings with pregnenolone, which does not act as a neuronal modulator of GABA, provides additional support for the importance of neuronal activity in the antiepileptic effects of this class of compounds. As a whole, these findings help to define neuroactive steroids as a novel class of anticonvulsant agents with potential clinical applicability.
The neuroactive steroids studied here shared with DZP and PB the ability to fully and dose-dependently prevent the clonic convulsions produced by PTZ (fig. 5, table 1). Because the PTZ test in rodents is predictive of anticonvulsant drug efficacy in absence (petit mal) (Swinyard, 1969) and myoclonic (Löscher and Schmidt, 1988) epilepsy in humans, the protective efficacy of neuroactive steroids against PTZ suggests their potential clinical value. The anti-PTZ effects of neuroactive steroids have been reported previously (Belelliet al., 1989; Kokate et al., 1994; Wielandet al., 1995; Carter et al., 1997). In addition, in our studies, the neuroactive steroids fully blocked the clonic convulsions induced by cocaine (fig. 2, table 1), convulsions that were insensitive to DZP or PB treatment as previously reported (Witkin and Tortella, 1991). DZP and PB are drugs of choice for the emergency treatment of seizures and/or status epilepticus resulting from cocaine intoxication (VanDette and Cornish, 1989). Unfortunately, status epilepticus following cocaine poisoning is often resistant to standard therapy and can be fatal (Dhuna et al., 1991). It is important to stress that in terms of total drug-related medical complications and deaths in the United States, cocaine has the highest morbidity and mortality (Benowitz, 1993). Therefore, this first report on the efficacy and favorable therapeutic window of neuroactive steroids against cocaine-induced seizures may open new clinical applications for this drug class.
The anticonvulsant profile of at least some neuroactive steroids was also extended to the protection against NMDA-induced seizures and lethality (figs. 3 and 4, table 1). Full protection against NMDA-induced seizures was produced by 3α,5β-P and by the NMDA receptor antagonist MK-801; nearly full blockade was produced by Co 2–1068. In terms of therapeutic window (PI), both of these neuroactive steroids demonstrated a better preclinical profile than MK-801. The anti-NMDA action of neuroactive steroids may be clinically important as NMDA-induced excitation is involved in seizure activity and postseizure pathological changes at the neuronal level (Bradford, 1995). Interpretation of data from the anticonvulsive action of drugs against NMDA-induced seizures is difficult, however, because drug effects can be non-specific (Leander et al., 1988). As a host of drugs can block NMDA-induced seizures (Leander et al., 1988;Palmer et al., 1993), NMDA-induced lethality appears to be more specific for evaluating in vivo efficacy of functional NMDA receptor blockade (Leander et al., 1988). In our experiment all neuroactive drugs tested produced significant protection against NMDA-induced lethality. However, only two of the neuroactive steroids displayed PIs that were notably greater than unity (3α,5α-P and Co 2–1068). Of these compounds, only Co 2–1068 demonstrated equivalent potency to block both the seizurogenic and the lethal effects of NMDA, a relationship demonstrated with selective NMDA receptor blockers, such as MK-801, reported here (Leander et al., 1988). Functional blockade of glutamatergic neurotransmission by select neuroactive steroids may offer a wide spectrum of antiepileptic effects.
Although the neuroactive steroids produced a common spectrum of anticonvulsant effects against both PTZ and cocaine, differences in the pharmacological effects of the neuroactive steroids were also uncovered. Differences in potency and PI values were observed, although these differences were not marked, with differences not generally exceeding 2-fold (tables 1 and 2). Potency differences in the anticonvulsant effects of neuroactive steroids also have been noted by others (cf. Kokate et al., 1994). Two additional differences across neuroactive steroids were more striking. First, the endogenous steroid 3α,5α-P was less effective in augmenting the effects of DZP (fig. 6, table 3) and its anticonvulsant effects were not augmented by DZP (fig. 7, table 4). The second major pharmacological difference observed among the neuroactive steroids was in their effects against NMDA. Although all of the neuroactive steroids significantly protected against the lethal effects of NMDA (fig. 4), only two of the compounds (3α,5β-P and Co 2–1068) were effective in preventing the convulsions induced by NMDA (fig. 3). Interestingly, both of these compounds are 5β-reduced, in which the steroid A-ring projects out of the general plane of the pregnane ring system. Whether the ability to block NMDA-induced convulsions is conferred by the cis-fused configuration of the steroid A-ring is a question that awaits experimental verification.
Endogenous as well as synthetic neuroactive steroids, like alphaxalone, (5α-pregnan-3α-ol-11,20-dione) potentiate the amplitude of membrane currents triggered by GABA in nanomolar concentrations, whereas in micromolar concentrations they can produce direct GABA-like effects (Barker et al., 1987; Callachan et al., 1987). Neuroactive steroids also share a molecular mechanism with benzodiazepines in their ability to increase the frequency of single channel openings in the GABAA receptor (Macdonald and Olsen, 1994). These common GABA mechanisms may explain the shared efficacy of the neuroactive steroids studied with that of PB and DZP against the GABAergic convulsant PTZ. The common GABAergic actions of neuroactive steroids and DZP or PB cannot, however, account for the differential effects on cocaine-induced convulsions. The low efficacy of the GABAergic drugs DZP and PB together with the high efficacy of an NMDA receptor antagonist (MK-801) to prevent cocaine seizures suggest that the GABAA receptor complex does not play a pivotal role. Instead, these findings favor the involvement of glutamatergic neurotransmission in cocaine-induced seizures under these conditions. This explanation is in line with earlier experiments showing that competitive and noncompetitive NMDA receptor antagonists blocked seizures induced by cocaine (Witkin and Tortella, 1991;Rockhold et al., 1991; Tortella et al., 1992). However, the interactions of neuroactive steroids with the NMDA receptor appear to be relevant only at micromolar concentrations and, given their qualitatively diverse effects on the NMDA-driven Ca++ flux (cf. Irwin et al., 1994), cannot account for the homogeneity of effects of the neuroactive steroids as anticonvulsants against cocaine. In addition, although only Co 2–1068 demonstrated NMDA antagonist activity in vivo, blocking both NMDA-induced convulsions and lethality at comparable doses (figs. 3 and 4, table 2), all of the neuroactive steroids were effective in preventing cocaine-driven convulsions.
Cocaine-, PTZ- or NMDA-induced seizures reflect summary disturbances of many neurotransmitter systems including adenosinergic (Murray et al., 1993), dopaminergic (Starr, 1996), glutamatergic and GABAergic systems (Bradford, 1995), as well as voltage-operated Ca++ channels (Heinemann and Hamon, 1986). The anticonvulsant efficacy of neuroactive steroids has been mainly attributed to their GABAA-enhancing action (Belelli et al., 1990; Kokate et al., 1994); however, other mechanisms may be at least partly involved in their anticonvulsant action. Some neuroactive steroids inhibit voltage-operated Ca++ channels in neurons isolated from the CA1 region of the guinea pig hippocampus and are similar to “classical Ca++ channel blockers” of the dihydropyridine class (ffrench-Mullen and Spence, 1991; Spenceet al., 1991). Enhanced, uncontrolled neuronal influx of Ca++ ions plays a critical role in the initiation and spread of seizure activity (Schwartzkroin and Wyler, 1980) which explains the protective efficacy of Ca++ channel antagonists in a variety of laboratory seizure models (Speckmannet al., 1993), including PTZ and NMDA tests (Meyer et al., 1987; Czuczwar et al., 1990; Palmer et al., 1993; Gasior et al., 1996). For convulsions induced by cocaine, however, Ca++ channel blockers do not appear to have efficacy (Derlet and Albertson, 1989;Derlet et al., 1994). Nonetheless, the inhibitory action of neuroactive steroids on voltage-operated Ca++channels may contribute to their anticonvulsant profiles.
In clinical practice, drug-resistant seizures are sometimes ameliorated with poly-drug therapy (Lammers et al., 1995). Success along this avenue is achieved when the drug combination improves control of epileptic symptoms without increasing toxic manifestations of the pharmacological treatments. Ideally, such adjunct therapy permits side-effect profiles to be reduced through the lowering of individual drug dosages. In addition to the in vitro findings of supra-additive effects of neuroactive steroids and PB in combination on GABAA receptor activity (Cottrell et al., 1987), the potential effects of neuroactive steroids on non-GABA targets (as discussed above) suggested that they may be suitable candidates for adjunct therapeutic application. Indeed, all four of the neuroactive steroids potentiated the anticonvulsant action of DZP against PTZ-induced convulsions with significant shifts to the left in the dose-effect function for DZP (fig. 6). The potency of DZP was improved 7- (3α,5α-P) to 50-fold (ganaxolone) by addition of a neuroactive steroid. Importantly, potentiation of the protective action of DZP was not accompanied by significant augmentation of the behavioral toxicity of DZP (table 3). As a result, the PIs of the drug combinations were markedly improved over that of DZP itself (table 3). Differences in the PI values of neuroactive steroid-DZP combinations cannot be accounted for by the dose selected for interaction. For all of the neuroactive steroids, none of the doses demonstrated impairment of performance in the inverted screen test or of ambulatory locomotor activity, except for PB which increased locomotor activity. Further, although doses of 3α,5α-P, 3α,5β-P, and ganaxolone used in combination with DZP were approximately equal to their ED50 values, the dose of both Co 2–1068 and PB was about half of the ED50. These data as a whole are in accord with in vitro findings that both neuroactive steroids and barbiturates in submicromolar concentrations enhanced binding of [H3]benzodiazepines to GABAA receptors (Majewska et al., 1986) and potentiated GABAACl− influx even in the presence of maximally effective concentrations of barbiturates and vice versa (Gee et al., 1987; Paul and Purdy, 1992). These in vitro data also suggest that neuroactive steroids might potentiate the anticonvulsive effects of barbiturates. The finding that neuroactive steroids potentiate the anticonvulsant potency of DZP raises the possibility of an important clinical implication thereby combined treatment (requiring markedly lower doses of DZP) might delay or eliminate tolerance to the anticonvulsive effects of DZP.
In conclusion, the neuroactive steroids, 3α,5α-P, 3α,5β-P, ganaxolone and Co 2–1068, offered a broad spectrum of protective activity against different experimental models of seizures. Differences in the pharmacological profiles of the neuroactive steroids suggest that specific anticonvulsant profiles may be achieved by structural variation. Nonetheless, the mechanisms accounting for the different effects of neuroactive steroids remain to be investigated. Experiments on the combined treatments with diazepam predicted that neuroactive steroids may be safely and effectively used in polytherapy to control drug-resistant seizures. The results obtained in this experiment rationalizes further evaluation of neuroactive steroids in the pharmacological management of epilepsy and other seizure-related disorders. Ganaxolone is currently under Phase II clinical investigation.
Acknowledgments
The authors thank Dr. Ravi Upasami and Kevin Tang for synthesis of Co 2–1068.
Footnotes
-
Send reprint requests to: Dr. M. Gasior, Preclinical Pharmacology Laboratory, NIDA Addiction Research Center, 5500 Nathan Shock Drive, Baltimore, MD 21224. U.S.A. e-mail:mgasior{at}irp.nida.nih.gov
-
↵1 Animals used in these studies were maintained in facilities fully accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). In conducting the research described in this report, the investigators adhered to the “Guide for the Care and Use of Laboratory Animals”, as promulgated by the Committee on the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council. Some of these results have appeared in abstract form (Witkin, J. M., Gasior, M., Goldberg, S. R. and Carter, R. B. Anticonvulsant profile of some neuroactive steroids. Soc. Neurosci. Abstracts 22: 2105, 1996).
-
↵2 A Visiting Fellow in the NIH Visiting Program granted from Fogarty International Center, Bethesda, MD. Permanent address: Department of Pharmacology, Medical University School, Lublin, Poland.
- Abbreviations:
- C.L.
- confidence limits
- CCD 1042
- 3α-hydroxy-3β-methyl-5α-pregnan-20-one (ganaxolone)
- Co 2–1068
- 3β-(4acetylphenyl) ethynyl-3α,21-dihydroxy-5β-20-one-21-hemisuccinate Na)
- DZP
- diazepam
- ED
- effective dose
- GABA
- γ-aminobutyric acid
- MK-801
- dizocilpine
- NMDA
- N-methyl-D-aspartate
- 3α
- 5α-P, 3α-hydroxy-5α-pregnan-20-one (allopregnanolone)
- 3α
- 5β-P, 3α-hydroxy-5β-pregnan-20-one (pregnanolone)
- PB
- phenobarbital
- PI
- protective index
- PTZ
- pentylenetetrazol
- S
- slope
- TD
- toxic dose
- Received January 22, 1997.
- Accepted April 14, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}