


Abstract
S-16924 is a potential antipsychotic that displays agonist and antagonist properties at serotonin (5-HT)1A and 5-HT2A/2C receptors, respectively. In a pigeon conflict procedure, the benzodiazepine clorazepate (CLZ) increased punished responses, an action mimicked by S-16924, whereas the atypical antipsychotic clozapine and the neuroleptic haloperidol were inactive. Similarly, in a Vogel conflict paradigm in rats, CLZ increased punished responses, an action shared by S-16924 but not by clozapine or haloperidol. This action of S-16924 was abolished by the 5-HT1A antagonist WAY-100,635. Ultrasonic vocalizations in rats were inhibited by CLZ, S-16924, clozapine, and haloperidol. However, although WAY-100,635 abolished the action of S-16924, it did not affect clozapine and haloperidol. In a rat elevated plus-maze, CLZ, but not S-16924, clozapine, and haloperidol, increased open-arm entries. Like CLZ, S-16924 increased social interaction in rats, whereas clozapine and haloperidol were inactive. WAY-100,635 abolished this action of S-16924. CLZ, S-16924, clozapine, and haloperidol decreased aggressive interactions in isolated mice, but this effect of S-16924 was not blocked by WAY-100,635. All drugs inhibited motor behavior, but the separation to anxiolytic doses was more pronounced for S-16924 than for CLZ. Finally, in freely moving rats, CLZ and S-16924, but not clozapine and haloperidol, decreased dialysis levels of 5-HT in the nucleus accumbens: this action of S-16924 was blocked by WAY-100,165. In conclusion, in contrast to haloperidol and clozapine, S-16924 possessed a broad-based profile of anxiolytic activity at doses lower than those provoking motor disruption. Its principal mechanism of action was activation of 5-HT1A (auto)receptors.
A hyperactivity of serotonergic networks was been implicated in the induction of anxious states (Eison and Eison, 1994; Coplan et al., 1995). Considerable interest has been focused on the role of inhibitory serotonin (5-HT)1A autoreceptors localized on raphe cell bodies, the engagement of which is associated with anxiolytic actions in a diversity of experimental paradigms (Cervo and Samanin, 1995b; Coplan et al., 1995; File et al., 1996; King et al., 1997). Furthermore, the activation of 5-HT1Aautoreceptors may be involved in the clinical control of anxious states by buspirone and related drugs (Gammans et al., 1992; Coplan et al., 1995). There remains, however, uncertainty as to which of the constellation of postsynaptic 5-HT receptors is principally responsible for the anxiogenic actions of 5-HT. Although 5-HT3 receptors have long attracted interest, the anxiolytic actions of 5-HT3 receptor antagonists are test dependent, modest, and variable (Costall et al., 1989; Cervo and Samanin, 1995a), and a more convincing body of evidence suggests that activation and blockade of 5-HT2 receptors are associated with anxiogenic and anxiolytic effects, respectively. In early studies, it proved difficult to distinguish the roles of 5-HT2A, 5-HT2B, and 5-HT2C receptors, although this has become possible with the availability of selective ligands differentiating these sites. Recent studies have thus indicated that 5-HT2C receptors play a more predominant role in the induction of anxiety than do their 5-HT2Acounterparts (Cervo and Samanin, 1995a; Kennett et al., 1996b; Griebel et al., 1997). Indeed, in a variety of experimental models, there is evidence that the activation and blockade of 5-HT2C receptors are associated with anxiogenic and anxiolytic actions, respectively (Kennett et al., 1996b; Griebel et al., 1997). In addition, activation of 5-HT2Creceptors similarly may provoke, or exacerbate, anxious states in humans (Kahn and Wetzler, 1991; Millan et al., 1992), although the potential anxiolytic actions of selective 5-HT2Cantagonists still require therapeutic evaluation. The role of the closely related 5-HT2B receptor remains unclear. Nevertheless, although their activation was suggested to act anxiolytically, their blockade does not elicit anxiety, and cerebral levels of 5-HT2B receptors are low (Kennett et al., 1996a). In light of these observations, it has been suggested that the simultaneous activation and blockade of 5-HT1A and 5-HT2C and of 5-HT2A receptors, respectively, may result in synergistic anxiolytic actions in the relative absence of disruptive side effects (Millan et al., 1992; Schreiber and De Vry, 1993).
Anxious symptoms are frequently encountered in many psychiatric disorders, such as drug abuse and schizophrenia. Indeed, anxiety can markedly exacerbate the intensity of psychotic symptoms and reduce drug compliance (Wirsching et al., 1995). This suggests that auxillary, anxiolytic properties would be useful for an antipsychotic agent. In this regard, the atypical antipsychotic clozapine is of interest because it possesses mild (agonist) and (potent) antagonist properties at 5-HT1A and 5-HT2Creceptors, respectively, in contrast to the neuroleptic haloperidol, which is devoid of activity at these sites (Brunello et al., 1995; Roth and Meltzer, 1995; Newman-Tancredi et al., 1996). Indeed, in contrast to haloperidol, clozapine possesses anxiolytic properties in certain experimental models in rodents and other species (Spealman et al., 1983; Mansbach et al., 1988; Wiley et al., 1993) (seeDiscussion).
We recently identified a novel benzodioxopyrrolidine and potential antipsychotic, S-16924 [(R)-2-{1-[2-(2,3-dihydrobenzo[1,4]dioxin-5-yloxy)-ethyl]-pyrrolidin-3yl}-1-(4-fluorophenyl)-ethanone], which shares the potent 5-HT2C(5-HT2A) antagonist properties of clozapine and behaves as a potent agonist at 5-HT1Aautoreceptors (Millan et al., 1998a,b). The purpose of the studies reported herein was to evaluate the potential anxiolytic properties of S-16924 in comparison with those of clozapine, haloperidol, and the benzodiazepine clorazepate (CLZ). To this end, we used diverse experimental paradigms based on both conditioned responses and natural, spontaneous behaviors. Furthermore, in view of the potential importance of a reduction in serotonergic transmission via the activation of 5-HT1A autoreceptors, we evaluated the influence of drugs on dialysate levels of 5-HT in freely moving rats. Finally, to directly evaluate the potential role of 5-HT1A(auto)receptors, we examined the influence of the selective 5-HT1A receptor antagonist WAY-100,635 [N-{2-[4-(2-me thoxyphenyl)-1-piperazinyl]ethyl}-N-(2-pyridinyl)cyclo-hexanecarboxamide 3HCl] on the actions of S-16924.
Materials and Methods
Animals.
The majority of studies were carried out with male Wistar rats (220–240 g b.wt.; Iffa-Credo, L’Arbresle, France). Male Sprague-Dawley rats (240–260 g) were used for the social interaction (SI) test and CD1 (ICR) BR mice (22–25 g) were used for the aggressive behavior and Rotarod models (Charles River, Saint-Aubin-les-Elbeuf, France). Animals were housed in sawdust-lined, standard Macrolon cages with, unless specified, free access to chow and water. The pigeon conflict test was performed with white Carneaux pigeons (500–600 g) of either sex (Grozek, Lewarde, France) individually housed in cages placed in a laminar flow unit. Laboratory temperature was 21 ± 1.0°C, and humidity was 60 ± 5%. There was a 12/12-h light/dark cycle with lights on at 7:30 AM. The light part of the cycle was low (3 lux) for the SI test.
Pigeon Conflict Test.
As described previously (Millan et al., 1997), food-restricted pigeons were trained to peck an illuminated (green or red) key for food in operant conditioning chambers (Coulbourn Instruments, Lehigh Valley, PA). Every 30th response during illumination of the green key produced access to food (unpunished component), whereas every 30th response during illumination of the red key produced both food and electric shock (punished component). The shock was delivered via an electrode (diameter, 0.5 mm) previously implanted with the animal under anesthesia with methoxyflurane (1.5–2.0%) in the region of each pubic bone, as described in detail by Arzin (1959). A 1-min time-out interval (no key light, no food, and no shock) separated each 3-min illumination period. Sessions were terminated after five cycles of alternating components and lasted 40 min in total. Drugs or saline were injected i.m. (1 ml/kg) 5 min before the test session. Responses during unpunished and punished components of test (drug) sessions were expressed as a percentage of control responses during the previous saline session. Data were analyzed using a paired Wilcoxon test (two-tailed for unpunished and one-tailed for punished responses).
Vogel Test in Rats.
The test was conducted in a polycarbonate cage (32 × 25 × 30 cm) with a perforated stainless grid floor, enclosed in a ventilated sound-attenuating chamber. The spout of a drinking bottle filled with water protruded into the cage at a height of 6 cm above the grid floor. Grid floor and spout were connected by two electrical wires to an Anxiometer (Columbus Instruments, Columbus, OH) that was used to record licks and deliver electrical shocks. During the 3 days preceding testing, rats were housed in groups of four and were restricted to 1-h-per-day access to tap water (from 9:00 to 10:00 AM). On day 4, they were isolated in cages with a grid floor just after water delivery. Testing took place on day 5. Each rat was administered with drug or vehicle and, 3 min later, placed in the test cage. The session was initiated after the animal had made 20 licks and received a first shock (a single, 0.5-s constant current pulse of 0.3-mA intensity) through the spout. Thereafter, a shock was delivered to the animal every 20th lick during a period of 3 min. Animals that did not initiate the session within 5 min were removed. Data analyzed were the number of licks emitted during the 3-min session. Some control (vehicle) animals did not receive shocks (NS) during the session and were used to evaluate free drinking behavior. Dose effects were analyzed using one-way ANOVA followed by Dunnett’s test (NS animals were not included in the analysis). The percentage of drug effect was computed as [(drug − vehicle)/(vehicle NS − vehicle)]. In antagonism studies, WAY-100,635 or vehicle was administered 60 min before testing, and data were analyzed by a two-way ANOVA, followed by Newman-Keuls test.
Fear-Induced Ultrasonic Vocalizations in Rats.
As previously described (Millan et al., 1997), the experimental procedure consisted of three different phases, separated by 24 h. On day 1 (training), rats were individually placed in the experimental chambers (Coulbourn Instruments) and received six randomly distributed footshocks (0.8 mA, 8 s) over a 7-min period. On day 2 (selection), each rat was placed in a chamber for 2 min and received a single shock; 30 min later, the rat was returned to the chamber, and the emission of ultrasonic vocalization (USV) was recorded over a 10-min session. Rats emitting less than 90 s of ultrasonic calls were eliminated from the study. On day 3, in dose-response studies, testing was performed as on the selection day, but rats were injected with drug or vehicle at the end of the 2-min period. In antagonism studies, 60 min was allowed between the 2-min period and the 10-min test session; WAY-100,635 or vehicle was administered just after the 2-min period and drug or vehicle 30 min was administered before the 10-min session. Data were expressed as total duration of USV. Dose effects were analyzed using ANOVA followed by Dunnett’s test, and ID50values [95% confidence limits (CLs)] were calculated from percent values relative to the scores of vehicle-treated animals (100%). In antagonism studies, data were analyzed by a two-way ANOVA, followed by Newman-Keuls test.
Elevated Plus-Maze Test in Rats.
As previously described (Millan et al., 1997), the experiments were performed in a white matt-painted plus-maze (PM) constructed of wood and elevated to a height of 50 cm. The apparatus consisted of two open arms (50 × 10 cm) and two enclosed arms of the same dimensions, with walls 40 cm high. The two open arms were opposite to each other. The rats were isolated in their individual home cages 24 h before testing. On the test day, each rat was administered with drug or vehicle and was placed, 30 min later, in the central square of the maze facing one of the enclosed arms. The number of entries and time spent in open and enclosed arms were recorded by an observer situated 2 m from the maze. An entry was counted only when the rat had its four limbs in an individual arm. Data analyzed were the total number of entries, the percentage entries, and the percentage time spent in open arms. Drug effects were analyzed using ANOVA, followed by Dunnett’s test.
SI Test in Rats.
The method was adapted from that of File et al. (1996). The animals were individually housed for 5 days before testing. On the test day, they were placed in weight-matched pairs (±5 g) in opposite corners of a highly illuminated (300 lux) open-topped arena (57 × 36 × 30 cm) for a 10-min session. A camera was mounted 2 m above the arena and was connected to a monitor and a videotape recorder in an adjacent room. The observer recorded from the screen the duration of active SI (i.e., the time spent in grooming, following, sniffing, biting, jumping, or crawling over or under the other animal). If animals remained adjacent to each other without any movement for longer than 10 s, scoring was discontinued until active SI resumed. At the end of each session, the test arena was carefully cleaned. Animals were administered drug or vehicle 30 min before testing, with each rat of the same pair receiving the same treatment. Dose effects were analyzed using a one-way ANOVA followed by Dunnett’s test. In antagonism studies, WAY-100,635 or vehicle was administered 60 min before testing, and data were analyzed by a two-way ANOVA, followed by Newman-Keuls test.
Aggressive Behavior in Isolated Mice.
As previously described (Millan et al., 1997), mice were isolated in individual, black-painted polycarbonate cages. After at least 1 month of isolation, test days were scheduled once a week. On a test day, an “intruder” mouse was placed into the cage of a “resident” mouse for 3 min. The number of fights and total fight duration were recorded. Both mice received the same treatment (drug or vehicle) 30 min before testing. Dose effects were analyzed by a one-way ANOVA followed by Dunnett’s test, and ID50 values (95% CLs) were calculated to estimate drug potency. In antagonism studies, WAY-100,635 or vehicle was administered 60 min before testing, and data were analyzed by a two-way ANOVA, followed by Newman-Keuls test.
Rotarod Test: Induction of Ataxia.
As described previously (Millan et al., 1997), 30 min after injection, mice were placed on the bar of a Rotarod apparatus (Ugo Basile, Varese, Italy), which gradually accelerated from 4 to 40 rpm over a period of 300 s. The latency of mice to fall was determined with a cutoff of 360 s. The ID50 values (95% CLs) were calculated from percent values relative to the vehicle-treated animals (100%). Dose effects were analyzed by a one-way ANOVA, followed by Dunnett’s test.
Spontaneous Locomotion in Rats.
As described previously (Millan et al., 1997), rats were individually placed into transparent activity chambers (45 × 29 × 20 cm and equipped with two IR beams 24 cm apart and 4 cm above the floor) immediately after drug or vehicle injection. Locomotion recording was initiated 30 min later and conducted over a 60-min session. The cells of the activity chambers were connected via an interface to a computer (Coulbourn Instruments). The consecutive interruption of two beams was taken as a locomotion count. Data were analyzed by one-way ANOVA followed by Dunnett’s test, and ID50 values (95% CLs) were calculated to estimate drug potency.
Determination of Extracellular Levels of 5-HT in Nucleus Accumbens and Striatum.
The procedure used was described in detail previously (Millan et al., 1997, 1998a). Under pentobarbital anesthesia (60 mg/kg i.p.), the rats were placed in a stereotaxic apparatus, and a guide cannula was implanted in both the nucleus accumbens and the contralateral striatum. Five days later, a Cuprophan CMA/11 probe of 4 mm length (striatum) or of 2 mm length (accumbens) and 0.24 mm o.d. was lowered into position and perfused at 1 μl/min with a phosphate-buffered Ringer’s solution (147.2 mM NaCl, 4 mM KCl, 2.3 mM CaCl2, pH 7.3). Two hours later, dialysis commenced, and samples were taken every 20 min. Three basal samples were taken, and then the drug injected. Samples were then taken for an additional 3 h. For interaction studies, WAY- 100,635 (0.16 mg/kg s.c.) was injected, followed 20 min later by S- 16924 (2.5 mg/kg s.c.). Levels of 5-HT were quantified using HPLC and coulometric detection with the following conditions: 20-μl dialysate samples were diluted with 20 μl of mobile phase (75 mM NaH2PO4, 20 μM ethylenediaminetetraacetic acid, 1 mM sodium decanesulfonate, 17.5% methanol, 0.01% triethylamine, pH 5.70) and 33-μl samples were analyzed by HPLC with a column (Hypersil ODS 5 μm, C18, 150 × 4.6 mm; CARSPECIAUX) maintained at 43°C for separation and a coulometric detector (ESA 5014, Coulochem II) for quantification. The first electrode of the detector was set at −70 mV (reduction), and the second was set at + 280 mV (oxidation). The mobile phase was delivered at a flow rate of 2 ml/min. The assay sensitivity was 0.1 pg/sample. Drug effects were expressed as a percentage of basal values (0%). Data were analyzed by ANOVA with drugs as between factor and time as within factor.
Drugs.
All drug doses are in terms of the base. Drugs were dissolved in sterile water [if necessary, a few drops of lactic acid were added, and pH adjusted to as close to neutrality (>5.0) as possible]. They were administered s.c. or i.m. (in pigeons) in an injection volume of 1 ml/kg (10 ml/kg in mice). In general, full dose-response curves were performed for all studies. However, in view of limitations in drug solubility, absolute upper limits of 80.0 s.c. were defined for S-16924 and clozapine. Clorazepate (Tranxene, 2 mg/2 ml ampullas) was from Sanofi Winthrop (Gentilly, France). Clozapine was from RBI (Natick, MA). Haloperidol was from Sigma (Chesnes, France). S-16924 and WAY-100,635 were synthesized by Servier chemists (G. Lavielle and J. L. Peglion).
Results
Influence of S-16924 Compared with Clorazepate, Clozapine, and Haloperidol in Pigeon Conflict Test.
CLZ evoked a robust increase in punished responses, although at the highest dose tested, unpunished responses were diminished (Fig. 1 and Table 1). Similarly, S-16924 elicited a dose-dependent and marked increase in punished responses but also decreased unpunished responses at a dose 4-fold greater than that increasing punished responses (Fig. 1 and Table 1). In contrast, although clozapine tended to increase punished responses, this effect did not attain statistical significance, and at the highest dose tested, both punished responses and unpunished responses were diminished (Fig. 1 and Table 1). Haloperidol also failed to elevate significantly punished responses and dose-dependently and markedly decreased unpunished responses (Fig. 1 and Table 1).
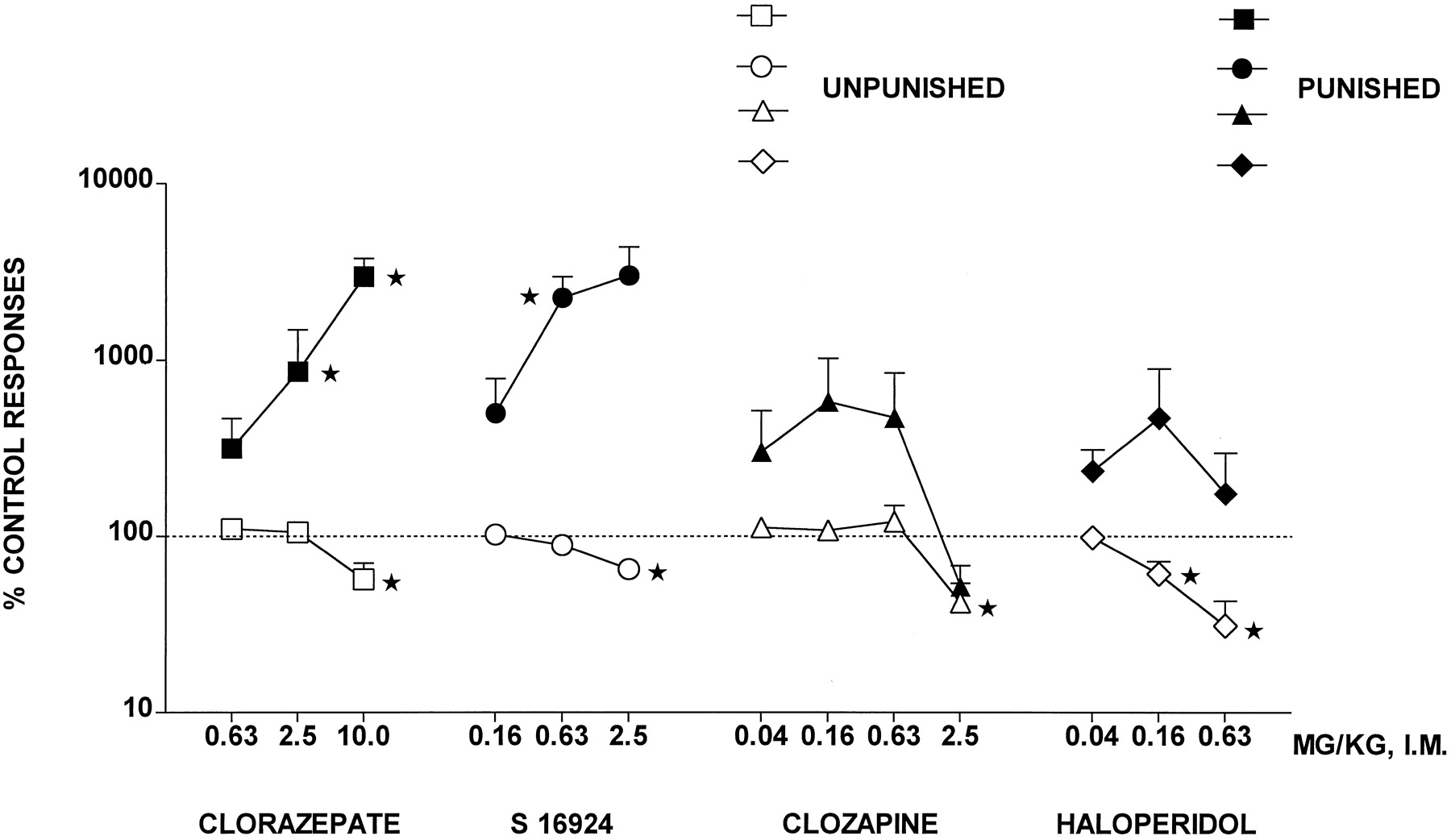
Actions of S-16924 compared with CLZ, clozapine, and haloperidol in the pigeon conflict test. Drug or vehicle was injected 5 min before testing. Data are means ± S.E.M. (n = 6–12 per value). Response rates are expressed as a percentage of those obtained during the previous vehicle session (animals were their own controls). Average control (vehicle sessions) values were 17.6 ± 5.9 punished responses and 1933.6 ± 47.2 unpunished responses. Asterisks indicate significant increases in punished responses and decreases in unpunished responses compared with control (vehicle sessions). Paired Wilcoxon test: one-tailed for punished and two-tailed for unpunished responses. *P < .05.
Actions of S-16924 compared with CLZ, clozapine, and haloperidol in various models of anxiolytic properties
Influence of S-16924 Compared with Clorazepate, Clozapine, and Haloperidol in Vogel Conflict Test.
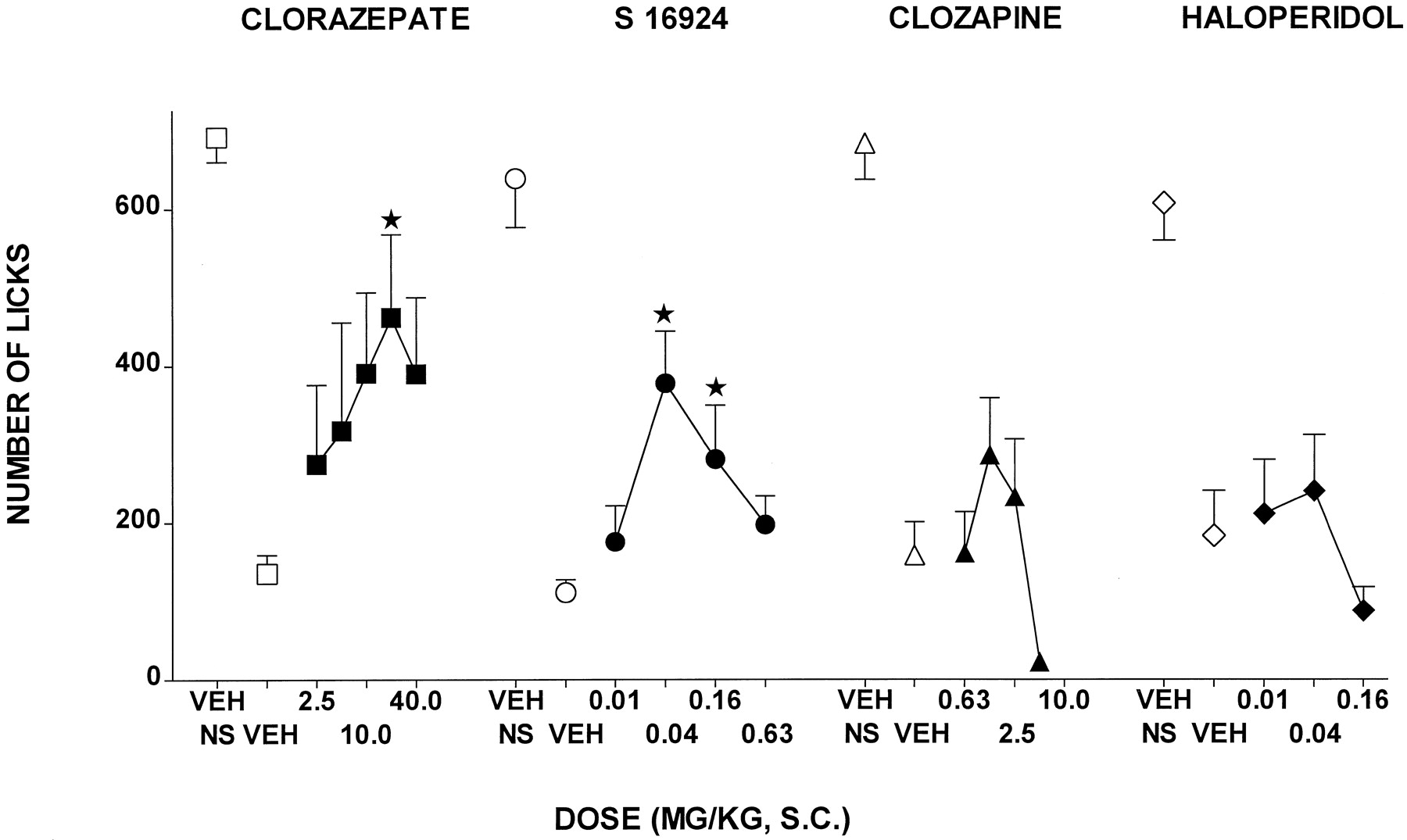
CLZ dose-dependently increased responding in the Vogel conflict test in rats, although its actions were expressed biphasically inasmuch as a further increase in dose resulted in a reduction in effect (Fig. 2and Table 1). S-16924 also increased responding, and like CLZ, its dose-response curve was biphasic (Fig. 2 and Table 1). The selective antagonist at 5-HT1A receptors, WAY-100,635 (0.16), abolished the increase in responding elicited by S-16924, without itself significantly modifying responding (Fig.3). In distinction to CLZ and S-16924, clozapine elicited only a mild and nonsignificant increase in responding, which was suppressed at the highest dose tested. Haloperidol did not increase responding, and like with clozapine, animals failed to respond at the highest dose examined (Fig. 2 and Table 1).

Actions of S-16924 compared with CLZ, clozapine, and haloperidol in the Vogel conflict test in rats. Drug or vehicle was administered 30 min before testing. Values represent means number of licks ± S.E.M. (n ≥ 5 per value). ANOVA as follows: CLZ, F(5,35) = 2.8,P < .05; S 16924,F(4,42) = 5.0, P < .01; clozapine, F(4,62) = 1.9,P > .05; and haloperidol,F(3,30) = 0.7, P > .05. Asterisks indicate significance of differences from vehicle values in Dunnett’s test after ANOVA. *P < .05.

Inhibition of the action of S-16924 (0.04 mg/kg) by WAY-100,635 (0.16 mg/kg) in the Vogel conflict test in rats. WAY-100,635 or vehicle was administered 60 min and S-16924 or vehicle was administered 30 min before testing. Data are means ± S.E.M. (n ≥ 7 per value). Two-way ANOVA as follows: S-16924, F(1,51) = 12.1,P < .001; WAY-100,635,F(1,51) = 6.1, P < .05; and interaction, F(1,51) = 4.7,P < .05. The closed asterisk indicates significance of differences between vehicle/S-16924 and vehicle/vehicle values and the open asterisk, WAY-100,635/S-16924 and vehicle/S-16924 values, in Newman-Keuls test after ANOVA. *P < .05.
Influence of S-16924 Compared with Clorazepate, Clozapine, and Haloperidol in Ultrasonic Vocalization Test.
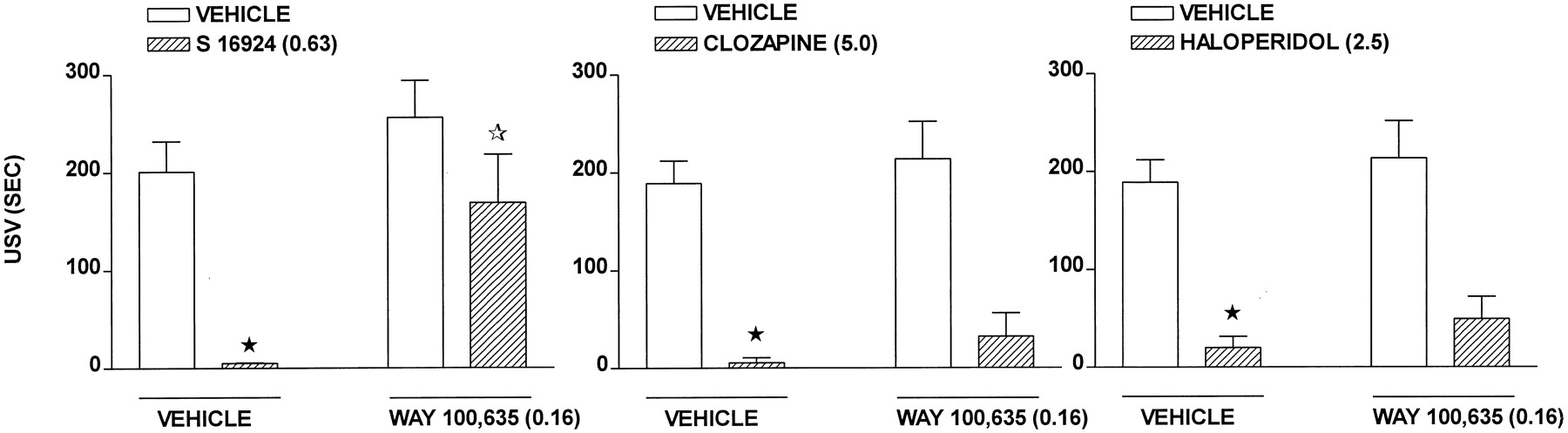
CLZ dose-dependently reduced, but did not abolish, USV in rats placed in an environment in which they had previously been exposed to an aversive stimulus (Fig.4 and Table 1). S-16924 dose-dependently and completely abolished USV (Fig. 4 and Table 1). This action of S-16924 was blocked by WAY-100,635, which did not itself modify USV (Fig. 5). Clozapine and, at high doses, haloperidol also dose-dependently blocked USV (Fig. 4 and Table 1), but in contrast to S-16924, their actions were not modified by WAY-100,635 (Fig 5).
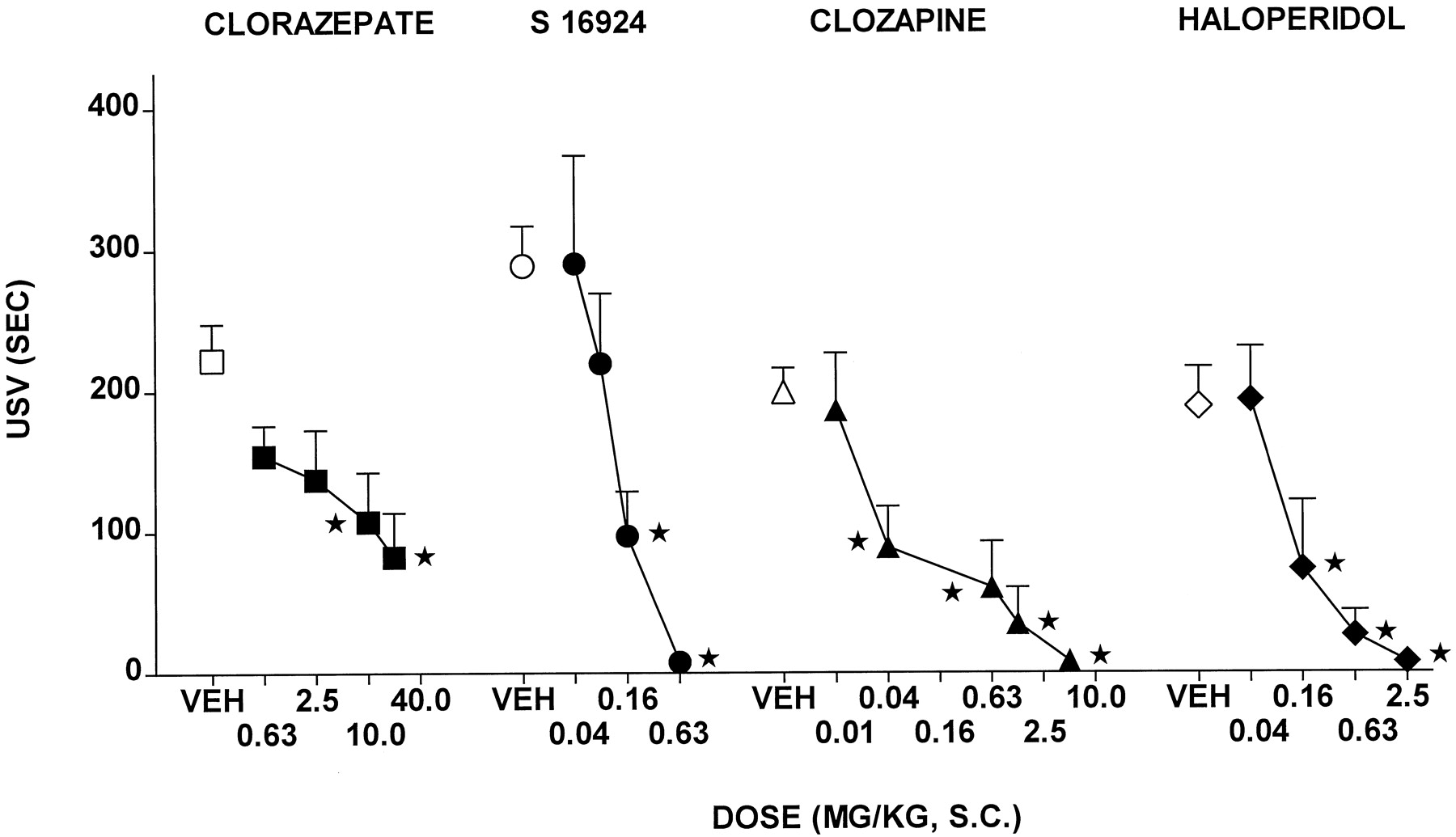
Actions of S-16924 compared with CLZ, clozapine, and haloperidol in the USV paradigm in rats. Drug or vehicle was administered 30 min before testing. Values represent means duration of USVs ± S.E.M. (n ≥ 4 per value). ANOVA as follows: CLZ, F(4,34) = 3.4,P < .05; S-16924,F(4,34) = 7.7, P < .001; clozapine, F(5,63) = 7.8,P < .001; and haloperidol,F(4,35) = 7.5, P < .001. Asterisks indicate significance of differences from vehicle values in Dunnett’s test after ANOVA. *P < .05.

Influence of WAY-100,635 (0.16 mg/kg) on the actions of S-16924 (0.63 mg/kg), clozapine (5.0 mg/kg), and haloperidol (2.5 mg/kg) in the USV model. WAY-100,635 or vehicle was administered 60 min and drug or vehicle was administered 30 min before testing. Data are means ± S.E.M. (n ≥ 5 per value). Two-way ANOVA as follows: S 16924, F(1,21) = 12.6,P < .01; WAY-100,635,F(1,21) = 4.6, P < .05; and interaction, F(1,21) = 2.1,P > .05. Clozapine,F(1,43) = 39.2, P < .001; WAY-100,635, F(1,43) = 0.5,P > .05; and interaction,F(1,43) = 0.01, P > .05. Haloperidol, F(1,42) = 31.5,P < .001. WAY-100,635,F(1,42) = 0.03, P > .05; and interaction, F(1,42) = 0.01,P > .05. The closed asterisk indicates significance of differences between vehicle/drug and vehicle/vehicle values and the open asterisk, WAY-100,635/S-16924 and vehicle/S-16924 values, in Newman-Keuls test after ANOVA. *P < .05.
Influence of S-16924 Compared with Clorazepate, Clozapine, and Haloperidol in PM Test.
CLZ evoked a dose-dependent increase in time spent in, and entries into, the open arms of a PM. Total entries were also elevated, but this effect was biphasic and did not reach statistical significance (Fig.6 and Table2). At the lowest doses tested, which did not increase overall entries, S-16924 tended to increase open arm time and entries. However, this action of S-16924 did not attain statistical significance. Clozapine and haloperidol had little effect (Fig. 6 and Table 2). A further increase in the doses of S-16924, clozapine, and haloperidol resulted, in each case, in a significant decrease in total entries (Fig. 6 and Table 2).

Actions of S-16924 compared with CLZ, clozapine, and haloperidol in the elevated PM test in rats. Drug or vehicle was administered 30 min before testing. Data are means ± S.E.M. (n ≥ 5 per value). ANOVA as follows. For percent entries in open arms (top): CLZ, F(4,31) = 4.3, P < .01; S- 16924,F(5,62) = 1.9, P > .05; clozapine, F(5,31) = 0.3,P > .05; and haloperidol,F(5,36) = 1.3, P > .05. For percent time in open arms (middle): CLZ,F(4,31) = 9.6, P < .001; S-16924, F(5,62) = 3.0,P < .05; clozapine,F(5,31) = 0.4, P > .05; and haloperidol, F(5,36) = 0.6,P > .05. For total entries (bottom): CLZ,F(4,31) = 1.9, P > .05; S-16924, F(5,62) = 7.1,P < .001; clozapine,F(5,31) = 5.6, P < .001; and haloperidol, F(5,36) = 9.8,P < .001. Asterisks indicate significance of differences from vehicle values in Dunnett’s test after ANOVA. *P < .05.
Actions of S-16924 compared with CLZ, clozapine, and haloperidol in various models of anxiolytic properties
Influence of S-16924 Compared with Clorazepate, Clozapine, and Haloperidol in SI Test.
CLZ elicited a dose-dependent and significant increase in active SI among pairs of unfamiliar rats placed in a novel environment, although this action was biphasic and a further increase in doses resulted in a loss of effect (Fig.7 and Table 2). S-16924 also increased active SI and, like CLZ, its dose-response curve was biphasic (Fig. 7and Table 2). The action of S-16924 was blocked by WAY-100,635, which was inactive alone (Fig. 8). In distinction to S-16924 and CLZ, neither clozapine nor haloperidol increased active SI (Fig. 7 and Table 2).
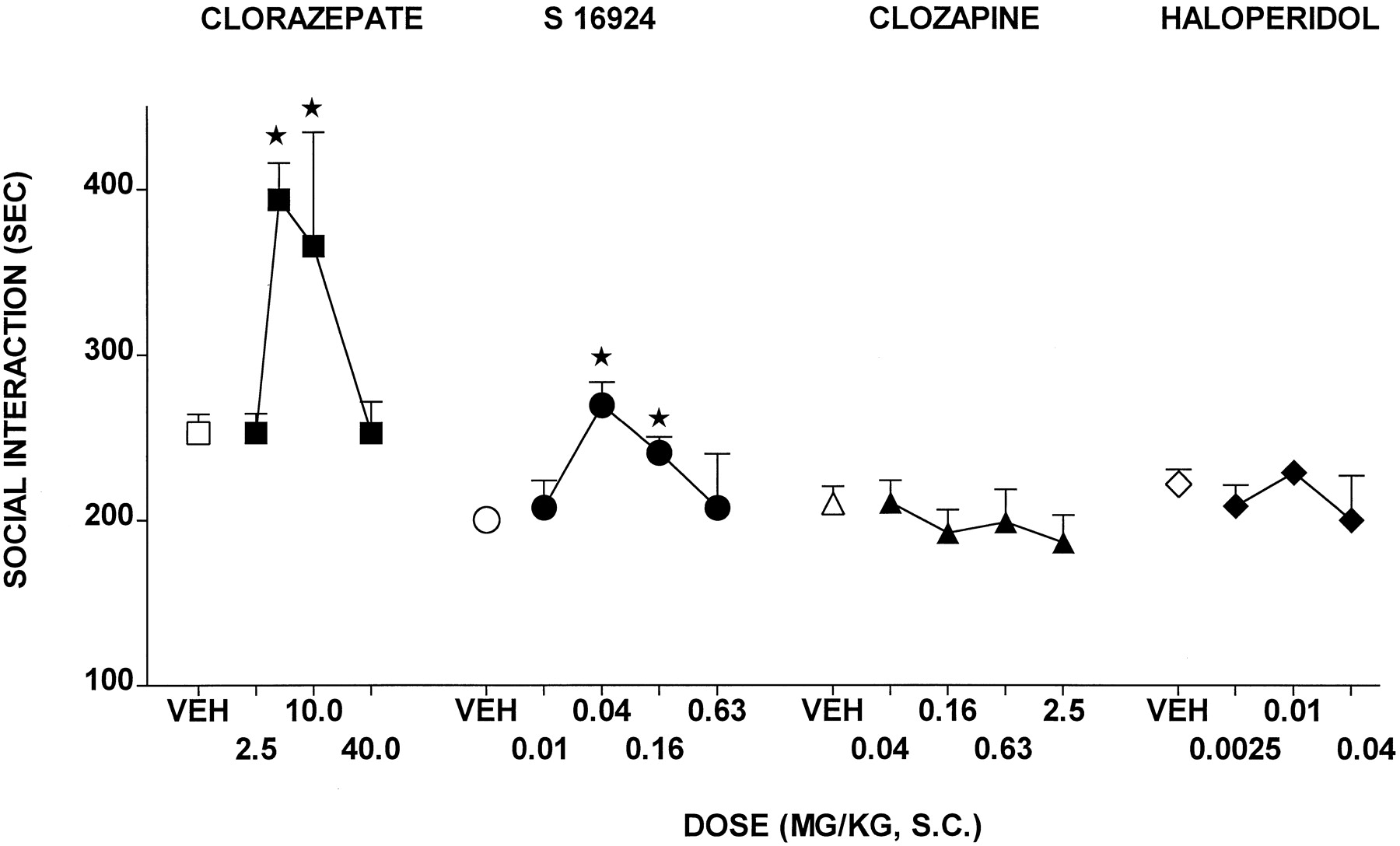
Actions of S-16924 compared with CLZ, clozapine, and haloperidol in the SI test in rats. Drug or vehicle was administered 30 min before testing. Values represent mean-duration of active SI ± S.E.M. (n ≥ 5 per value). ANOVA as follows: CLZ,F(4,52) = 7.7, P < .001; S- 16924, F(4,42) = 6.1,P < .001; clozapine,F(4,27) = 0.6, P > .05; and haloperidol, F(3,22) = 0.7,P > .05. Asterisks indicate significance of differences from vehicle values in Dunnett’s test after ANOVA. *P < .05.

Inhibition of the action of S-16924 (0.16 mg/kg) by WAY-100,635 (0.16 mg/kg) in the SI test in rats. WAY-100,635 or vehicle was administered 60 min and S-16924 or vehicle was administered 30 min before testing. Data are means ± S.E.M. (n = 9–12 per value). Two-way ANOVA as follows; S 16924,F(1,35) = 0.5, P > .05; WAY-100,635, F(1,35) = 6.1,P < .05; and interaction,F(1,35) = 9.4, P < .01. The closed asterisk indicates significance of differences between vehicle/S-16924 and vehicle/vehicle values and the open asterisk, WAY-100,635/S-16924 and vehicle/S-16924 values, in Newman-Keuls test after ANOVA. *P < .05.
Influence of S-16924 Compared with Clorazepate, Clozapine, and Haloperidol on Aggression in Isolated Mice.
CLZ, S-16924, clozapine, and haloperidol, in each case, dose-dependently and markedly decreased aggressive interaction in isolated mice, although over the dose range tested, the action of CLZ was submaximal (Fig.9 and Table 2). WAY-100,635, which did not modify aggressive interactions alone, failed to block the action of S-16924 in the procedure, even at a higher dose (0.63) than those evaluated in the other models (Fig. 9).

Influence of S-16924 compared with CLZ, clozapine, and haloperidol on aggressive behavior in isolated mice and lack of inhibition of the action of S-16924 (0.63 mg/kg s.c.) by WAY-100,635 (0.16 and 0.63 mg/kg). Top and middle, dose-response curves for number of fights and fight duration, respectively. Drug or vehicle was administered 30 min before testing. Data are means ± S.E.M. (n ≥ 4 per value). ANOVA as follows. For number of fights (top): CLZ, F(3,33) = 3.1,P < .05; S-16924,F(4,31) = 11.0, P < .001; clozapine, F(4,39) = 20.6,P < .001; and haloperidol,F(3,27) = 9.3, P < .001. For fight duration (middle): CLZ,F(3,33) = 4.2, P < .05; S-16924, F(4,31) = 11.6,P < .001; clozapine,F(4,39) = 10.3, P < .001; and haloperidol, F(3,27) = 6.2,P < .01. Asterisks indicate significance of differences from vehicle values in Dunnett’s test after ANOVA. *P < .05. Bottom, lack of inhibition of the action of S-16924 by WAY-100,635. WAY-100,635 or vehicle was administered 60 min and S-16924 or vehicle was administered 30 min before testing. Data are means ± S.E.M. (n = 4–9 per value). Two-way ANOVA as follows. For number of fights: S-16924,F(1,30) = 73.0, P < .001, WAY-100,635, F(2,30) = 0.1,P > .05; and interaction,F(2,30) = 0.9, P > .05. For fight duration: S-16924, F(1,30) = 31.0,P < .001; WAY-100,635,F(2,30) = 0.5, P > .05; and interaction, F(2,30) = 0.5,P > .05. Asterisks indicate significance of differences between vehicle/S-16924 and vehicle/vehicle values in Newman-Keuls test after ANOVA. *P < .05.
Influence of S-16924 Compared with Clorazepate, Clozapine, and Haloperidol on Motor Behavior.
CLZ, S-16924, clozapine, and haloperidol all dose-dependently elicited ataxia in a Rotarod procedure in mice and decreased spontaneous locomotor activity in rats (Table3). Furthermore, as described elsewhere, haloperidol elicits catalepsy in rats with an ED50 of 0.15 mg/kg s.c., whereas up to doses of 80.0 mg/kg s.c., S-16924 and clozapine do not provoke catalepsy (Millan et al., 1998b). Table 4 shows a comparison of drug potency in these paradigms (and other parameters reflecting disruption of motor behavior) to potency for the expression of anxiolytic actions: CLZ displayed only a modest (2.5-fold) dose separation for motor versus anxiolytic actions, whereas S-16924 expressed its anxiolytic properties at doses about 6-fold inferior to those over which it modified motor behavior. From Table 4, it is also apparent that S-16924 shared the anxiolytic properties of CLZ in five of the six procedures used. In contrast, clozapine and haloperidol were active only in the USV and aggressive behavior models. Furthermore, haloperidol was active only at doses markedly disrupting motor behavior.
Influence of S-16924 compared with CLZ, clozapine, and haloperidol on motor and locomotor behavior
Overall summary of anxiolytic compared with motor-disruptive properties based on minimal effective doses in all procedures
Influence of S-16924 Compared with Clorazepate, Clozapine, and Haloperidol on Extracellular, Dialysate Levels of 5-HT.
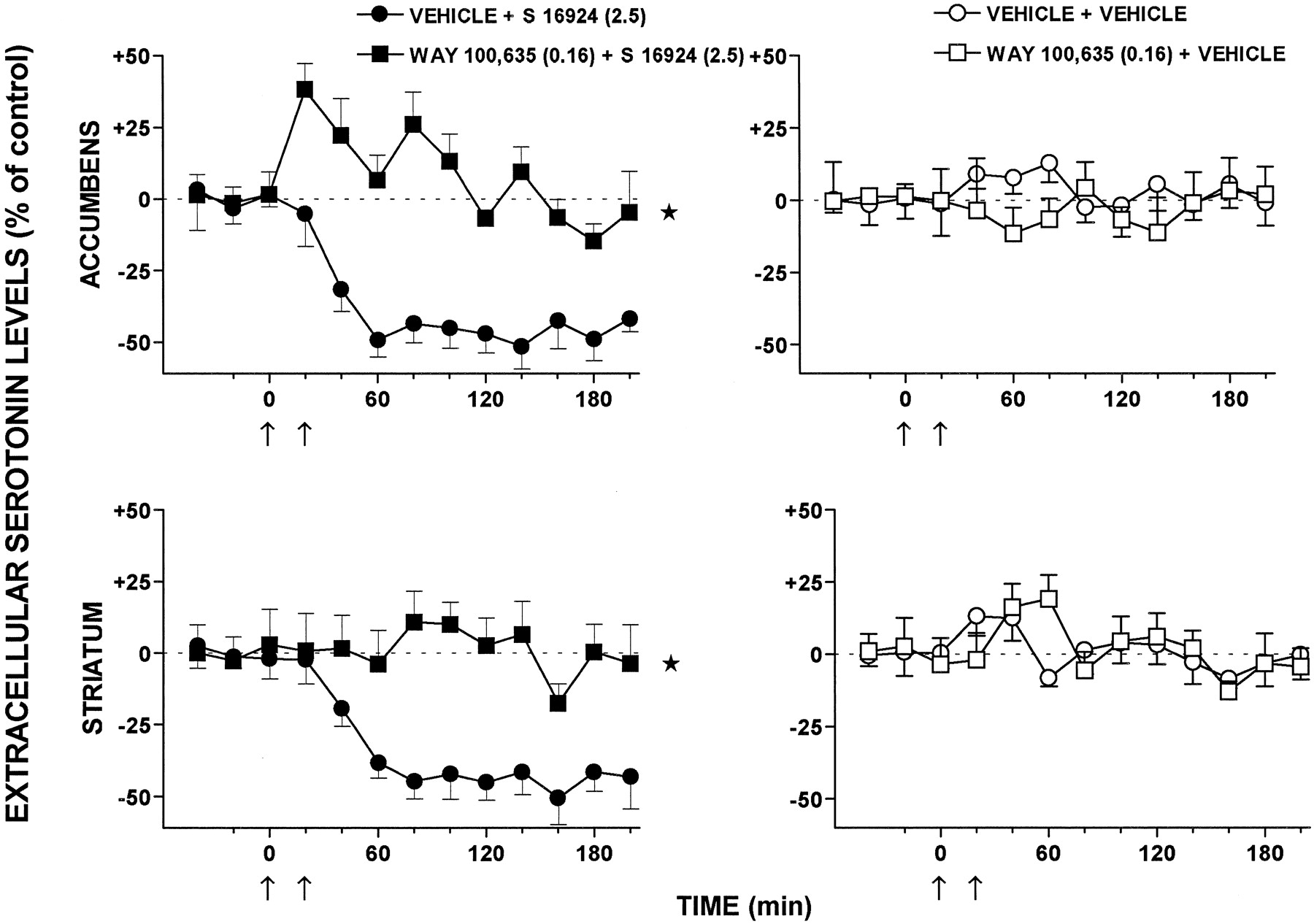
CLZ and S-16924 elicited a marked and sustained reduction in dialysate levels of extracellular 5-HT in the nucleus accumbens and striatum (Figs.10 and11). The action of S-16924 was abolished by WAY-100,635, which did not itself modify levels of 5-HT (Fig. 10). In distinction to CLZ and S-16924, clozapine only slightly reduced 5-HT levels in the striatum and did not affect these in the nucleus accumbens (Fig. 11). Haloperidol did not significantly modify dialysate levels of 5-HT in either the nucleus accumbens or the striatum (Fig. 11).

Influence of WAY-100,635 on the modulation by S 16924 of extracellular levels of 5-HT in both the nucleus accumbens and striatum of freely moving rats. Data are means ± S.E.M. (n = 7 or 8 per value). They are expressed as a percentage of basal, preinjection values which were defined as 0%. These were 1.00 ± 0.16 and 0.96 ± 0.12 pg/20 μl of dialysate for 5-HT in the nucleus accumbens and striatum, respectively, in vehicle-treated animals. ANOVA was performed over 40 to 200 min. Accumbens: WAY-100,635, F(1,13) = 1.1,P > .05; S 16924,F(1,12) = 64.3, P < .01; and WAY- 100,635 × S-16924,F(1,12) = 70.7, P < .01. Striatum: WAY-100,635, F(1,14) = 0.2,P > .05; S-16924,F(1,13) = 59.5, P < .01; and WAY-100,635 × S-16924,F(1,13) = 50.9, P < .01. Asterisks indicate significance of the WAY-100,635-treated group compared with the vehicle-treated group. *P < .05.

Influence of CLZ, clozapine, and haloperidol on extracellular levels of 5-HT in the nucleus accumbens and striatum of freely moving rats. Data are means ± S.E.M. (n = 6–9 per value). They are expressed as a percentage of basal, preinjection values, which were defined as 0%. These were 0.65 ± 0.09 and 0.55 ± 0.07 pg/20 μl of dialysate for 5-HT in the nucleus accumbens and striatum, respectively, in vehicle-treated animals. For comparison of individual values with the vehicle-treated group, ANOVA was performed over 20 to 180 min. Influence of CLZ: nucleus accumbens, F(1,14)= 28.0, P < .01; and striatum,F(1,11) = 21.8, P < .01. Influence of clozapine: nucleus accumbens,F(1,16) = 0.1, P > .05; and striatum, F(1,13) = 8.0,P < .05. Influence of haloperidol: nucleus accumbens, F(1,16) = 0.1,P > .05; and striatum,F(1,11) = 1.0, P > .05. Asterisks indicate significance of drug-treated groups compared with the vehicle-treated group. *P < .05.
Discussion
Comparative Anxiolytic Profiles of CLZ, S-16924, Clozapine, and Haloperidol.
In line with previous studies of benzodiazepines, CLZ was active in all models used and showed modest separation between dose ranges required for expression of its anxiolytic versus sedative properties. With the exception of the PM procedure (see below), S-16924 mimicked this broad-based pattern of anxiolytic activity. In addition, S-16924 was more potent than CLZ and displayed a more marked separation between its anxiolytic versus motor-disruptive properties. In distinction to S-16924, haloperidol lacked significant anxiolytic properties in the majority of paradigms. Furthermore, its apparent “anxiolytic activity” in the USV and aggressive mice models can likely be attributed to disruption of motor behavior. In contrast to haloperidol, however, clozapine expressed actions in the USV and aggressive mice models at doses lower than those eliciting motor disruption; similar findings have been documented in abstract form bySanchez and Arnt (1995). In the PM model, clozapine was inactive despite a decrease in total entries, an observation similar to that reported by Cao and Rodgers (1997). In a further model based on untrained behavior, the SI paradigm, clozapine was likewise ineffective. Although Corbett et al. (1993) reported that a high dose of clozapine (10.0) increases SI, these findings were obtained in a procedural variant reflecting activity against the negative symptoms of schizophrenia rather than anxiolytic actions. Regarding models of conditioned behavior, the mild (400%) and biphasic influence of clozapine in the pigeon conflict test herein did not reach statistical significance, a result mimicking the findings of Rigdon et al. (1996). However, the effect (500%) of clozapine did attain statistical significance in the study of Mansbach et al. (1988). A quantitatively similar, anxiolytic effect of clozapine has been reported in monkey and rat Geller conflict procedures, although dose-response curves were similarly biphasic (Spealman and Katz, 1980; Spealman et al., 1983;Wiley et al., 1993; Moore et al., 1994). In line with these findings, herein, the Vogel test also revealed a tendency toward an anxiolytic action of clozapine, although statistical significance was not attained. Procedural variables may well underlie such differences in the anxiolytic actions of CLZ among various studies. Nevertheless, overall, the present data, from six paradigms examined in parallel, globally coincide with previous reports that clozapine can exert anxiolytic actions in certain paradigms. Its sedative actions at higher doses, however, likely interfere with the expression of its potential anxiolytic properties.
Mechanisms Underlying Anxiolytic Actions of S-16924: 5-HT1A Autoreceptors.
Several complementary lines of evidence suggest that activation of 5-HT1Aautoreceptors plays a major role in the anxiolytic properties of S-16924. First, selective agonists at 5-HT1Aautoreceptors, such as S-15535, flesinoxan, 8-hydroxy-2-dipropylaminotetralin, and ipsapirone, are consistently active in all the procedures used herein (Schreiber and De Vry, 1993) with the exception of the PM model, for which a highly variable pattern of results has been obtained with both these and other 5-HT1A receptor ligands (Schreiber and De Vry, 1993; Coplan et al., 1995; File et al., 1996; Collinson and Dawson, 1997; Millan et al., 1997). Thus, the profile of activity of S-16924 corresponds to that of selective agonists at 5-HT1A autoreceptors. Second, in line with its 5-HT1A agonist properties (Millan et al., 1998a), S-16924 markedly suppressed dialysate levels of 5-HT in the nucleus accumbens and striatum. This observation mimics a similar reduction by S-16924 in extracellular levels of 5-HT in the frontal cortex at doses of 0.04 to 2.5 mg/kg s.c. (Millan et al., 1998a): that is, S-16924 activates 5-HT1A autoreceptors at doses comparable to those active in models of anxiolytic activity. Third, both the reduction in extracellular levels of 5-HT and the anxiolytic actions of S-16924 in several procedures were abolished by the highly selective 5-HT1A antagonist WAY-100,635.
Although clozapine behaves as a partial agonist at cloned, human 5-HT1A receptors with an efficacy comparable to that of S-16924, it is considerably (50-fold) less potent than S-16924, and in vivo, the 5-HT1A agonist actions of clozapine are less pronounced than those of S-16924 (Millan et al., 1998a,b; Newman-Tancredi et al., 1996). Indeed, in contrast to S-16924, WAY-100,635 failed to modify the actions of clozapine in the USV procedure. In addition, clozapine only slightly decreased dialysate levels of 5-HT in the striatum, an action exerted independently of 5-HT1A sites (Lejeune et al., 1994), and failed to modify these in the accumbens (see below). These data suggest that 5-HT1A autoreceptors are not involved in the anxiolytic actions of clozapine. Regarding CLZ, its inhibitory influence on 5-HT levels supports observations suggesting that reinforcement of γ-aminobutyric acidergic inhibitory tone on serotonergic pathways contributes to the anxiolytic properties of benzodiazepines (Pan and Williams, 1989; Miczek et al., 1995;Huidobro-Toro et al., 1996).
Mechanisms Underlying Anxiolytic Actions of S-16924: Other Receptors.
In addition to 5-HT1Aautoreceptors, several other mechanisms may be involved in the anxiolytic actions of S-16924. First, stimulation of postsynaptic 5-HT1A receptors may mediate anxiolytic effects under certain conditions (Schreiber and De Vry, 1993; Coplan et al., 1995; File et al., 1996). However, an anxiogenic role has also been proposed for postsynaptic 5-HT1A receptors, and their significance remains controversial (Schreiber and De Vry, 1993;File et al., 1996). In any case, S-16924 possesses low efficacy at postsynaptic 5-HT1A sites, and it is unlikely that they are of major importance in its anxiolytic actions. Second, 5-HT2C receptor antagonists are active in all the procedures used herein, with the exception of the USV test (Schreiber and De Vry, 1993; Kennett et al., 1996b; Griebel et al., 1997). In this model, WAY-100,635 abolished the action of S-16924. It was not, however, effective in modifying its effects in aggressive mice, suggesting that inasmuch as selective antagonists at 5-HT2C receptors reduce aggressive behavior at nonsedative doses, the potent 5-HT2C antagonist properties of S-16924 may be involved in this paradigm. In addition, blockade of 5-HT2C receptors, or 5-HT2A receptors, for which S-16924 also exhibits high potency, may play a facilitatory role in enhancing the anxiolytic actions of S-16924 in other paradigms. This possibility is supported by the similar affinity of S-16924 for 5-HT1A(Ki = 3.8 nM) and 5-HT2C sites (7.5 nM) sites as well as functional studies in vivo suggesting that the 5-HT1Aagonist and 5-HT2C antagonist properties of S-16924 are expressed over a similar dose range (Millan et al., 1998a; M. J. Millan, unpublished observations). Third, there is evidence for a role of mesocortical (and other) dopaminergic systems in the response to and modulation of stress and anxiety, and depletion of frontocortical pools of dopamine is associated with anxiogenic effects (Borowski and Kokkinidis, 1996; Espejo, 1997). Thus, the ability of S-16924 to modulate dopaminergic transmission might also be involved in its anxiolytic properties. Finally, although there is little evidence that selective α1 adrenoceptor antagonists possess selective anxiolytic properties, they inhibit the activity of raphe-localized serotonergic neurons (Lejeune et al., 1994;Söderpalm and Engel, 1988). Thus, via this mechanism, the potent α1 antagonist properties of S-16924 (Millan et al., 1998a) might also contribute to its anxiolytic actions.
This latter mechanism is of relevance to clozapine, which is a potent antagonist at α1 adrenoceptors, the blockade of which underlies its (weak) inhibition of the electrical activity of serotonergic neurons and likely accounts for the mild reduction in extracellular 5-HT levels in striatum (Lejeune et al., 1994). Nevertheless, clozapine is also a potent antagonist at 5-HT2C receptors (Roth and Meltzer, 1995), and this mechanism may underlie its anxiolytic effects in aggressive mice. On the other hand, 5-HT2C antagonists are ineffective in the USV model (Schreiber and De Vry, 1993), and the mechanisms involved in the anxiolytic actions of clozapine in this procedure remain to be elucidated.
Comparison with Other Antipsychotic Agents.
Several other antipsychotics have been examined in individual procedures of potential anxiolytic properties. The clozapine congener seroquel inhibited USV in rats and aggression in mice in the absence of motor disruption (Ellenbroek et al., 1996). A further clozapine analog, olanzapine, displayed anxiolytic properties in conflict tests (Moore et al., 1994). The phenylindole derivative sertindole was active in the two-compartment and SI tests in rats but, curiously, was 20 million-fold more potent than diazepam in these procedures while being inactive in the Vogel and USV paradigms (Sanchez et al., 1991). Although the mechanism or mechanisms of action of olanzapine, clozapine, and sertindole remain unclear, they express antagonist properties at 5-HT2C/5-HT2Areceptors yet show little affinity for 5-HT1Asites. Notably, then, several other potential antipsychotics showing significant affinity for 5-HT1A sites may possess anxiolytic activity. Thus, umespirone was active in the rat SI and marmoset threat models (Barnes et al., 1991), and 11292U90 was active in pigeon and rat conflict procedures (Rigdon et al., 1996). However, for these drugs, no mechanistic studies were performed with 5-HT1A antagonists to confirm an involvement of 5-HT1A receptors. In contrast, the present study provides compelling pharmacological and neurochemical evidence that an activation of 5-HT1A autoreceptors is involved in the anxiolytic actions of S-16924.
Conclusions.
In contrast to clozapine and haloperidol, S-16924 exerted a broad pattern of anxiolytic actions, which, with the exception of the PM test, mimicked that of the benzodiazepine CLZ. Furthermore, the actions of S-16924 were expressed with a greater separation to doses disrupting motor behavior than those of CLZ. The distinctive, high-potency agonist action of S-16924 at (presynaptic) 5-HT1A receptors likely plays a predominant role in its anxiolytic properties, but a facilitatory role of 5-HT2C or other receptor types cannot be excluded. The dose range over which S-16924 exerted its anxiolytic actions was similar to that for expression of its potential antipsychotic properties in rodents (Millan et al., 1998b). Clearly, it would be of interest to extend the present data to other models and to other species. Furthermore, it would be of interest to compare the actions of S16924 with those of other classes of anxiolytic agent for a fuller appreciation of its potential anxiolytic properties. Nevertheless, should the potential anxiolytic actions of S-16924 be expressed in humans, they would likely prove to be of use in the treatment of schizophrenia, for which indication S-16924 is under development.
Acknowledgments
We thank L. Cistarelti, B. Denorme, S. Giradon, H. Gressier, C. Melon, S. Monneyron, and S. Veiga for technical support and C. LeRoy for secretarial assistance.
Footnotes
-
Send reprint requests to: Dr. Mark J. Millan, Institut de Recherches Servier, Centre de Recherches de Croissy, Psychopharmacology Department, 125 Chemin de Ronde, 78290 Croissy-sur-Seine, Paris, France.
- Abbreviations:
- CLZ
- clorazepate
- 5-HT
- serotonin
- PM
- plus-maze
- SI
- social interaction
- S-16924
- (R)-2-{1-[2-(2,3-dihydro-benzo[1,4]dioxin-5-yloxy)-ethyl]-pyrrolidin-3yl}-1-(4-fluorophenyl)-ethanone
- USV
- ultrasonic vocalization
- WAY-100
- 635,N-{2-[4-(2-methoxyphenyl)-1-piperazinyl] ethyl}-N-(2- pyridinyl)cyclo-hexanecarboxamide 3HCl
- Received June 29, 1998.
- Accepted October 6, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}