


Abstract
S18327 was dose-dependently active in several models of potential antipsychotic activity involving dopaminergic hyperactivity: inhibition of apomorphine-induced climbing in mice, of cocaine- and amphetamine-induced hyperlocomotion in rats, and of conditioned avoidance responses in rats. Furthermore, reflecting its high affinity at serotonin2A sites, S18327 potently blocked phencyclidine-induced locomotion and 1-[2,5-dimethoxy-4-iodophenyl]-2-aminopropane-induced head-twitches in rats. In models of glutamatergic hypoactivity, S18327 blocked hyperlocomotion and spontaneous tail-flicks elicited by theN-methyl-d-aspartate antagonist dizocilpine. The actions of S18327, together with its binding profile at multiple monoaminergic receptors (15 parameters in total), were compared with those of clozapine, haloperidol, and 11 other antipsychotics by multiparametric analysis, and the resulting dendrogram positioned S18327 close to clozapine. Consistent with a clopazine-like profile, S18327 generalized to a clozapine discriminative stimulus and evoked latent inhibition in rats, blocked aggression in isolated mice, and displayed anxiolytic properties in the ultrasonic vocalization and Vogel procedures in rats. Relative to the above paradigms, only markedly (>20-fold) higher doses of S18327 were active in models predictive of potential extrapyramidal side effects: induction of catalepsy and prolactin secretion, and inhibition of methylphenidate-induced gnawing in rats. S18327 showed only modest affinity for histaminic and muscarinic receptors. Multiparametric analysis of these data distinguished S18327 from both haloperidol (high extrapyramidal potential) and clozapine (high histaminic and muscarinic affinity). In conclusion, S18327 displays a broad-based pattern of potential antipsychotic activity at doses appreciably lower than those eliciting extrapyramidal side effects. In this respect, S18327 closely resembles clozapine, but it is chemically distinct and displays weak affinity for histaminic and muscarinic receptors.
As described in the accompanying paper, the novel ligand S18327 (1-{2-[4-(6-fluoro-1,2-benzisoxazol-3-yl)piperid-1-yl]ethyl}3-phenyl imidazolin-2-one) displays pronounced antagonist properties at α1-adrenergic receptors (ARs), serotonin [5-hydroxytryptamine (5-HT)2A], and D4 receptors, as well as less potent antagonist actions at α2-AR and D1and D2 receptors. Interestingly, the α2-AR antagonist properties of S18327 underlie a generalized enhancement of cerebral adrenergic transmission and a preferential facilitation of the activity of frontocortical compared with subcortical dopaminergic pathways. On the other hand, the inhibitory influence of S18327 on the activity of serotonergic neurons originating in the dorsal raphe nucleus may be attributed to its antagonist actions at α1-ARs. Notably, S18327 only weakly accelerates striatal turnover of dopamine (DA). In light of these observations, in the present study we examined the activity of S18327 in paradigms predictive of the control of positive and negative-cognitive symptoms of schizophrenia compared with the induction of extrapyramidal and other side effects. To this end, the following models were used.
First, because hyperactive, mesolimbic dopaminergic pathways are implicated in the induction of positive symptoms, the influence of S18327 on amphetamine- and cocaine-induced locomotion in rats, apomorphine-induced climbing in mice, and conditioned avoidance responses (CARs) in rats was evaluated (Wirsching et al., 1995; Millan et al., 1998a,b). Second, inasmuch as a deficiency in glutamatergic/N-methyl-d-aspartate (NMDA) receptor-mediated transmission may be involved in psychotic states (Sokoloff, 1998; Faustman et al., 1999), we examined the influence of S18327 on the hyperlocomotion and spontaneous tail-flicks (STFs) elicited by the selective open channel blocker at NMDA receptors, dizocilpine (Millan, 1991; Martin et al., 1997; Brocco et al., 1999). The STF response is of particular interest inasmuch as α2-AR receptors are involved in its mediation (Brocco et al., 1999; Millan et al., submitted). Third, an additional open channel blocker at NMDA receptors, phencyclidine (PCP), inhibits 5-HT reuptake and elicits several behavioral responses via serotonergic mechanisms (Hiramatsu et al., 1989; Steinpreis, 1996). Notably, PCP-induced locomotion in rats involves the (indirect) activation of 5-HT2A receptors in the nucleus accumbens (Maurel-Rémy et al., 1995, 1998). Thus, the influence of S18327 on PCP-induced locomotion was determined. In addition, we examined the ability of S18327 to block head-twitches (HTWs) provoked by the hallucinogen and 5-HT2A agonist 1-[2,5-dimethoxy-4-iodophenyl]-2-aminopropane (DOI; Schreiber et al., 1995; Willins and Meltzer, 1997). Fourth, deficits in cognitive-attentional function displayed by psychotic patients may be modeled by a latent inhibition (LI) paradigm in rats (Weiner and Feldon, 1997). In this model, we thus determined the ability of S18327 to enhance the ability to ignore irrelevant sensory input. Fifth, we examined the ability of S18327 to generalize to a discriminative stimulus (DS) generated by the “atypical” antipsychotic clozapine (Goudie and Taylor, 1998, Millan et al., 1999b). Sixth, anxiolytic properties may enhance patient compliance, suppress the fear triggered by productive crises, and counter the social withdrawal associated with negative symptoms (Wiley et al., 1993; Wirsching et al., 1995; Millan et al., 1999a). Thus, the activity of S18327 in models of potential anxiolytic activity was examined in rats: fear-conditioned ultrasonic vocalizations (USVs) and a Vogel conflict test. The influence of S18327 on aggression in isolated mice was likewise studied. Seventh, regarding potential extrapyramidal side effects, the ability of S18327 to evoke catalepsy and to inhibit stereotyped gnawing elicited by methylphenidate, as well as its influence on prolactin (PRL) secretion, was determined in rats. Finally, the affinity of S18327 for histaminic and muscarinic receptors was quantified because blockade of these sites is involved in the autonomic/cardiovascular side effects of clozapine (Cunningham-Owens, 1996).
This broad-based pattern of functional models, together with the receptorial analyses of the accompanying paper, was intended to facilitate comparisons of the phenylimidazoline S18327 with clozapine (a dibenzodiazepine) and with the neuroleptic haloperidol (a butyrophenone derivative), both of which we previously examined in these paradigms (Millan et al., 1998b, 1999a,b). In addition, for several key receptor types and functional paradigms (see Tables 2 and5), we examined the actions of 11 other antipsychotic agents, for most of which “atypical”/“clozapine-like” profiles have been suggested (Brunello et al., 1995; Meltzer, 1995; Arnt and Skarsfeldt, 1998). The generation of this homogeneous and extensive database facilitated a rigorous assessment through “multiparametric” (hierarchical) analysis (Gordon, 1987; Ward, 1963) of similarities and differences among S18327, clozapine, haloperidol, and the other antipsychotic agents. This approach permits the simultaneous analysis in a “polydimensional” space of multiple parameters. In the present case, two complementary analyses were performed: 1) parameters (a total of 15) related to antipsychotic properties and 2) parameters (a total of 5) contributing to side effects. These analyses yielded “dendrograms” hierarchically classifying drugs in accordance with their overall homology.
Binding affinities and functional actions of S18327 as compared to other antipsychotic agents
Binding affinities at histaminic and muscarinic receptors, and extrapyramidal actions for S18327 as compared with other antipsychotic agents
Materials and Methods
Animals.
Unless otherwise specified, we used male Wistar rats of 220 to 250 g (Iffa-Credo, L'Arbresle, France) and CD1 mice of 22 to 25 g (Charles River, Saint-Aubin-les-Elbeuf, France) housed in sawdust-filled cages with unlimited access to food and water. Laboratory temperature was 21 ± 1°C, and humidity was 60 ± 5%. There was a 12/12-h light/dark cycle, with lights on at 7:30 AM. All animals were adapted for at least 1 week to laboratory conditions before use.
Apomorphine-Induced Climbing in Mice.
As previously described (Millan et al., 1998a), climbing was evaluated in mice placed individually in steel cylinders (14 cm diameter) with walls (14 cm high) of vertical bars (1 cm apart and 2 mm diameter). Climbing behavior was assessed according to a score (0–4) both 10 and 20 min after apomorphine (0.75 mg/kg s.c.). Vehicle or S18327 was administered 30 min before apomorphine. Data were analyzed by ANOVA followed by Dunnett's test, and the ID50 values with 95% confidence limits (CLs) were calculated.
Spontaneous Cocaine-, Amphetamine-, PCP-, and Dizocilpine-Induced Locomotion in Rats.
In accordance with a procedure detailed previously (Maurel-Rémy et al., 1995), locomotion was quantified in transparent polycarbonate cages (45 × 30 × 20 cm) placed in activity chambers (Lablinc System; Coulbourn, Lehigh Valley, PA). Locomotion was determined for 60 min after the administration of vehicle (spontaneous locomotion), cocaine (20 mg/kg i.p.), amphetamine (2.5 mg/kg i.p.), PCP (20.0 mg/kg s.c.), or dizolcipine (0.16 mg/kg s.c.). S18327 or vehicle was administered 30 min before drug. The consecutive interruption (within 3 s) of two infrared beams 4 cm above the cage floor and 24 cm apart was computed as a “movement.” Data were analyzed by ANOVA followed by Dunnett's test, and ID50 values (95% CLs) were calculated.
Conditioned Avoidance Procedure in Rats.
As described in detail previously (Millan et al., 1998a), rats were trained to avoid an electric shock (560 μA and 5 s) by changing compartments of a shuttle box (Letica, Barcelona, Spain) after the illumination of a stimulus light. Each trial (10 performed daily) consisted of a 10-s period with the light “on” followed or not by the period with shock depending on the response to the light. The trial was ended when the rat moved into the other compartment. Data examined were the number of CARs emitted in the presence of the light (maximum possible, 10). S18327 or vehicle was administered 30 min before the avoidance session. Data were analyzed with a paired Wilcoxon's signed rank test, and the ID50 values (95% CLs) were calculated.
DOI-Induced HTWs in Rats.
As described previously (Schreiber et al., 1995), after the injection of DOI (2.5 mg/kg i.p.), rats were placed in transparent observation cages (33.5 × 23.5 × 19 cm), and 5 min later, HTWs were counted for 5 min. S18327 or vehicle was administered 30 min before DOI. Data were analyzed by ANOVA followed by Dunnett's test, and the ID50 values (95% CLs) were calculated.
Dizolcipine-Induced STFs in Rats.
STFs were evaluated in rats loosely restrained in horizontal, opaque plastic cylinders with the tail hanging freely over the laboratory bench surface. One STF was defined (Millan, 1991) as the raising of the tail to a level higher than that of the body axis. The number of STFs displayed was quantified over 5 min after a 5-min adaptation period to the cylinder. Dizolcipine (0.08 mg/kg s.c.) was administered 30 min before the evaluation of STFs, and S18327 or vehicle was injected 10 min before dizolcipine. Data were analyzed by ANOVA followed by Dunnett's test, and the ID50 values (95% CLs) were calculated.
LI in Rats.
The procedure used was described in detail elsewhere (Millan et al., 1998b). Briefly, water-restricted rats were submitted to four successive phases (training, preexposure, conditioning, and test) in chambers equipped with lickometers (model ENV 251 M; Med Associates Inc., St. Albans, VT). During training (days 1–6), animals were allowed to drink for 5 min, and rats completing more than 600 licks in the last two sessions were included in the study. During the preexposure (day 7) and the conditioning (day 8) sessions, animals were placed for 15 min in the chambers with the water spout removed. Preexposure consisted of either 0 or 10 tones (2.5 kHz, 10-s duration, 90-s interstimulus interval) within the session. During conditioning, animals received two tones 5 and 10 min after the beginning of the session. Each tone was immediately followed by a scrambled footshock (50 μA and 3 s). For the test session (day 9), the water spouts were replaced in the chambers, and each animal allowed to drink freely until 100 licks had been made. Then, a tone was presented until either the animal took an additional 10 licks or 300 s elapsed. A suppression ratio (SR) was calculated according to the formula: SR = t1 + t2, with t1 and t2 as the times to complete licks 90 to 100 and 100 to 110, respectively. Animals were administered S18327 or vehicle 60 min before the preexposure and conditioning sessions. Data were analyzed with a two-way ANOVA followed by Newman-Keuls test. Induction of LI was inferred when the SR of a 10-tone preexposed group was significantly higher than that of the group receiving the same treatment and not preexposed (Millan et al., 1998b).
Drug Discrimination in Rats.
As described previously (Millan et al., 1999b), rats were trained to discriminate clozapine (5.0 mg/kg i.p.) from saline with a two-lever, fixed-ratio 10 food-reinforced operant design. Sessions, which commenced 30 min after the injection of clozapine or vehicle, were of 15-min duration and were performed daily except on weekends. On the test days (Wednesday and Friday), S18327 or vehicle (administered s.c.) was substituted for clozapine. The percentage of generalization was determined, and the ED50 values (95% CLs) were calculated.
USV in Rats.
As detailed previously (Millan et al. 1999a), there were three different experimental phases performed at intervals of 24 h. On day 1 (training), rats were placed in a chamber equipped with a grid floor and were exposed to six randomly distributed electric shocks (800 μA and 8 s) over a 7-min period. On day 2 (selection), they were placed in the chamber for 2 min and received a single shock. They were returned to the chamber 30 min later, and USVs were recorded for 10 min. Only rats emitting USVs for a total duration of at least 90 s were further examined. On day 3, the procedure was identical to that of day 2, but rats were treated with S18327 or vehicle immediately after the 2-min session. The total duration of USV was recorded over the 10-min session. In the antagonist study with WAY100,635, there was a 60-min interval between the 2-min and 10-min sessions. WAY100,635 (0.16 mg/kg s.c.) or vehicle was administered just after the 2-min session, and S18327 or vehicle was administered 30 min before the 10-min session. The dose effect was analyzed with ANOVA followed by Dunnett's test, and the ID50 values (95% CLs) were calculated. In the antagonism study, data were analyzed with a two-way ANOVA, followed by Newman-Keuls test.
Vogel Test in Rats.
As previously described (Millan et al., 1999a), the test was performed in a polycarbonate cage (32 × 25 × 30 cm) with a grid floor and with the spout of a water bottle located 6 cm above the floor. Both the grid and the spout were connected to an Anxiometer (Columbus Instruments, Columbus, OH). Rats were restricted to water for 1 h over 4 days and then placed in the cages on day 4. On day 5, S18327 or vehicle was administered, and the session was initiated after the rat had made 20 licks and received a single shock (300 μA and 0.5 s) via the spout. Over 3 min, shock was then delivered every 20 licks. In one group of vehicle-treated animals, no shock was delivered to provide a baseline. The percentage drug effect was computed as [(S18327 − vehicle)/(nonshocked − vehicle)]. Data were analyzed with ANOVA, followed by Dunnett's test.
Aggression in Isolated Mice.
As previously described (Millan et al., 1999a), pairs of mice were isolated in black cages for 1 month. On the test day, one mouse (intruder) was placed into the cage of the other (resident), and the total number and duration of fights were determined. Both mice were treated 30 min before the test with either S18327 or vehicle. Data were analyzed with ANOVA followed by Dunnett's test, and the ID50 values (95% CLs) were calculated.
PRL Levels in Rats.
As described previously (Millan et al., 1998a), PRL levels were determined in systemic plasma 30 min after the administration of S18327 or vehicle with the use of a radioimmunoassay and a highly selective antibody directed against rat PRL displaying less than 0.1% cross-reactivity to all other hormones (Amersham International, Buckinghamshire, England). Data were analyzed by ANOVA followed by Dunnett's test.
Catalepsy in Rats.
As described previously (Millan et al., 1998a), the left and right hindpaws of rats were placed over the ipsilateral forepaw, and the time over which this position was maintained was determined. Three separate measures were made, which were separated by 1-min intervals. The maximal possible duration was 30 s. S18327 or vehicle was administered 30 min before testing. The mean of the three determinations was calculated, and data were analyzed with ANOVA followed by Dunnett's test. The AD50 values (95% CLs) were calculated.
Methylphenidate-Induced Gnawing in Rats.
As previously described (Millan et al., 1998a), rats were administered methylphenidate (40.0 mg/kg i.p.) and placed in observation cages (33.5 × 23.5 × 19 cm). The number of gnawing periods was recorded over 10 min (one 10-s observation period/min), 30 min after methylphenidate administration. S18327 or vehicle was injected 30 min before methylphenidate. Data were analyzed with ANOVA followed by Dunnett's test, and the ID50 values (95% CLs) were calculated.
Binding Studies.
The affinities of S18327 at various sites were determined according to conventional procedures as previously described (Millan et al., 1998b) and summarized in Table1. Isotherms were analyzed by nonlinear regression using Prism (GraphPAD Software, San Diego, CA) to generate IC50 values. TheKi values were derived according to the Cheng-Prussof equation (Kenakin, 1997):Ki = IC50/(1 + L/Kd), where L is the concentration of radioligand, and Kd is the dissociation constant of the radioligand.
Actions of S18327 in diverse models of potential antipsychotic activity
Multiparametric Analysis.
In addition to S18327, the neuroleptic haloperidol, the atypical agent clozapine, and 11 additional antipsychotic agents were evaluated in several of the procedures that were predictive of antipsychotic properties. In addition, their affinities were determined at multiple monoaminergic receptors previously implicated in the distinctive antipsychotic profile of clozapine compared with haloperidol (accompanying paper;Brunello et al., 1995; Arnt and Skarsfeldt, 1998). This generated a substantial database (15 parameters, 14 antipsychotic agents, and 210 data points) permitting a comparison of respective antipsychotic profiles. In an independent analysis, the activity of antipsychotic agents was examined in several models predictive of extrapyramidal motor and endocrine side effects that characterize haloperidol. Affinities at histaminic (H) and muscarinic (M) receptors, of which the blockade by clozapine results in autonomic and cardiovascular side effects (Cunningham-Owens, 1996), were also integrated into this analysis of potential side effects. Multiparametric analyses of the database (hierarchical classification) were undertaken according toGordon (1987) and Ward (1963) with the Program SPAD 3 (Center International de Statistiques et d'Informatiques Appliquées, St. Mandé, France). The hierarchical classification generates a dendrogram based on the simultaneous analysis in a polydimensional space of all parameters. Thus, the positions of individual drugs in the dendrograms reflect their “global” profile across all parameters examined. Correspondingly, the dendrograms indicate overall interdrug homology; that is, the length of the line connecting two specific drugs is inversely proportional to their degree of homology. Pairs of drugs widely separated differ markedly, whereas those positioned close together are similar.
Drugs.
All drug doses are given in terms of the base. Drugs were dissolved in sterile water. If necessary, a few drops of lactic acid were added, and pH adjusted as close to neutrality as possible (>5.0). Drugs were injected in an injection volume of 1 ml/kg (rats s.c. or i.p.) or 10 ml/kg (mice, s.c. or p.o.; rats, p.o.).d-Amphetamine sulfate was obtained from Calaire Chimie (Calais, France). Cocaine HCl was obtained from Coopérative Pharmaceutique Française (Melun, France). Clozapine, dizocilpine maleate, and (±)-DOI HCl were obtained from Research Biochemicals Inc. (Natick, MA). Apomorphine HCl and PCP HCl were obtained from Sigma Chimie (St. Quentin-Fallavier, France). Methylphenidate HCl was obtained from Ciba-Geigy (Basel, Switzerland). S18327 base and WAY100,635 (N-{2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl}-N-(2-pyridinyl)cyclo-hexanecarboxamide) 3 HCl were synthetized by J.-L.P. (Institut de Recherches Servier).
Results
Activity in Models Predictive of Antipsychotic Activity: Paradigms Implicating Dopaminergic Mechanisms.
As shown in Fig.1, S18327 dose-dependently blocked the induction of climbing behavior in mice by the dopaminergic agonist apomorphine (Fig. 1A), with an ID50 value of 0.2 mg/kg s.c. S18327 likewise exhibited a marked and dose-dependent reduction in CAR (Fig. 1B), with an ID50 value of 0.8 mg/kg s.c. (Table 1). Similarly, the locomotor behavior in rats elicited by the psychostimulants and catecholamine releasers amphetamine (Fig. 1C) and cocaine (Fig. 1D) was dose-dependently antagonized by S18327 with ID50 values of 1.3 and 0.7 mg/kg s.c., respectively.

Activity of S18327 in models of potential antipsychotic activity involving dopaminergic mechanisms. A, inhibition of apomorphine (0.75 mg/kg)-induced climbing in mice. B, inhibition of CARs in rats. C, inhibition of amphetamine (2.5 mg/kg i.p.)-induced locomotion in rats. D, inhibition of cocaine (20.0 mg/kg i.p.)-induced locomotion in rats. Data are mean ± S.E. (n = 5–8 per value). On ANOVA: A, F4,31 = 14.2, P < .001; C,F4,40 = 3.9, P < .01; and D, F3,28 = 7.9,P < .001. Asterisks indicate significance of differences from vehicle values in Dunnett's test. B, *P < .05, significance of differences from vehicle values in the Wilcoxon signed rank test.
Activity in Models Predictive of Antipsychotic Activity: Paradigms Implicating Serotonergic Mechanisms.
The 5-HT2A agonist and hallucinogen DOI evoked HTWs in rats, and this behavior was potently blocked by S18327 (Fig.2A), with an ID50value of 0.1 mg/kg s.c. (Table 1). In analogy, the locomotor behavior in rats elicited by PCP was antagonized by S18327 over a low dose range (Fig. 2B), yielding an ID50 value of 0.1 mg/kg s.c.

Activity of S18327 in models of potential antipsychotic activity involving serotonergic (5-HT2Areceptor-mediated) mechanisms. A, inhibition of HTWs elicited by the hallucinogen and 5-HT2A agonist DOI (2.5 mg/kg i.p.) in rats. B, inhibition of PCP (20.0 mg/kg s.c.)-induced locomotion in rats. Data are mean ± S.E. (n = 5–8 per value). On ANOVA: A, F3,17 = 4.6,P < .02; and B,F5,70 = 4.2, P < .05. *P < .05, significance of differences from vehicle values in Dunnett's test.
Activity in Models Predictive of Antipsychotic Activity: Blockade of Actions of NMDA Antagonist Dizocilpine.
The open channel blocker at NMDA receptors, dizocilpine, elicited a pronounced locomotor response in rats that was dose-dependently prevented by S18327 (Fig.3A), with an ID50value of 0.9 mg/kg s.c. (Table 1). Dizolcipine also elicited, in an independent experiment, STFs in rats, and this response was similarly blocked by S18327 in a dose-dependent fashion (Fig. 3B), yielding an ID50 value of 1.7 mg/kg s.c.

Activity of S18327 in models of potential antipsychotic activity reflecting blockade of responses provoked by the NMDA antagonist, dizocilpine. A, inhibition of dizocilpine (0.16 mg/kg s.c.)-induced locomotion in rats. B, inhibition of dizolcipine (0.08 mg/kg s.c.)-induced STF in rats. Data are mean ± S.E. (n = 5–8 per value). On ANOVA: A,F4,35 = 9.2, P < .01; and B, F4,20 = 6.1,P < .05. *P < .05, significance of differences from vehicle values in Dunnett's test.
Multiparametric Analysis of Antipsychotic Actions of S18327 in Comparison with Those of Other Antipsychotic Agents.
In most of the above paradigms, we evaluated the influence of the neuroleptic haloperidol, the atypical antipsychotic clozapine, and a diversity of antipsychotic agents (Table 2). Their affinities were also determined at several dopaminergic, adrenergic, and serotonergic receptor types that have been strongly implicated in 1) the expression of clinical antipsychotic properties, 2) the distinctive profile of clozapine, and 3) the above-described models of antipsychotic activity (Brunello et al., 1995; seeDiscussion) (Table 2). It may be seen in Fig.4 that haloperidol and clozapine are well separated from each other. Raclopride was the only antipsychotic that migrated adjacent to haloperidol. A distinct cluster of drugs was composed of ORG5222, ocaperidone, tiospirone, and risperidone, whereas another group was composed of MDL100,907, quetiapine, and amperozide. Sertindole, ziprasidone, and olanzapine were placed in a cluster containing clozapine, to which S18327 was positioned the closest.

Dendrogrammic representation generated by multiparametric analysis (hierarchical classification) of overall homology between S18327 and other drugs regarding their monoaminergic receptor profiles and antipsychotic properties. See Table 2 for database.
Activity in Models Predictive of Extrapyramidal Activity.
Inasmuch as haloperidol and other neuroleptics elicit extrapyramidal motor and endocrine side effects, the activity of S18327 was determined in models predictive of such actions (Table3. Although S18327 dose-dependently elicited catalepsy, this response was seen over a dose range markedly higher than those active in the above-discussed models of potential antipsychotic activity (Fig. 5A). Compared with median potency (ID50) in models of antipsychotic activity (Table 1), there was a 30-fold separation to the AD50 for induction of catalepsy. In line with this weak potency in eliciting catalepsy, S18327 did not block induction of stereotyped gnawing by the catecholamine releaser methylphenidate (Fig. 5B), a response that likewise involves striatal populations of D2 receptors. Moreover, S18327 only weakly increased circulating levels of PRL (Fig. 5C), a response reflecting blockade of tonically active, hypophyseal populations of D2 receptors.
Actions of S18327 in models predictive of extrapyramidal side effects

Activity of S18327 in models predictive of extrapyramidal motor and endocrine side effects. A, induction of catalepsy. B, inhibition of sterotyped gnawing elicited by the DA releaser methylphenidate (40.0 mg/kg i.p.). C, elevation in circulating levels of PRL. Data are mean ± S.E. (n = 5–8 per value). On ANOVA: A, F4,26 = 4.4,P < .01; B, F3,16= 1.0, P > .05; and C,F4,29 = 9.9, P < .05. *P < .05, significance of differences from vehicle values in Dunnett's test.
Interaction of S18327 with Histaminic and Muscarinic Receptors.
Inasmuch as the potent blockade by clozapine of histaminic and muscarinic receptors contributes to its undesirable, autonomic/cardiovascular impact, the interaction of S18327 was evaluated at these sites. S18327 showed modest affinity for native H1 receptors compared with clozapine (compare Table 4 with Table5). Furthermore, S18327 showed low affinity for H2 sites. S18327 also manifested low affinity for cloned human (h)M1, hM2, hM3, and hM4 receptors.
Interaction of S18327 with histaminic and muscarinic receptors
Multiparametric Analysis of Side Effects of S18327 in Comparison with Those of Other Antipsychotic Agents.
In each of the above-discussed models of potential, extrapyramidal motor and endocrine side effects and binding at H1 and hM1receptors, the activity of reference antipsychotics was examined (Table5). This permitted the construction through multiparametric analysis of the dendogram illustrated in Fig. 6. It may be seen that haloperidol and clozapine were clearly differentiated. Raclopride was positioned adjacent to haloperidol, consistent with their preferential D2 antagonist activity and marked extrapyramidal impact compared with their lack of affinity at histaminic/muscarinic receptors. On the other hand, olanzapine migrated close to clozapine, in line with its high affinity at histaminic and muscarinic receptors. A further cluster consisted of risperidone, ocaperidone, ORG5222, and tiospirone, which showed marked activity in extrapyramidal models and high affinity at histaminic receptors. Finally, based on their limited actions in models of potential extrapyramidal activity and their comparatively modest affinity at histaminic and muscarinic sites, S18327, quetiapine, sertindole, ziprasidone, MDL100,907, and amperozide were differentiated from both haloperidol and clozapine.

Dendrogrammic representation generated by multiparametric analysis (hierarchical classification) of overall homology between S18327 and other drugs regarding their side effect profiles. See Table 5 for database.
Generalization of S18327 to a Clozapine DS.
In rats trained to recognize a DS generated by clozapine (5.0 mg/kg i.p.), S18327 showed dose-dependent and significant generalization (Fig.7A and Table6). Over the dose range at which it generalized to clozapine, S18327 did not significantly modify response rates (Fig. 7B).

Generalization of S18327 to a DS mediated by clozapine (5.0 mg/kg i.p.). A, generalization to clozapine (lever selection); data show percentage of rats selecting drug lever (n = 4–7 per value). *P < .05, significance of difference from control training values (0% drug lever selection; Fisher's exact probability test). B, influence on response rates; data are mean ± S.E. of percentage of values obtained during the previous vehicle training session (n = 4–7 per value). Response rates were not significantly modified at any dose (P > .05 in a paired Wilcoxon test) compared with the control, training session.
Generalization of S18327 to a clozapine discriminative stimulus and actions of S18327 in models of potential anxiolytic activity
Induction of LI by S18327.
SRs in vehicle-treated rats were higher, but not significantly (P > .05), in the preexposed than in the nonpreexposed groups, which is consistent with a tendency toward the induction of LI (Fig.8). This difference was amplified and became statistically significant (P < .05) in the presence of S18327 inasmuch as SRs of the nonpreexposed, but not preexposed, animals were increased. That is, according to our criteria (see Millan et al., 1998b), S18327 induced LI. The difference of preexposed values in S18327 versus vehicle groups just failed to reach significance (P > .05). This latter observation is in line with our previous studies of clozapine (0.16 mg/kg s.c.) in this LI model (Millan et al., 1998b). Indeed, clozapine induces an LI response of a similar magnitude (preexposed values, 2.4-fold higher than nonpreexposed values) as that seen herein with S18327 (factor, 3.6-fold).

Induction by S18327 of LI in rats. Values are mean ± S.E. (n = 7–10 per value) and represent SRs for rats either preexposed to 10 tones or not (NPE;F1,32 = 19.9, P < .001). *P < .05, significant difference between values of the NPE/S18327 group and the 10 tones/S18327 group (Newman-Keuls test).
Anxiolytic Properties of S18327.
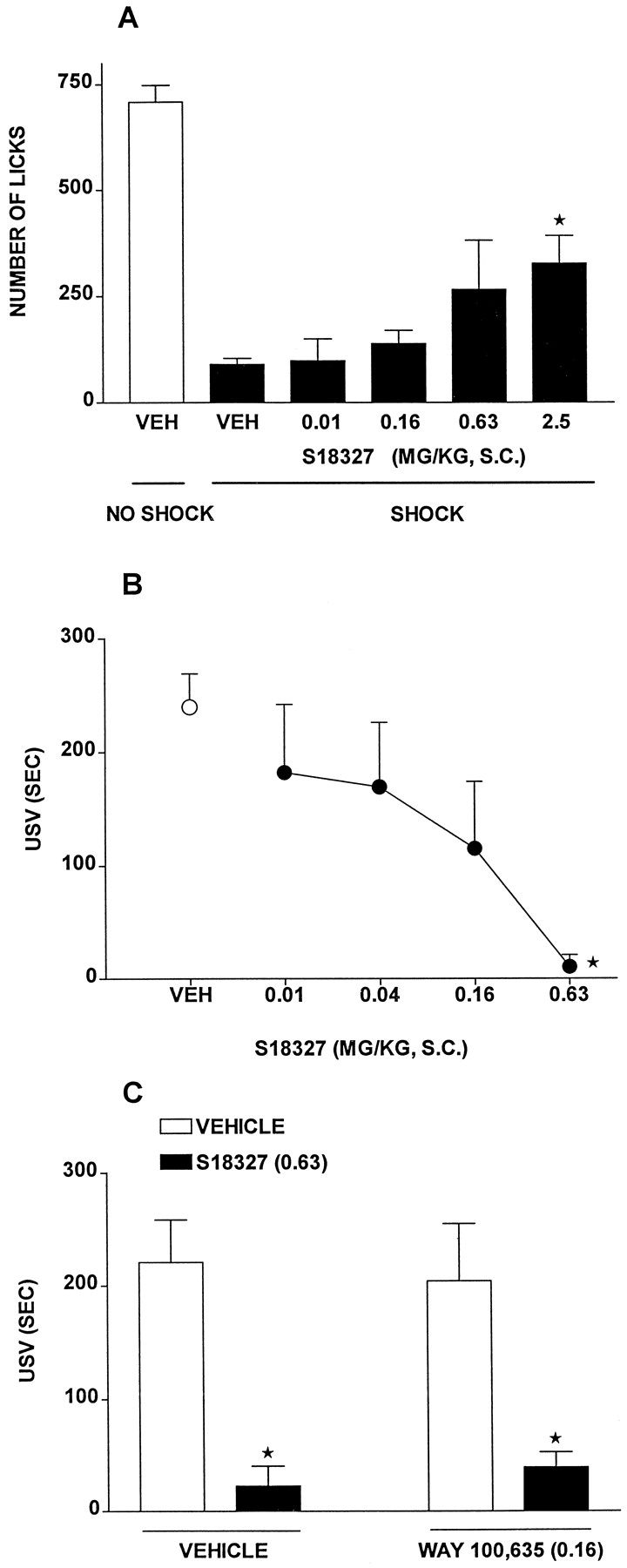
In the Vogel test in rats, S18327 elicited a dose-dependent increase in punished responses (Fig.9A and Table 6). Similarly, S18327 dose-dependently and markedly decreased the duration of fear-induced USVs in rats (Fig. 9B). In the latter paradigm, the anxiolytic action of S18327 was resistant to the selective 5-HT1Aantagonist WAY100,635 (0.16 mg/kg s.c.), which itself did not significantly modify USV (Fig. 9C). S18327 dose-dependently reduced aggressive behavior in isolated mice, as concerns both the number of attacks made by the dominant on the submissive mouse and the total duration of attacks (Fig. 10).

Activity of S18327 in the Vogel test and the USV procedure. A, actions in the Vogel procedure. B, actions in the USV paradigm. C, lack of influence of the selective 5-HT1Areceptor antagonist WAY100,635 (0.16 mg/kg s.c.) on the action of S18327 (0.63 mg/kg s.c.) in the USV test. Data are mean ± S.E. (n = 5–14 per value). On ANOVA: A,F4,32 = 3.2, P < .05; and B, F4,29 = 4.2, P < .01. Asterisks indicate significance ofdifferences from vehicle values in Dunnett's test. C, two-way ANOVA as follows: influence of S18327:F1,23 = 35.0, P < .05; influence of WAY100,635: F1,23 = 0.1, P > .05; and interaction:F1,23 = 0.3, P > .05. *P < .05, significance of differences between S18327/vehicle and vehicle/vehicle values and between WAY100,635/S18327 and WAY100,635/vehicle values (Newman-Keuls test).

Influence of S18327 on aggressive behavior in isolated mice. A, influence on duration of aggressive encounters. B, influence on number of aggressive encounters. Data are mean ± S.E. (n = 5–8 per value). On ANOVA: A,F4,32 = 5.4, P < .005; and B, F4,32 = 11.6,P < .001. *P < .05, significance of differences from vehicle values in Dunnett's test.
Influence of S18327 on Spontaneous Locomotor Behavior.
S18327 dose-dependently reduced spontaneous locomotor behavior in rats with an ID50 value (95% CLs) of 3.8 (1.3–11.3) mg/kg s.c.
Activity of S18327 on Oral Administration.
As shown in Table7, S18327 displayed marked activity on oral administration in models of potential antipsychotic and anxiolytic activity.
Actions of S18327 upon oral administration
Discussion
Models Predictive of Control of Positive Symptoms.
In accordance with its antagonist properties at D2receptors, S18327 displayed marked activity in several classic dopaminergic models predictive of the control of positive symptoms of schizophrenia (Brunello et al., 1995; Wirsching et al., 1995; Millan et al., 1998a,b). Although S18327 also blocks hD3and hD4 receptors, selective D3 and D4 antagonists are inactive in these models, and the contribution of D3 and D4 receptor blockade to antiproductive properties remains under discussion (Roth et al., 1995; Levant, 1997; Millan et al., 1998a; Wilson et al., 1998). Similarly, it remains to be demonstrated that selective D1 receptor antagonists exert antipsychotic properties clinically. Nevertheless, inasmuch as they are potently active in the present paradigms (Gerlach and Hansen, 1992; Brunello et al., 1995; Josselin et al., 1997; Darracq et al., 1998), blockade of D1 receptors may also participate in these actions of S18327. Furthermore, inasmuch as blockade of limbic and/or frontocortical α1-ARs attenuates the locomotor actions of psychostimulants (Berthold et al., 1992; Mathé et al., 1996; Darracq et al., 1998), the potent α1-AR antagonist actions of S18327 may also be involved. On the other hand, under the conditions used here, antagonists at diverse serotonergic receptors are not effective (Maurel-Rémy et al., 1995; M. Brocco, unpublished observations), although the potential ability of selective 5-HT2A receptor antagonists to control positive symptoms remains under clinical investigation (Lieberman et al., 1998). As proposed for clozapine (Prinssen et al., 1994), the combined blockade by S18327 of multiple receptor types may account for its profile of potential antipsychotic activity in these (and other) protocols. Finally, in light of the inhibitory influence of S18327 on the locomotor response to cocaine and amphetamine, it would be of interest to undertake additional studies of its potential modulation of their rewarding properties with the use of other functional paradigms.
Inhibition of Actions Elicited by NMDA Receptor Antagonist Dizocilpine.
Mechanisms underlying the locomotor response to dizocilpine and the complex pattern of functional interrelationships among glutamatergic and monoaminergic pathways remain under intense investigation (Meltzer et al., 1997; Morari et al., 1998). In fact, a role for D1, D2, α1-AR, and/or 5-HT2Areceptors has been variously evoked in the mediation of dizocilpine locomotion and its inhibition by clozapine (Mathé et al., 1996;Martin et al., 1997; Ninan and Kulkarni, 1998). Thus, the respective importance of specific receptor types in the blockade of dizocilpine-induced locomotor activity by S18327 remains to be elucidated. Interestingly, the STF response to dizocilpine, which reflects the blockade of NMDA receptors in nucleus accumbens, is dependent on D1 receptors for its expression. Furthermore, it is resistant to 5-HT2A and α1-AR antagonists, yet abolished by α2-AR antagonists (Brocco et al., 1999). Thus, the antagonist actions of S18327 at D1 receptors and α2-AR likely account for its blockade of dizocilpine-induced STF. Regardless of underlying mechanisms, the inhibition by S18327 of these actions of dizocilpine is of particular interest because a deficit in glutamatergic transmission (and activity at NMDA receptors) is implicated in the pathogenesis of schizophrenia (Sokoloff, 1998; Faustman et al., 1999).
Importance of 5-HT2A Antagonist Properties: Inhibition of Actions of PCP and DOI.
Like dizocilpine, PCP is an open channel blocker at NMDA receptors. However, neuronal substrates underlying their functional actions differ, and serotonergic mechanisms play a more prominent role in the motor actions of PCP (Hiramatsu et al., 1989; Ögren and Goldstein, 1994; Martin et al., 1997;Maurel-Rémy et al., 1998). Indeed, under the present conditions, PCP- versus amphetamine-induced locomotion is abolished by selective 5-HT2A versus D2 receptor antagonists, a distinction that underlies potent blockade by clozapine of PCP, but not amphetamine, locomotion (Maurel-Rémy et al., 1995, 1998). S18327 shares the preference of clozapine for 5-HT2A versus D2 receptors (Millan et al., 1998b) and, similarly, more potently blocked the locomotor response to PCP than to amphetamine. This observation is of considerable interest inasmuch as in contrast to amphetamine, PCP reproduces both positive and negative symptoms of schizophrenia (Steinpreis, 1996). In analogy to PCP locomotion, a key role of 5-HT2A receptors in HTWs induced by the hallucinogen and 5-HT2 agonist DOI has been demonstrated (Schreiber et al., 1995; Willins and Meltzer, 1997), and S18327, like clozapine, also potently blocked this response (Millan et al., 1998b). These observations emphasize the importance of 5-HT2A antagonist properties to the potential antipsychotic profile of S18327.
Generalization to a Clozapine DS.
Clozapine elicits a “compound” DS that cannot be attributed to any specific receptor type (Goudie and Taylor, 1998; Millan et al., 1999b). The majority of drugs (e.g., olanzapine) that generalize to clozapine are chemically related (Goudie and Taylor, 1998; Millan et al., 1999b). It is thus of particular interest that S18327 generalized to a clozapine DS. This observation underlies similarities in their receptorial and functional profiles, despite their chemical distinctiveness, an aspect accentuated by the multiparametric analysis discussed below.
Influence on Cognitive-Attentional Function.
Psychotic patients poorly filter irrelevant sensory information, a deficit modeled by the paradigm of LI (Weiner and Feldon, 1997; Friedman et al., 1999). Although haloperidol is only variably active, clozapine is consistently effective (Weiner and Feldon, 1997; Millan et al., 1998b), and here, S18327 significantly enhanced LI, suggesting that it may improve cognitive-attentional performance. Several receptorial mechanisms may potentially be involved. First, the antagonist properties of S18327 at 5-HT2A receptors may be involved; thus, activation and blockade of 5-HT2Areceptors disrupts and elicits, respectively, LI (Moser and Moran, 1994). Second, regarding blockade by S18327 of D4receptors, these sites are concentrated in the hippocampus, frontal cortex, and other structures implicated in LI and are involved in the modulation of cognitive-attentional processes (Lahoste et al., 1996; Tallman, 1997;Weiner and Feldon, 1997). Furthermore, selective D4 antagonists display procognitive effects and induce LI (Tallman, 1997; Dekeyne et al., 1998). Third, the antagonist actions of S18327 at α1-AR receptors may be involved; thus, stimulation and blockade of α1-ARs disrupted and improved, respectively, performance in a prepulse inhibition model of disrupted cognitive-attentional function (Bakshi and Geyer, 1997; Carasso et al., 1998). The population of α1-AR involved is possibly implicated in thalamic gating of sensory input to the cortex (McCormick and Pape, 1995). Nevertheless, adrenergic mechanisms in the cortex itself may also be implicated (Arnsten et al., 1999; Friedman et al., 1999). Finally, α2-AR antagonist actions of S18327 may be relevant inasmuch as α2-ARs are involved in control of cognitive-attentional function (Nutt, 1994;Camacho et al., 1996; Smith et al., 1998; Friedman et al., 1999). Furthermore, mesocortical dopaminergic transmission facilitates processes of LI (Weiner and Feldon, 1997), and S18327 increases DA release in frontal cortex by the blockade of α2-AR heteroceptors on dopaminergic terminals (see accompanying paper).
Potential Anxiolytic Properties.
In analogy to clozapine and certain other antipsychotic agents (Wiley et al., 1993; Millan et al., 1999a), S18327 displayed anxiolytic properties in several paradigms. Its activity in the USV test was expressed at doses well below those eliciting motor disruption, whereas increases in response rates in the Vogel test clearly cannot reflect motor-suppressant properties. Furthermore, over dose ranges active in the Vogel and USV models, S18327 did not show significant effects in classic algesiometric models, such as the rat tail-flick test, excluding the potential involvement of antinociceptive properties (M.J.M., M.B., and A.D., unpublished observations). Thus, the anxiolytic actions of S18327 are likely specific. Although the underlying mechanisms are unclear, S18327 is a potent antagonist of 5-HT2A receptors, the blockade of which is associated with anxiolytic properties (Stutzmann et al., 1991; Mora et al., 1997). Conceivably, the α1-AR antagonist actions of S18327 are of pertinence because this mechanism underlies its inhibition of serotonergic transmission (see accompanying paper), a hyperactivity of which contributes to anxious states (Eison and Eison, 1994). However, anxiolytic actions of selective α1-AR antagonists have only rarely been documented (López-Rubalcava and Fernández-Guasti, 1994). Furthermore, the selective α1-AR antagonist prazosin, even at a high dose (0.63 mg/kg s.c.), was inactive in the Vogel and USV procedures (A.D. and M.B., unpublished observations).
Potential Extrapyramidal and Other Side Effects.
Only high doses of S18327 elicited catalepsy, and it failed to block methylphenidate gnawing, responses reflecting blockade of striatal D2 receptors and predictive of extrapyramidal side effects (Cunningham-Owens, 1996). These observations, together with its weak induction of striatal DA synthesis (see accompanying paper), suggest that S18327 possesses a substantial therapeutic window regarding antipsychotic versus motor side effects. Several features of the receptorial profile of S18327 may be involved. First, S18327 expresses α2-AR antagonist properties, and a role of α2-AR blockade has been evoked in the limited, extrapyramidal impact of clozapine (Nutt, 1994; Herberg et al., 1995; Montgomery et al., 1997; Kalkman et al., 1998). Second, balanced D1 and D2 receptor blockade, as displayed by S18327 (and clozapine), may limit extrapyramidal motor symptoms (Gerlach and Hansen, 1992). Third, S18327 mimics the pronounced 5-HT2A versus D2 receptor preference of clozapine, a characteristic associated with improved separation of antipsychotic actions versus extrapyramidal side effects (Roth et al., 1995). Furthermore, although S18327 did significantly elevate PRL levels, the magnitude of this effect is substantially less than that of neuroleptics evaluated under similar conditions: notably, haloperidol elicits a maximal increase in circulating levels of PRL of approximately 211 ng/ml (0.63 mg/kg s.c.; Millan et al., 1998b) compared with S18327 (23 ng/ml, 40.0 mg/kg s.c.). Thus, the endocrine (PRL) impact of S18327 should be relatively modest, providing an interesting analogy to clozapine (Cunningham-Owens, 1996; Millan et al., 1998b). The efficacy (agonist or antagonist) of S18327 at multiple muscarinic receptors remains to be determined. Nevertheless, compared with clozapine (for a discussion, see Michal et al., 1999; Olianas et al., 1997; and Zorn et al., 1994), the considerably lower affinity of S18327 (Table 5) suggests that cardiac, gastrointestinal, and other types of anticholinergic side effects should be relatively weak (Cunningham-Owens, 1996). The affinity of S18327 for histamine1 sites was also less marked than that for clozapine (Table 5), suggesting that side effects involving histaminic receptor blockade, such as somnolence and weight gain, should be less pronounced (Cunningham-Owens, 1996). However, additional comparative in vitro and in vivo functional studies are required to corroborate this assertion.
Multiparametric Analysis: Multireceptorial, “Clozapine-Like” Antipsychotic Profile of S18327.
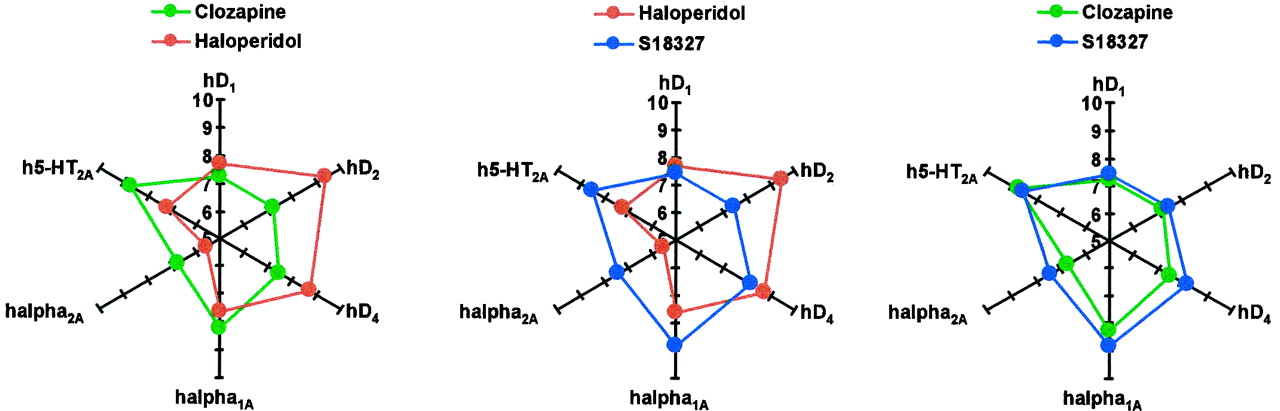
The above discussion suggests that S18327 may moderate both positive and negative-cognitive symptoms of schizophrenia and that it possesses potential anxiolytic properties. Furthermore, it has a low extrapyramidal potential. In this respect, the functional profile of S18327 resembles that of clozapine (Millan et al., 1998b), and its multireceptorial profile at monoaminergic receptors is very similar (Table 2). This similarity to clozapine was underpinned via multiparametric analysis. Although detailed comments on all agents (Brunello et al., 1995; Meltzer, 1995; Arnt and Skarsfeldt, 1998) incorporated into this extensive database are beyond the scope of the present paper, two major points may be emphasized. First, regarding functional properties and monoaminergic receptorial profiles, S18327 was clearly separated from haloperidol and positioned adjacent to clozapine. Furthermore, the radar plots of Fig.11 exemplify the similar receptorial profile of S18327 and clozapine versus haloperidol at those specific sites implicated, as discussed above, in the functional actions of S18327. Second, regarding potential side effects, S18327 can be discriminated from both haloperidol (low extrapyramidal impact) and clozapine (modest affinity for histaminic/muscarinic sites; Table 5;Cunningham-Owens, 1996).
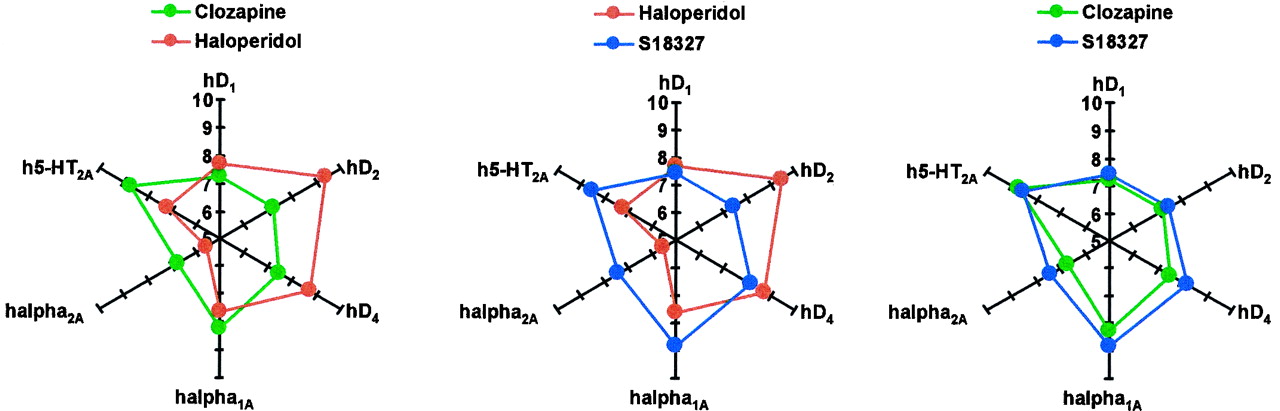
“Radar” representation of the comparative patterns of interaction (affinities) of S18327, clozapine, and haloperidol at cloned hD1, hD2, hD4, h5-HT2A, hα1A, and hα2A receptors, which, as indicated in the text, are implicated in the functional actions of S18327. Affinities are indicated as pKi values.
Conclusions.
The present data reinforce receptorial, neurochemical, and electrophysiological analyses (see accompanying paper) in suggesting that the multireceptorial monoaminergic profile of S18327 is associated with antipsychotic properties in the relative absence of extrapyramidal side effects. This “atypical,” multireceptorial, and functional profile of S18327 strongly resembles that of clozapine, an interpretation reinforced by the innovative approach of multiparametric analysis. Furthermore, modest histaminic and muscarinic properties of S18327, and its contrasting chemical structure, differentiate S18327 from clozapine as far as its potential side effect profile. This, S18327 possesses a distinctive and promising profile for the improved treatment of schizophrenia, an indication for which it is currently under clinical evaluation.
Acknowledgments
We thank C. Langaney and K. Dutartre for secretarial assistance and B. Denorme, S. Girardon, H. Gressier, S. Monneyron, and S. Veiga for technical assistance.
Footnotes
-
Send reprint requests to: Dr. Mark J. Millan, Institut de Recherches Servier, Centre de Recherches de Croissy, Psychopharmacology Department, 125 Chemin de Ronde, 78290-Croissy-sur-Seine, France.
- Abbreviations:
- AR
- adrenergic receptor
- 5-HT
- 5-hydroxytryptamine (serotonin)
- CAR
- conditioned avoidance response
- CLs
- confidence limits
- DOI
- 1-[2,5-dimethoxy-4-iodophenyl]-2-aminopropane
- DA
- dopamine
- DS
- discriminative stimulus
- HTW
- head-twitch
- LI
- latent inhibition
- NMDA
- N-methyl-d-aspartate
- PCP
- phencyclidine
- PRL
- prolactin
- SR
- suppression ratio
- STF
- spontaneous tail-flick
- USV
- ultrasonic vocalization
- Received June 7, 1999.
- Accepted September 22, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}