


Abstract
S 16924 antagonized locomotion provoked by dizocilpine and cocaine, reduced conditioned avoidance responses and blocked climbing elicited by apomorphine, models predictive of control of the positive symptoms of schizophrenia: its median inhibitory dose (ID)50 was 0.96 mg/kg, s.c. vs. 1.91 for clozapine and 0.05 for haloperidol. Rotation elicited in unilateral, substantia nigra-lesioned rats by the D1 agonist, SKF 38393, and by the D2 agonist, quinpirole, was blocked equipotently by S 16924 (0.8 and 1.7) and clozapine (0.6 and 2.0), whereas haloperidol preferentially blocked quinpirole (0.02)vs. SKF 38393 (1.8). S 16924 more potently inhibited the head-twitches elicited by 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI) and the locomotion provoked by phencyclidine than it inhibited the locomotion elicited by amphetamine (ID50s = 0.15 and 0.02 vs. 2.4). Clozapine showed a similar preference (0.04 and 0.07 vs. 8.6), but not haloperidol (0.07 and 0.08 vs. 0.04). The discriminative stimulus (DS) properties of DOI were also blocked by S 16924 (ID50 = 0.17) and clozapine (0.05) but not by haloperidol (>0.16). S 16924 fully (100%) generalized [effective dose (ED)50 = 0.7] to a clozapine DS and clozapine (0.23) fully generalized to a S 16924 DS whereas haloperidol (≥0.08) only partially generalized (≤50%) to their DS in each case. Power spectra analysis of electroencephalograms from frontal cortex showed that both S 16924 (2.0) and clozapine (5.0) reinforced frequencies in the 7 to 8 Hz range whereas haloperidol (0.5) preferentially reinforced frequencies in the 10 to 14 Hz range. In a model of perturbation of cognitive-attentional function, significant latent inhibition was obtained with S 16924 (0.08) and clozapine (0.16), but not haloperidol (0.0063 and 0.04): higher doses of S 16924 (2.5), clozapine (5.0) and haloperidol (0.1) all blocked disruption of latent inhibition by amphetamine (1.5). Catalepsy was provoked by haloperidol (0.04–0.63) but not by S 16924 (≥80.0) or clozapine (≥80.0). Further, S 16924 (ID50 = 3.2) and clozapine (5.5) inhibited induction of catalepsy by haloperidol. This action of S 16924 was abolished by the 5-HT1A receptor antagonist, WAY 100,635 (0.16), which less markedly attenuated the anticataleptic action of clozapine. Further, although gnawing elicited by methylphenidate was inhibited by S 16924 (ID50 = 8.4), clozapine (19.6) and haloperidol (0.04), only the action of S 16924 was blocked by WAY 100,635 (0.16). Haloperidol potently (0.01–0.16, ∼24-fold) increased prolactin levels whereas they were less markedly affected by S 16924 (2.5–40.0, 4-fold) and clozapine (10.0–40.0, 3-fold). Clozapine displayed high affinity at cloned, human, muscarinic (M1) and native, histamine (H1) receptors (Ki s = 4.6 and 5.4 nM, respectively), whereas S 16924 (>1000 and 158) and haloperidol (>1000 and 453) displayed low affinity. In conclusion, S 16924 displays a profile of activity in diverse models of potential antipsychotic and extrapyramidal properties similar to that of clozapine and different to that of haloperidol. In particular, reflecting its partial agonist actions at 5-HT1A receptors, S 16924 inhibits rather than induces catalepsy in rats. However, in contrast to clozapine, S 16924 displays only low affinity for muscarinic and histaminic receptors.
In the companion paper, it was shown that the novel benzodioxopyrrolidine, S 16924, possesses a pattern of interaction at multiple monoaminergic receptors similar to that of the atypical antipsychotic, clozapine and different to that of the neuroleptic, haloperidol, in particular as concerns its more pronounced affinity at D4,alpha-1 adrenergic, 5-HT2A and 5-HT2Cvs. D2 receptors. In addition, S 16924 possesses more potent partial agonist properties at 5-HT1A receptors than clozapine, although haloperidol has negligible affinity for these sites. Correspondingly, via activation of 5-HT1A autoreceptors, S 16924 diminishes the activity of cerebral serotonergic pathways and selectively facilitates dopaminergic transmission in mesocortical as compared to mesolimbic and nigrostriatal pathways. This potent interaction of S 16924 with 5-HT1A receptors is of particular interest in light of the important role of pre- and postsynaptic 5-HT1A receptors in the control of mood and cognition, and their high concentration in the FCX, hippocampus, septum and other corticolimbic regions (Schreiber and De Vry, 1993). Further, recent studies have shown alterations in the density of 5-HT1A receptors in the FCX of schizophrenic patients (Burnet et al., 1996; Gurevich and Joyce, 1997). Certain studies have suggested that 5-HT1A agonists may potentiate the antipsychotic properties of dopaminergic antagonists (Evenden, 1992; see “Discussion”). Further, the selective facilitation of frontocortical as compared to subcortical dopaminergic transmission by S 16924 (companion paper) may contribute to a reduction of the “hypofrontality” which is implicated in deficit symptomatology (Knable and Weinberger, 1997). In addition, activation of 5-HT1A receptors has been associated with both anxiolytic and antidepressive properties, which may be of adjunctive use in the (long-term) management of schizophrenic patients (Schreiber and De Vry, 1993). As concerns extrapyramidal symptoms, studies in rodents have indicated that stimulation of 5-HT1A receptors inhibits the induction of catalepsy by haloperidol and other neuroleptics (Andersen and Kilpatrick, 1996; Invernizzi et al., 1988). In light of these observations, the aim of our present studies was to evaluate the activity of S 16924 in several, functional paradigms suggestive of the control of positive and deficit-cognitive symptoms of schizophrenia, as compared to the induction of extrapyramidal and other side-effects. The models used and their conceptual bases are outlined below.
As concerns the positive symptoms of schizophrenia, these have been attributed to an overactivity of mesolimbic dopaminergic pathways and, correspondingly, blockade of postsynaptic D2 receptors in limbic structures is associated with their moderation (Kahn and Davis, 1995). In line with this reasoning, in several experimental models, antipsychotic (antipositive) activity is correlated with dopamine D2 receptor antagonist properties at mesolimbic D2 receptors: for example, blockade of the locomotor activity elicited by the DA-reuptake inhibitors/releasers and psychostimulants, amphetamine and cocaine, and by the open channel blocker at N-methyl-d-aspartate receptors, dizocilpine, which indirectly activates mesolimbic dopaminergic transmission (Delfset al., 1990; Jackson et al., 1994; Narayananet al., 1996; Svensson et al., 1995). Antagonism of stereotyped climbing elicited by the direct dopaminergic agonist, apomorphine, in mice and a reduction of CARs in rats, have also proven robust and reproducible models sensitive to D2 receptor antagonists for the prediction of therapeutic efficacy against positive symptoms (Kahn and Davis, 1995; Wirsching et al., 1995).
There are currently no straightforward approaches for the prediction of activity against negative-cognitive symptoms. However, they appear to involve a “hypofrontality” of the prefrontal and FCX and a deficient mesocortical dopaminergic input to this region (Goldberg and Gold, 1995; Knable and Weinberger, 1997). In contrast to haloperidol and in analogy to dopamine, S 16924 reinforces frontocortical dopaminergic transmission (vide supra). Further, based on these findings and the fact that negative symptoms are more effectively managed by clozapine, than haloperidol, several complementary strategies may be defined. First, the influence of drugs upon EEG power spectra from the FCX (Shvaloff et al., 1988) may be characterized. Second, in an approach which integrates internal cues from various corticolimbic and other regions, drug discrimination procedures may be used (Carey and Bergman, 1997; Nielsen, 1988). Herein, thus, rats were trained to recognize the DS properties of clozapine or S 16924. Third, in contrast to amphetamine, which elicits principally the positive symptoms of schizophrenia, PCP reproduces both positive and negative symptoms (Gorelick and Balster, 1995; Steinpreis, 1996). Further, in distinction to haloperidol, and reflecting its more potent antagonist actions at 5-HT2Avs.D2 receptors (Brunello et al., 1995; Roth and Meltzer, 1995), clozapine preferentially blocks the locomotion provoked by PCP as compared to amphetamine (Maurel-Remy et al., 1995aand b). Thus, the relative potency of S 16924 in blocking PCP- as compared to amphetamine-evoked locomotion was determined. In addition, the antagonism of HTW elicited by the hallucinogen and serotonergic agonist, DOI, was examined since this behavior is also mediated via activation of 5-HT2A receptors, probably in the FCX (Willins and Meltzer, 1997; Schreiber et al., 1995). Fourth, a facilitation of cognitive-attentional function would likely contribute to an improvement in negative symptoms inasmuch as schizophrenics display a compromised processing (filtering) of sensory information (Gray et al., 1995; Weiner and Feldon, 1997). This inability to ignore irrelevant input may be modelled by a latent inhibition (LI) paradigm whereby pre-exposure to a stimulus loses its capacity to control behavior (Gray et al., 1995; Weiner and Feldon, 1997).
Regarding side-effects, neuroleptics elicit an extrapyramidal motor syndrome by interruption of activity at striatal D2(or D1) receptors (Josselin et al., 1997; Keks, 1996; Millan et al., 1995a). Drug propensity to induce dystonias and Parkinson-like symptoms is reflected by the induction of catalepsy in rats (Hoffman and Donavan, 1995). Further, the hyperprolactinaemia and associated endocrinological problems evoked by neuroleptics is due to blockade of tonically-active D2receptors on adenohypophyseal lactotrophs (Cunningham-Owens, 1996;Millan et al., 1995a). Thus, we determined the ability of S 16924 to elicit catalepsy and PRL secretion in rats. The activity of S 16924 was also evaluated in a further model reflecting activity at striatal populations of D2 receptors: inhibition of the stereotyped gnawing elicited by the DA uptake inhibitor/releaser, methylphenidate (Kleven et al., 1996; Millan et al., 1995b). Finally, the marked activity of clozapine—but not haloperidol—at muscarinic and histaminic receptors results in pronounced autonomic, cardiovascular and other side-effects (Cunningham-Owens, 1996; Zorn et al., 1994). Thus, the affinity of S 16924 was measured at these receptor types. As in the companion paper, the actions of S 16924 were compared throughout to those of clozapine and haloperidol.
Methods
Animals.
Male Wistar rats (220–240 g body weight, Iffa-Credo, L’Arbresle, France) and CD1 (ICR) BR mice (22–25 g, Charles River, Saint-Aubin-les-Elbeuf, France) were used. The animals were housed in sawdust-lined cages with, unless specified, free access to food and water. Laboratory temperature was 21 ± 1°C and humidity 60 ± 5%. There was a 12 hr/12 hr light-dark cycle with lights on at 7:30.
Influence on PRL levels.
PRL levels were determined in systemic plasma using a radioimmunoassay and a specific antibody against rat PRL (Amersham, Buckingham, England) as described previously (Millan et al., 1995a). Data were analyzed by ANOVA followed by Dunnett’s test. Drug potency was expressed in terms of the minimal effective dose (P < .05 to vehicle) derived from Dunnett’s test.
Inhibition of rotation elicited by quinpirole and SKF 38393.
The procedure employed was described in detail previously (Millanet al., 1995b). Briefly, rats anesthetized with pentobarbital (45 mg/kg, i.p.) were placed in a stereotaxic apparatus and the left substantia nigra pars compacta injected over 4 min with 4.0 μl of 6-hydroxydopamine (2 μg/μl). After 3 wk recovery, rats showing a pronounced contralateral turning response to apomorphine (0.04 mg/kg, s.c.) were selected for further study. Separate groups of rats were trained either with quinpirole (0.02 mg/kg, s.c.) or with SKF 38393 (0.63 mg/kg, s.c.). Rotation was recorded over 20 to 50 min and 45 to 60 min after administration of quinpirole and SKF 38393, respectively. Rotation was monitored automatically using a harness coupled to a microcomputer (Rotacount 8, Columbus Instruments, OH). An alternate design (ABACADA…) was used such that, in every other session, rats received vehicle rather than a test drug prior to quinpirole or SKF 38393. Rotation was expressed as a percentage of the mean of the (A) sessions preceding and after drug treatment. Drugs were given 25 min before quinpirole or SKF 38393. Data were analyzed by a paired Student’s test and ID50s (95% CLs) calculated to estimate drug potency.
Inhibition of apomorphine-induced climbing.
As previously (Millan et al., 1995b), mice were administered drug or vehicle and placed individually in upturned, steel cylinders (14 cm diam., 14 cm h.) with walls of vertical bars (2 mm diam., 1 cm apart). Thirty min later, they received injections with apomorphine (0.75 mg/kg, s.c.) and replaced in the cylinders. Each animal was observed for climbing behavior (total score: 0 to 4) at 10 and 20 min after the injection of apomorphine. Data (percentages of animals with a total—summed for 10 and 20 min—climbing score of <2) were analyzed by Fisher’s exact probability test and effective dose (ED)50s (95% CLs) calculated to estimate drug potency.
Inhibition of spontaneous amphetamine-, cocaine-, dizocilpine- and PCP-induced locomotion in rats.
The procedure used was essentially as described previously by Maurel-Remy et al.(1995b). Rats were administered drug or vehicle and placed in individual transparent polycarbonate cages (45 × 30 × 20 cm). Thirty min later, they received injections with amphetamine (2.5 mg/kg, i.p.), cocaine (20 mg/kg, i.p.), dizocilpine (0.16 mg/kg, s.c.) or PCP (20.0 mg/kg, s.c.) and the cages placed in activity chambers (Lablinc System, Coulbourn, Lehigh Valley, PA). For measures of spontaneous locomotion, no further treatment was given. Chambers were equipped with two infrared beams 4 cm above the floor and 24 cm apart. The consecutive interruption of two beams within 3 sec was computed as a movement (locomotor activity). Activity was monitored over 60 min after injection of amphetamine, cocaine, dizocilpine or PCP. Data were analyzed by ANOVA followed by Dunnett’s test and ID50s (95% CLs) calculated to estimate drug potency.
Inhibition of spontaneous activity in mice.
Thirty min after treatment with drug (mg/kg, s.c.) or vehicle, mice were removed from their individual housing cages and placed in a white, plexiglass recording chamber (27 × 27 × 27 cm) equipped with four photocells (6 cm apart and 2 cm above the cage floor) located on each of two facing walls. The photocells were connected through an interface to a microcomputer (OSYS/ORGA System, Changé, France). Interruption of two adjacent photocell beams was counted as a movement. Locomotor activity, i.e., number of movements, was recorded during a 10-min session.
Inhibition of CAR.
The procedure employed was similar to that described by Evenden (1992). Rats used for the study were pretrained to move from one compartment of a shuttle-box (Letica, Barcelona, Spain) to the other when a stimulus light was “on” to avoid an electric shock through the grid-floor. They were submitted to a daily session of 10 trials, separated one another by a 30-sec intertrial interval. Each trial consisted of a 10-sec period (maximal duration) with stimulus light “on,” followed or not by a 5-sec period (maximal duration) with electric shock (560 μA), depending on the response of the animal to the stimulus light. The trial terminated once the rat had moved into the other compartment, either during the light “on” period (CAR) or during the shock period (escape response). Data were the number of CARs (maximal value: 10) per session. The animals served as their own controls with the vehicle session performed on the day before the test (drug) session. Drug or vehicle were injected 30 min before the session. Data were analyzed by a paired Wilcoxon signed rank test and ID50s (95% CLs) calculated to estimate drug potency.
Inhibition of PCP-induced locomotion after chronic administration of S 16924.
S 16924 (0.16 or 2.5 mg/kg, s.c.) or vehicle were administered once a day for 14 days. On test day (day 15), the effects of S 16924 (0.16 or 2.5 mg/kg, s.c.) upon PCP-induced locomotion were evaluated as described above. The effects of S 16924 in rats treated for 2 wk with S 16924 were compared to those obtained in rats treated for 2 wk with vehicle using ANOVA followed by Newman-Keuls test.
Inhibition of DOI-induced HTWs.
As described previously (Schreiber et al., 1994), rats received injections with DOI (2.5 mg/kg, i.p.) and placed in transparent, plexiglass observation cages (33.5 × 23.5 × 19.0 cm) without a sawdust lining. Five min after the administration of DOI, the number of HTWs was counted over 5 min. Drugs were given 30 min before DOI. Data were analyzed by ANOVA followed by Dunnett’s test and ID50s (95% CLs) calculated to estimate drug potency.
Latent inhibition.
The protocol used was based on that described by Weiner and Feldon (1997). The animals were housed in groups of four with free access to food during the entire study. Commencing 72 hr before the first training session, all rats had their access to water restricted to 1 hr per day in the morning. The LI-CER procedure used consisted of four successive phases: training, PE, conditioning and test. They were conducted in standard operant chambers (Med Associates Inc., St Albans, VT; model ENV 007) equipped with lickometers (Med Associates Inc., model ENV 251M). During training (days 1–6), animals were allowed to drink in the chambers for 5 min and only rats completing more than 600 licks in the last two sessions were included in the study. During the PE (day 7) and the conditioning (day 8) sessions, animals were placed for 15 min in the chambers with the water spout removed. For half of the animals, PE consisted of a series of tone presentations within the session. Each tone had a frequency of 2.5 kHz, an intensity of 30 dB above background and a duration of 10 sec. The number of tones was either 10 or 40 using interstimulus intervals of 80 or 12.5 sec, respectively. The other half of the animals (NPE rats) were placed in the operant chambers without being exposed to the tones. The expression of LI covaries with the number of tones during PE (Moran and Moser, 1992). Thus, in studies of drug-induced LI, we used a 10 tone PE paradigm to avoid a limitation of drug actions by ceiling effects. In contrast, studies of amphetamine-induced disruption of LI used a 40 tone PE as stable and reproducible LI effects were obtained under these conditions in vehicle-treated animals. During conditioning, animals were exposed to two tones 5 and 10 min after the beginning of the session. Each tone was immediately followed by a scrambled footshock (0.5 mA, 3 sec; shocker: Med Associates Inc; model ENV 410). For the test session (day 9), the water spouts were replaced in the chambers and each animal allowed to drink freely until 100 licks had been made. Then, a tone presentation occurred and continued until either the animal made a further 10 licks or 300 sec had elapsed. A SR was calculated according to the formula SR = t1/(t1 + t2) with t1 and t2 as the times to complete licks 90 to 100 and 100 to 110, respectively. Usually, t1 was low (ranging between 1.5 and 2 sec). The NPE animals, which reliably learned the tone-shock association, generally stopped drinking upon presentation of the tone; t2 was high and consequently, SR was low. In contrast, the PE animals, with blunted tone-shock learning, usually continued to drink during the tone presentation; t2 was low and the SR was high. Induction of LI was concluded when the SR of the PE group was significantly higher (P < .05 after a two-way ANOVA) than that of the NPE group receiving the same drug treatment. For studies of drug-induced LI, animals were administered with drugs or vehicle 60 min before the PE and conditioning sessions. For studies of the blockade of amphetamine-induced disruption of LI, both drug (or vehicle) injections were followed 45 min later by an amphetamine (1.5 mg/kg, s.c.) or vehicle administration. Data were analyzed by ANOVA.
Electroencephalographic power spectra.
The method employed was essentially as that detailed previously (Shvaloff et al., 1988). Male Wistar rats (400–450 g) were anaesthetized with chloral hydrate (350 mg/kg, i.p.) and placed in a stereotaxic apparatus. Two (left and right) bipolar, transcortical electrodes were implanted in the prefrontal cortex (A + 4 mm, L = ±2.5 mm, with bregma as 0). After a 10-day recovery period and progressive habituation to the restraining device in the EEG recording chamber, the animals were used for EEG recording. The rats were administered either vehicle (day 1) or drug (day 2) and placed in the recording chambers. The EEG was recorded over 5 min, 60 min postadministration. The EEG signals were amplified, low-pass filtered (−90 dB/30 Hz), digitized (64 pts/sec) and transformed (Fourrier analysis on 2-sec epoch samples) resulting in an average power spectrum over the 18 to 30 Hz frequency range. The EEG profile of drugs was expressed (ratio) relative to that of their vehicle controls. EEGs obtained from left and right cortical electrodes were averaged for presentation.
Drug discrimination.
Food-deprived rats were trained to discriminate DOI (0.63 mg/kg i.p.), S 16924 (2.5 mg/kg i.p.) or clozapine (5.0 mg/kg i.p.) from saline with a standard two-lever, fixed-ratio 10, food-reinforced operant procedure according to the method of Schreiber et al. (1994). Each daily session started 15 min (DOI and S 16924 groups) or 30 min (clozapine group) after drug or saline injection and was terminated after 15 min. Drug or saline sessions alternated randomly. The discrimination criterion consisted of 10 consecutive sessions with correct responding,i.e., no more than 13 responses on both the reinforced and the nonreinforced levers before the first reinforcement was obtained. Then, test sessions were conducted every Wednesday and Friday, whereas training sessions continued on the other days (5 days/wk). Rats that responded incorrectly on the two most recent training days were submitted to an additional training session instead of a test session. During testing, responding on the selected lever, i.e., the lever on which 10 responses were recorded first, was reinforced for the remainder of the session. For agonist testing, compounds were substituted for the training drug and administered at the corresponding time before the session. For antagonist testing (DOI group), compounds were injected 45 min before the training drug, which was given 15 min before the session. Data recorded during a test session were lever selection and response rates, i.e., the total number of presses on both levers. Lever selection data were expressed as the percentage of rats selecting the drug lever and were analyzed by a Fisher exact probability test, with control values defined as 0% (agonist testing) or 100% (antagonist testing) drug lever selection. ED50s (95% CLs) were also calculated to estimate drug potency. Response rates were compared by a paired Wilcoxon signed rank test (P < .05) to those obtained during the preceding saline (agonist testing) or drug (antagonist testing) training session: percentages of inhibition of response rates and ID50s (95% CLs) were also calculated.
Induction of catalepsy.
Catalepsy was measured as previously (Millan et al., 1995a). Rats were placed in a position whereby the left and right hind-paw were placed over the ipsilateral fore-paws. The time over which this position was maintained was determined over a maximum of 30 sec (100% effect). Data were the mean of three measures separated by 1-min intervals. Drugs were injected 60 min before testing. Data were analyzed by ANOVA followed by Dunnett’s test and Active Dose (AD)50s (95% CLs) calculated to estimate drug potency.
Inhibition of haloperidol- or SCH 39166-induced catalepsy.
S 16924, clozapine or vehicle were administered to rats 30 min before the injection of either haloperidol (2.5 mg/kg, s.c.) or SCH 39166 (10.0 mg/kg, s.c.), which were given 30 min before testing. Catalepsy was measured as described above. Data were analyzed by ANOVA followed by Dunnett’s test and ID50s (95% CLs) calculated to estimate drug potency. In interaction studies of haloperidol-induced catalepsy, the 5-HT1A antagonist, WAY 100,635 or vehicle were injected 30 min before the administration of S 16924 or clozapine (10.0 mg/kg, s.c.). Data were analyzed by two-way ANOVA, followed by Newman-Keuls test.
Inhibition of haloperidol-induced catalepsy after chronic treatment with S 16924 or clozapine.
Rats were administered S 16924 (2.5 mg/kg, s.c.), clozapine (10.0 mg/kg, s.c.) or vehicle once a day for 14 days. On test day (day 15), these treatments were followed 30 min later by the injection of haloperidol (2.5 mg/kg, s.c.) and, after a further 30 min, by measurement of catalepsy. Data were analyzed by two-way ANOVA, followed by Newman-Keuls test.
Inhibition of methylphenidate-induced gnawing.
As previously (Millan et al., 1995b), rats were administered with methylphenidate (40.0 mg/kg, i.p.) and placed in transparent plexiglass observation cages (33.5 × 23.5 × 19.0 cm) with a grid-floor. Thirty min later, the number of periods (of 10) of gnawing was evaluated over 10 min (one 10-sec observation period/min). Under such conditions, methylphenidate yields a maximal response of 10. Drugs were administered 30 min before methylphenidate. Data were analyzed by ANOVA followed by Dunnett’s test and ID50s (95% CLs) calculated to estimate drug potency. In interaction studies, the 5-HT1A antagonist WAY 100,635 (0.63 mg/kg, s.c.) or vehicle were injected 30 min before the administration of S 16924 (10.0) or clozapine (40.0 mg/kg, s.c.). Data were analyzed by a two-way ANOVA, followed by Newman-Keuls test.
Induction of serotonergic behaviors.
Two behaviors typically elicited by selective and high efficacy 5-HT1A agonists, STFs and FBP, a component of the “serotonin syndrome,” were measured. As described previously (Millan et al., 1994), one STF was defined as the raising of the tail to a level higher than that of the body axis in rats loosely-restrained in horizontal cylinders and FBP was defined as a position with the hind-legs in extension and the abdomen closely apposed to the cage floor. The number of STFs elicited over 5 min and the number of rats per treatment group showing FBP were evaluated 30 min after s.c. injection of 8-OH-DPAT, S 16924, clozapine or haloperidol.
Binding.
Competition binding studies were performed at multiple histaminergic, muscarinic receptor types, as well as at NMDA, AMPA (dl-α-NH2-2,3-dihydro-5-methyl-3-oxo-4-isoxazolepropanoic acid), γ-amino butyric acid and benzodiazepine binding sites. Assay conditions are summarized in table 7 (see also Millan et al., 1995a). Isotherms were analyzed by nonlinear regression, using the program “PRISM” (Graphpad Software Inc., San Diego, CA) to yield IC50 values. Ki s were derived from IC50 values according to the Cheng-Prusoff equation: Ki = IC50/(1 + L/Kd ) where L is the concentration of radioligand and Kd is the dissociation constant of the radioligand.
Binding protocols and drug affinities (Ki) at multiple histaminergic (H), muscarinic (M) and other binding sites as compared to dopamine D2 receptors
Drugs.
All drugs were dissolved in sterile water with a few drops of lactic acid. The pH was adjusted to as close to neutrality as possible (>5.0). Unless otherwise specified, drugs were injected s.c. in a volume of 1 ml/kg. In general, full dose-response curves were performed for all studies. However, in view of limitations in drug solubility, absolute upper limits of 80.0 mg/kg were defined for S 16924 and clozapine. Drug structures, sources and salts were as follows: d-amphetamine sulfate (Calaire Chimie, Calais, France); apomorphine HCl, PCP HCl and haloperidol (Sigma Chimie, St Quentin-Fallavier, France); cocaine HCl (Coopérative Pharmaceutique Française, Melun, France); dizocilpine hydrogen maleate, (±)-8-OH-DPAT ((±)-8-dihydroxy-2-(di-n-propylamino) tetralin) HCl, (±) DOI (1-[2,5-dimethoxy-4-iodophenyl]-2-aminopropane) HCl, clozapine, quinpirole HCl and SKF 38393 [(±)-1-phenyl-2,3,4,5-tetrahydro-(1H)-3-benzazepine-7,8-diol] HCl (Research Biochemicals International, Natick, MA); methylphenidate HCl (Ciba-Geigy) and SCH 39166 ((−)-trans-6,7,7a,8,9,13b-hexahydro-3-chloro-2-hydroxyl-N-methyl-5H-benzo[d]-naphto-[2,1-benzazepine]) HCl (Schering Plough Corp., Bloomfield, IL). WAY 100,635 fumarate and S 16924 HCl were synthesized by J.-L. Peglion and G. Lavielle (Servier), respectively.
Results
Influence of S 16924, compared with clozapine and haloperidol, upon rotation in unilateral substantia nigra pars compacta-lesioned rats.
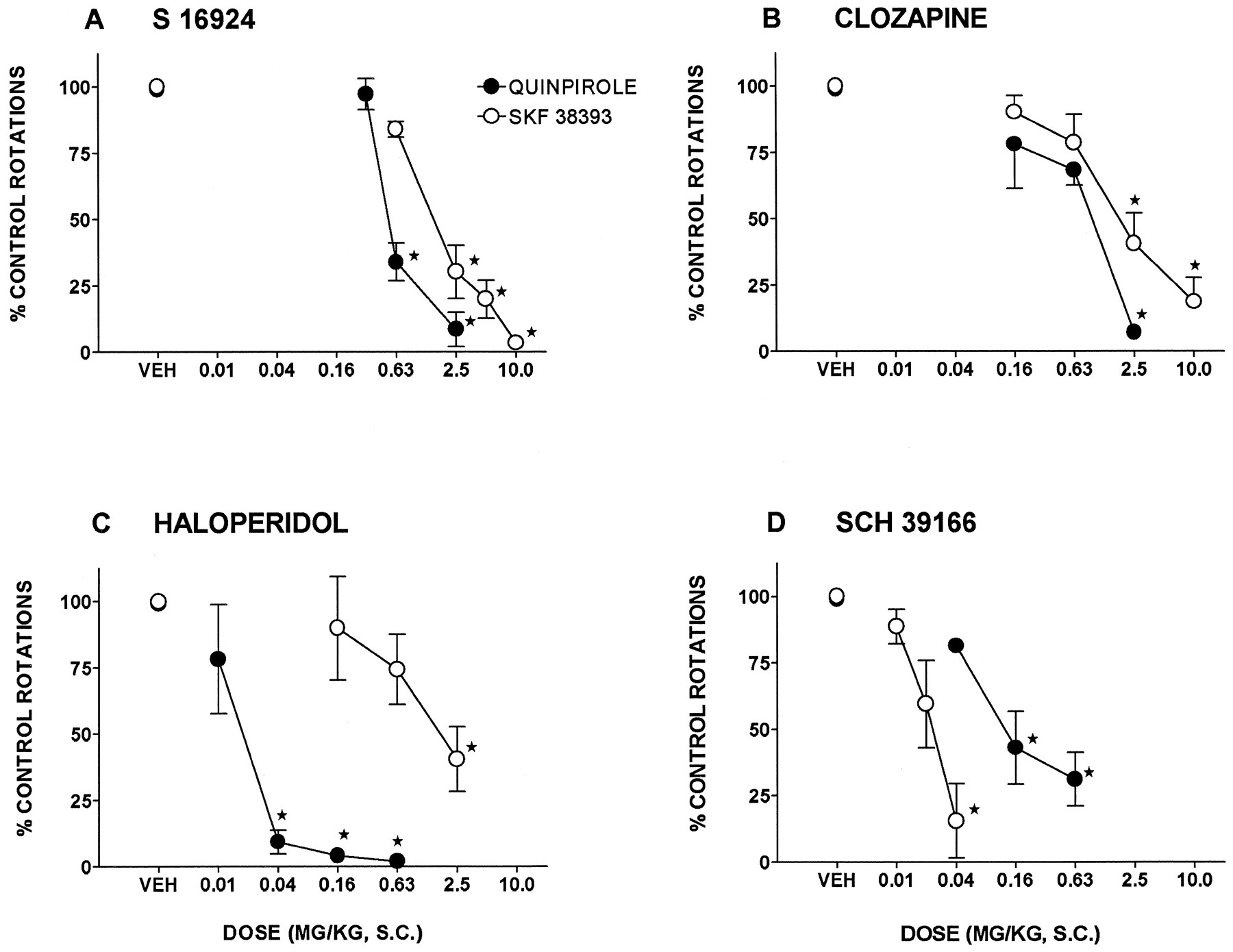
The preferential D2 receptor agonist, quinpirole, and the preferential D1 receptor agonist, SKF 38393, elicited a pronounced, contralateral rotation in rat sustaining unilateral lesions of the substantia nigra pars compacta. The selective D1 receptor antagonist, SCH 39166, potently blocked the action of SKF 38393 as compared to that of quinpirole with ID50s (95% CLs) of 0.02 (0.01–0.03) and 0.2 (0.04–0.9) mg/kg, s.c., respectively (fig. 1). In contrast, haloperidol preferentially and potently antagonized the action of quinpirole (fig. 1; table 1). S 16924 and clozapine, however, both dose-dependently and fully inhibited the induction of rotation by quinpirole and SKF 38393 with similar potencies (fig. 1; table 1).

Influence of S 16924 as compared to clozapine, haloperidol and SCH 39166 on the rotation elicited by quinpirole (0.02 mg/kg, s.c.) and SKF 38393 (0.63 mg/kg, s.c.) in rats sustaining unilateral lesions of the substantia nigra, pars compacta. A, S 16924; B, clozapine; C, haloperidol and D, SCH 39166. Data are means ± S.E.M.s, N ≥ 6 per value. Asterisks indicate significance of differences to respective vehicle values using a two-tailed, paired Student’s t test. *P < .05.
Influence of S 16924 as compared to clozapine and haloperidol in various procedures predictive of antipsychotic (antiproductive) properties
Influence of S 16924, compared with clozapine and haloperidol, in diverse models of potential antipsychotic (antipositive) activity.
The locomotion provoked by the psychostimulants, cocaine and dizocilpine, was dose-dependently and potently inhibited by haloperidol with clozapine exerting its activity over a much higher dose range (fig. 2; table 1). S 16924 was also dose-dependently active in these procedures with an intermediate potency (fig. 2; table 1). Similarly, in analogy to haloperidol and clozapine, and with intermediate potency, S 16924 both reduced the induction of climbing behavior by the dopaminergic agonist, apomorphine, and reduced avoidance responses in a CAR paradigm (fig. 2; table 1). Across all of these procedures, S 16924, haloperidol and clozapine exerted their actions with similar maximal effects (fig. 2; table 1).

Influence of S 16924 as compared to clozapine and haloperidol in various models of potential antipsychotic (antiproductive) activity. A, Inhibition of cocaine- (20 mg/kg, i.p.) induced hyperlocomotion; B, inhibition of dizocilpine- (0.16 mg/kg, s.c.) induced hyperlocomotion; C, inhibition of apomorphine- (0.75 mg/kg, s.c.) induced climbing and D, inhibition of conditioned avoidance responses. Data are means ± S.E.M.s, N≥ 6 per value. ANOVA as follows. A, S 16924, F (4,33) = 9.1, P < .001; clozapine, F (5,60) = 7.5, P < .001 and haloperidol, F (3,43) = 15.0, P < .001. B, S 16924, F (3,29) = 12.9, P < .001; clozapine, F (4,58) = 14.2, P < .001 and haloperidol, F (4,55) = 11.3, P < .001. Asterisks indicate significance of differences to vehicle values in Dunnett’s test following ANOVA (A and B), in the Fisher exact probability test (C) and in a paired Wilcoxon test (D). *P < .05.
Influence of S 16924, compared with clozapine and haloperidol, upon the locomotion elicited by amphetamine as compared to PCP.
The locomotion elicited by the psychostimulant, amphetamine, was blocked by haloperidol with a potency marginally greater than that required to inhibit the action of PCP, whereas clozapine displayed markedly greater potency against PCP than amphetamine (fig.3; and tables 1 and2). Similarly, S 16924 antagonized the action of PCP with far greater potency than that of amphetamine (fig.3; tables 1 and 2). After chronic, 2-wk administration of S 16924, its capacity to inhibit the locomotion elicited by PCP was not reduced; that is, no tolerance developed (table3).

Influence of S 16924 as compared to clozapine and haloperidol upon the hyperlocomotion elicited by amphetamine (2.5 mg/kg, i.p.) as compared to phencyclidine (PCP) (20.0 mg/kg, s.c.). A, S 16924; B, clozapine and C, haloperidol. Data are means ± S.E.M.s, N ≥ 6 per value. ANOVA as follows. Hyperlocomotion, amphetamine: S 16924, F (3,28) = 18.2, P < .001; clozapine F (3,63) = 8.6, P < .001 and haloperidol, F (4,67) = 13.5, P < .001. Hyperlocomotion, PCP: S 16924, F (4,34) = 8.4, P < .001; clozapine, F (4,40) = 8.3, P < 0.001 and haloperidol, F (5,53) = 5.5, P < .001. Asterisks indicate significance of differences to vehicle values in Dunnett’s test following ANOVA. *P < .05.
Influence of S 16924 as compared to clozapine and haloperidol in various tests of antipsychotic properties involving serotonergic and/or dopaminergic activity
Influence of chronic administration of S 16924 upon its ability to inhibit the locomotion provoked by phencyclidine (PCP)
Influence of S 16924, compared with clozapine and haloperidol, upon the actions of DOI.
S 16924, clozapine and haloperidol all potently inhibited the induction of HTWs by the hallucinogen, DOI (fig.4; and table 2). Over a similar dose-range, both S 16924 and clozapine also inhibited the DS properties of DOI, whereas haloperidol was inactive in this model (fig. 4; table2). However, haloperidol, in distinction to S 16924 and clozapine, produced a marked reduction in response rates and doses higher than 0.16 could not be tested (table 2). The doses of S 16924 and clozapine active in the DOI-induced HTW and DS procedures were, like those blocking PCP-locomotion, low as compared to those required to inhibit amphetamine-induced locomotion (figs 2 and 4; tables 1 and 2). This was not the case for haloperidol (figs 2 and 4; tables 1 and 2).

Influence of S 16924 as compared to clozapine and haloperidol upon the head-twitches and a discriminative stimulus elicited by DOI. A, Head-twitches elicited by DOI (2.5 mg/kg, i.p.). Data are means ± S.E.M.s, N ≥ 6 per value. ANOVA as follows: S 16924, F (3,46) = 7.9, P < .001; clozapine, F (5,44) = 9.8, P < .001 and haloperidol, F (4,30) = 6.8, P < .001. Asterisks indicate significance of differences to vehicle values in Dunnett’s test following ANOVA. *P < .05. B, Discriminative stimulus elicited by DOI (0.63 mg/kg, i.p.). Data are percentage of animals selecting the DOI lever. N = 4 to 10 per value. Asterisks indicate significance of differences to control values (100% drug lever selection) in the Fisher exact probability test. *P < .05.
Influence of S 16924, compared with clozapine and haloperidol, in S 16924 and clozapine drug discrimination paradigms.
In rats trained to recognize a DS elicited by clozapine (5 mg/kg, i.p.), dose-dependent and full generalization was obtained with clozapine itself, as well as with S 16924 at somewhat lower doses (fig.5; table 2). In contrast, haloperidol did not generalize to clozapine up to doses that suppressed response rates (fig. 5; table 2). A similar pattern of data was acquired in rats trained to recognize S 16924 (2.5 mg/kg, i.p.) where S 16924 and, less potently, clozapine dose-dependently and fully generalized (fig. 5; table 2). In contrast, haloperidol only partially generalized even up to doses which very markedly depressed response rates (fig. 5; table2). The potent motor-suppressant actions of haloperidol were underpinned by studies of its influence upon spontaneous motor behavior in rats and mice. This was decreased by haloperidol with ID50s (95% CLs) of 0.03 (0.01–0.06) and 0.2 (0.1–0.5) mg/kg, s.c., respectively. S 16924 and clozapine also decreased spontaneous motor activity, although only over higher doses ranges. S 16924, rats: 0.9 (0.4–2.3) and mice: 0.6 (0.1–5.0) and clozapine: rats, 6.0 (2.5–14.1) and mice: 1.1 (0.3–3.5) mg/kg, s.c. These active doses in rats are similar to their rate-suppressant doses in the clozapine DS model (table 2).

Generalization of S 16924 as compared to clozapine and haloperidol to discriminative stimuli elicited by clozapine (5 mg/kg, i.p.) and S 16924 (2.5 mg/kg, i.p.). N = 5 to 7 per value. Doses are in mg/kg, s.c. (filled symbols) or i.p. (open symbols). Upper panels, Drug lever selection. Data are percentage of animals selecting drug lever. Asterisks indicate significance of differences to control values (100% drug lever selection) in the Fisher exact probability test. *P < .05. Lower panels, Response rates. Data are means ± S.E.M.s of percentage of vehicle response values obtained during the most recent training session. Asterisks indicate significant decreases in response rates as compared to vehicle in the paired Wilcoxon test. *P < .05.
Influence of S 16924, compared with clozapine and haloperidol, upon EEG power spectra obtained from the FCX.
There was a marked similarity in the EEG power spectra obtained from the FCX as concerns S 16924 and clozapine, which both elicited a pronounced and distinctive peak over 7 to 8 Hz (fig. 6). In distinction, haloperidol produced a modest and broad shoulder over 10 to 14 Hz (fig. 6).

Influence of S 16924 as compared to clozapine and haloperidol upon electroencephalographic (EEG) power spectra from the frontal cortex. Data are the means of the percentages «spectral energy drug/vehicle» (ordinates) over the 1 to 30 Hz range of frequency (abscissae) obtained 1 hr after drug or vehicle administration, from a 5-min EEG recording period.
Influence of S 16924, compared with clozapine and haloperidol, in a LI paradigm.
Under the 10 tone PE condition (fig.7), SRs in vehicle-injected rats were slightly, but not significantly, higher in the PE as compared to NPE groups. In the presence of S 16924 (0.08 mg/kg, s.c.) and clozapine (0.16), but not haloperidol (0.0063), the difference between NPE and PE conditions became significant inasmuch as the SRs of the NPE animals was increased without an alteration in the scores of PE rats. That is, the conditioning of the emotional response was not modified per se. Haloperidol was also inactive at a dose of 0.04 mg/kg (not shown). In the 40 tone PE condition (fig. 8), significant LI was obtained across all treatments and in S 16924- and vehicle-treated animals individually, although significance was just missed for the clozapine and haloperidol groups. Amphetamine (1.5 mg/kg) disrupted LI by reducing SRs of the PE group almost to the level of the NPE group. At high doses, corresponding to those active in the models of antipositive activity described above (table 1), S 16924, clozapine and haloperidol inhibited disruption of LI by amphetamine (fig. 8).
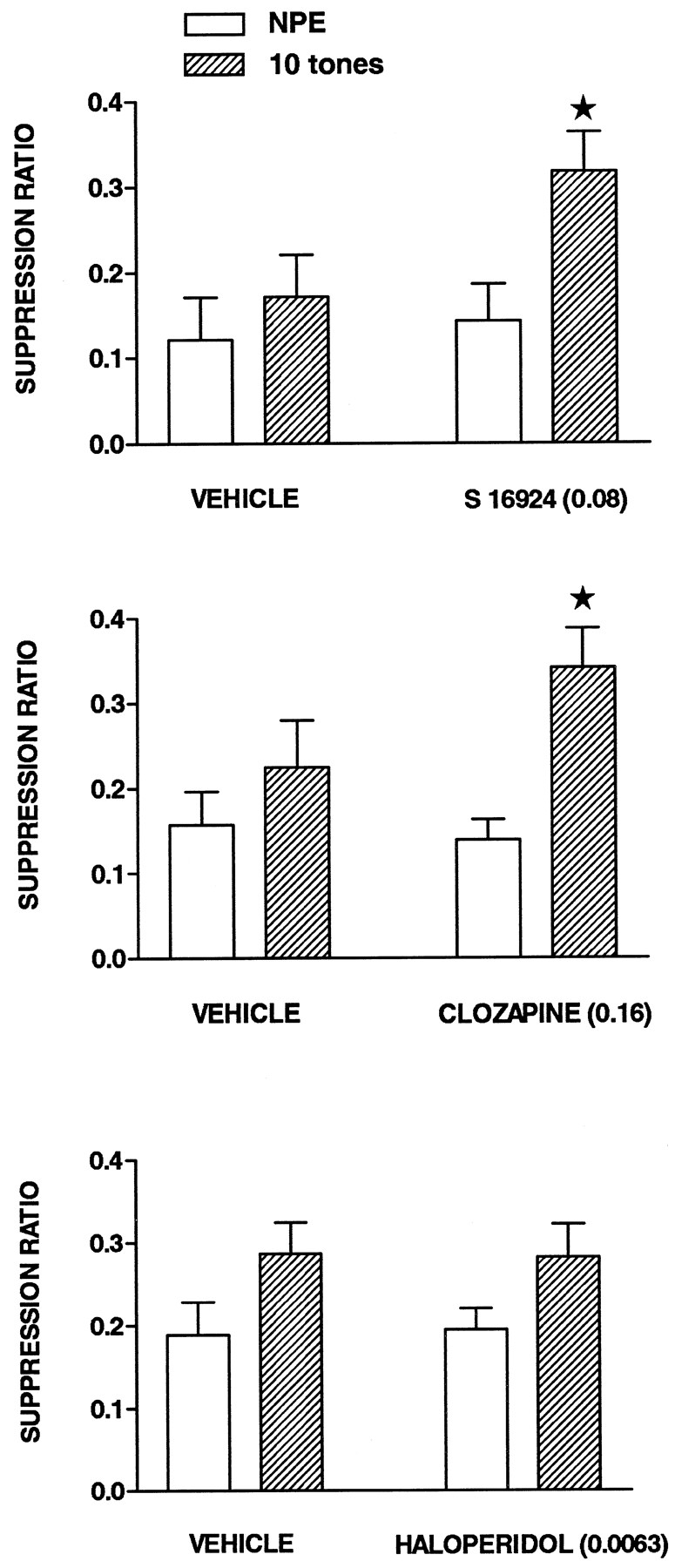
Induction of latent inhibition by S 16924 as compared to clozapine and haloperidol. Drugs were administered 60 min before pre-exposure and conditioning, in each case. Values represent suppression ratios for rats either pre-exposed to 10 tones or not (NPE) and are means ± S.E.M.s, N = 6 to 20 per value. Two-way ANOVA revealed a significant influence of pre-exposure: S 16924, F (1,31) = 5.7, P < .05; clozapine, F (1,34) = 9.1, P < .01 and haloperidol, F (1,74) = 6.5, P < .05. Asterisks indicate significance of differences, in a post hoccomparison, between values of NPE and 10 tone groups for a common drug treatment. *P < .05.

Inhibition by S 16924 as compared to clozapine and haloperidol of the disruption of latent inhibition by amphetamine (1.5 mg/kg). A, Vehicle and B, amphetamine. Drugs and amphetamine were injected 60 and 15 min, respectively, before the preexposure and the conditioning sessions. Values represent suppression ratios for rats either preexposed to 40 tones or not (NPE) and are means ± S.E.M.s, N > 9 per value. A two-way ANOVA performed on control (vehicle) groups (first two columns of A and B) yielded a significant influence of preexposure: F (1,182) = 14.9, P < .001; amphetamine F (1,182) = 62.9, P < .001 and a significant preexposure × amphetamine interaction: F (1,182) = 5.2, P < .05. Post hoc comparisons between the NPE and 40 tone groups yielded, in the presence of vehicle, significant LI (P < .01) but not in the presence of amphetamine (P > .05). A two-way ANOVA (preexposure × drug treatment) yielded a significant influence of pre-exposure: A, F (1,151) = 15.9, P < .001 and B, F (1,158) = 24.1, P < .001. Asterisks indicate significance of differences, in a post hoc comparison, between values of NPE and 40 tones groups for vehicle, S 16924, clozapine or haloperidol groups receiving vehicle or amphetamine. *P < .05.
Induction and inhibition of catalepsy by S 16924, compared with clozapine and haloperidol.
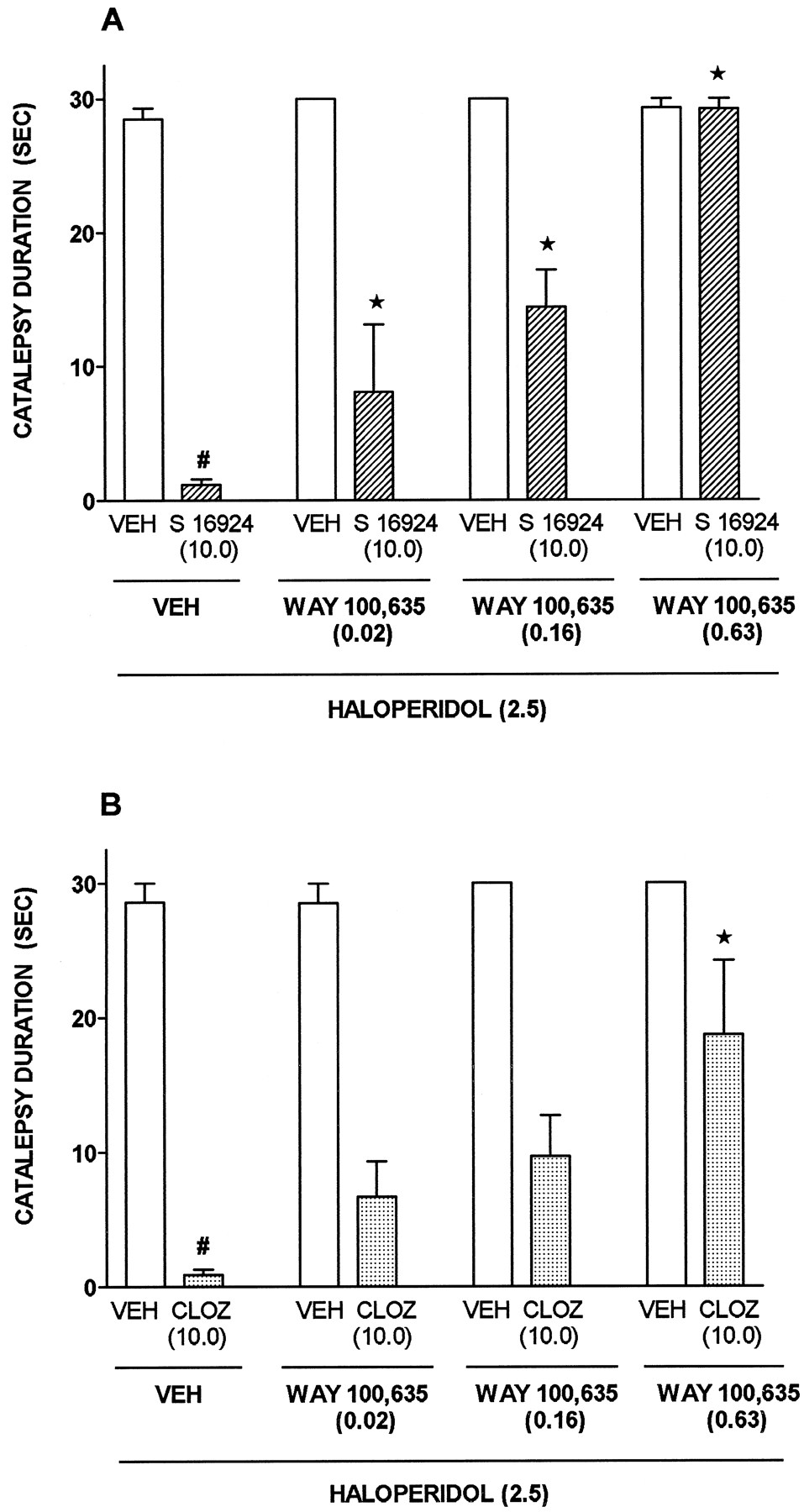
Whereas haloperidol and SCH 39166 [AD50 = 0.8 (0.2–3.2 mg/kg, s.c.)] elicited catalepsy, both S 16924 and clozapine were inactive (fig.9; table4). Indeed, S 16924 and clozapine antagonized induction of catalepsy by haloperidol with ID50s (95% CLs) of 3.2 (1.5–6.7) and 5.5 (1.4–21.4 mg/kg, s.c.) respectively (fig. 9). Similarly, both S 16924 [1.5 (0.9–2.6)] and clozapine [1.8 (0.3–0.4)] blocked induction of catalepsy by SCH 39166 (fig. 9). The influence of S 16924 against haloperidol-induced catalepsy was dose-dependently prevented by the selective 5-HT1A receptor antagonist, WAY 100,635, whereas it only partially interfered with the action of clozapine (fig.10). After 2-wk chronic administration of S 16924 or clozapine, their ability to interfere with the induction of catalepsy by haloperidol was not diminished (table5).

Induction and modulation of catalepsy by S 16924 as compared to clozapine, haloperidol and SCH 39166. A, Induction of catalepsy; B, Inhibition of the cataleptic action of haloperidol (2.5 mg/kg, s.c.) and C, inhibition of the cataleptic action of SCH 39166 (10.0 mg/kg, s.c.). Data are means ± S.E.M.s, N≥ 6 per value. ANOVA as follows: A, haloperidol, F (4,43) = 58.3, P < .001; SCH 39166, F (5,34) = 21.0, P < .001; S 16924, F (3,20) = 0.18, P > .05 and clozapine, F (3,20) = 0.10, P > .05. B, S 16924, F (4,39) = 19.6, P < .001 and clozapine, F (5,39) = 6.6, P < .001. C, S 16924, F (4,35) = 19.1, P < .001 and clozapine, F (4,35) = 6.2, P < .001. Asterisks indicate significance of differences in Dunnett’s test following ANOVA. *P < .05.
Induction by S 16924 as compared to clozapine and haloperidol of catalepsy and PRL secretion

Attenuation by WAY 100,635 of the ability of S 16924 (10.0 mg/kg, s.c.) and clozapine (10.0 mg/kg, s.c.) to inhibit the cataleptic action of haloperidol (2.5 mg/kg, s.c.). A, S 16924 and B, Clozapine (CLOZ). Data are means ± S.E.M.s, N ≥ 5 per value. Two-way ANOVA as follows. A, WAY 100,635, F (3,50) = 31.9, P < .001; S 16924, F (1,50) = 330.8, P < .001 and interaction, F (3,50) = 27.8, P < .001. B, WAY 100,635, F (3,28) = 5.08, P < .01; CLOZ, F (1,28) = 104.4, P < .001 and interaction, F (3,0) = 2.95, P = 0.05. Symbols (#) indicate significance of differences of VEH/S 16924 and VEH/CLOZ to VEH/VEH values and asterisks indicate significance of differences to respective VEH/S 16924 values in Newman-Keuls test after ANOVA. # and *P < .05.
Influence of chronic administration of clozapine or of S 16924 upon their ability to inhibit the induction of catalepsy by haloperidol
Inhibition of methylphenidate-induced gnawing by S 16924, compared with clozapine and haloperidol.
The stereotyped gnawing elicited by methylphenidate (40.0) was dose-dependently and completely blocked by haloperidol and, less potently, by S 16924 and clozapine (fig.11), with ID50s (95% CLs) of 0.04 (0.02–0.06), 8.4 (5.4–13.1) and 19.6 (9.9–38.5) mg/kg, respectively. The ability of S 16924 to block gnawing was prevented by WAY 100,635 (fig. 11). In contrast, WAY 100,635 did not modify the inhibition of gnawing by clozapine (fig. 11).

Inhibition of methylphenidate-induced gnawing by S 16924 as compared to clozapine, haloperidol and SCH 39166, and blockade of the action of S 16924 but not clozapine by WAY 100,635. A, Inhibition of methylphenidate- (40.0 mg/kg, i.p.) induced gnawing and B, Inhibition of the antignawing action of S 16924 (10.0 mg/kg, s.c.) as compared to clozapine (10.0 mg/kg, s.c.) by WAY 100,635 (0.63 mg/kg, s.c.). Data are means ± S.E.M.s, N ≥ 5 per value. A, ANOVA as follows. S 16924, F (4,37) = 17.1, P < .001; clozapine, F (3,31) = 9.97, P < .001 and haloperidol, F (3,25) = 87.4, P < .001. B, Two-way ANOVA as follows. S 16924, F (1,16) = 34.7, P < .001; WAY 100,635, F (1,16) = 42.6, P < .001 and interaction, F (1,16) = 34.7, P < .001. Clozapine, F (1,16) = 176.1, P < .001; WAY 100,635, F (1,16) = 0.09, P = .76 and interaction, F (1,16) = 0.09, P = .76. In A, asterisks denote significance of differences to respective vehicle values. In B, symbols (#) denote significance of differences of vehicle/S 16924 to vehicle/vehicle values and asterisks denote differences of WAY 100,635/S 16924 to vehicle/S 16924 values in Newman-Keuls test following ANOVA. # and *P < .05.
Influence of S 16924, compared with clozapine and haloperidol, upon PRL levels in the systemic circulation.
Haloperidol dose-dependently and potently elicited a marked increase in circulating levels of PRL (table 4). In distinction, clozapine elicited only a mild increase in PRL levels, even at a high dose (fig. 12; table4). S 16924 also only increased PRL levels at a high dose relative to that of haloperidol and its maximum effect was inferior to that of haloperidol, although greater than that of clozapine (fig. 12; table4).

Influence of S 16924 as compared to colzapine and haloperidol upon levels of PRL in the systemic circulation. Data are means ± S.E.M. N > 6 per value. ANOVA as follows: S 16924, F (6,75) = 13.4, P < .01; clozapine, F (6,86) = 6.2, P < .01 and haloperidol, F (5,47) = 18.7, P < .01. Asterisks indicate significance of differences in Dunnett’s following ANOVA. *P < .05.
Influence of S 16924, compared with clozapine and haloperidol, upon 5-HT1A receptor-mediated motor behaviors.
In contrast to 8-OH-DPAT, a high efficacy 5-HT1A receptor agonist, S 16924, clozapine and haloperidol all failed to elicit either FBP or STFs (table 6). S 16924, clozapine and haloperidol all potently inhibited the induction of STFs by 8-OH-DPAT (table 6).
Influence upon spontaneous tail-flicks (STFs) and flat-body posture (FBP) of S 16924 as compared to clozapine and haloperidol
Interaction of S 16924, compared with clozapine and haloperidol, at muscarinic and histaminic receptors.
Clozapine displayed marked affinity for native histamine1 receptors and for native and cloned, human M1, M2, M3 and M4 receptors (table 7). Analogous to haloperidol, the affinity of S 16924 for muscarinic receptors was negligible and its affinity at H1 receptors was much lower than that of clozapine. The affinity of S 16924 for benzodiazepine, gabaergic (GABAA and GABAB), NMDA and AMPA receptors was also negligible (>1 μM) (table 7).
Discussion
Receptor profiles of S 16924 as compared to clozapine and haloperidol in relationship to their functional actions.
As described in the companion paper, S 16924 possesses a pattern of interaction at multiple dopaminergic, serotonergic and adrenergic receptors that differs markedly to that of haloperidol and that closely resembles that of clozapine (table 8). In particular, modest and equilibrated affinity for D2 and D1 receptors and more pronounced affinity at D4, α1-AR, 5-HT2A and 5-HT2Cvs. D2 receptors. In addition, S 16924 may be distinguished from both haloperidol and clozapine by its potent, partial agonist properties at 5-HT1A receptors. The following discussion illustrates the importance of the distinctive receptorial profile of S 16924 in determining its functional actions in vivo.
Affinities of S 16924 as compared to clozapine and haloperidol at specific monoaminergic receptors
Potential antipositive properties of S 16924: significance of D2 receptor blockade.
In line with previous studies, and reflecting its potent blockade of postsynaptic D2receptors in limbic structures, haloperidol was highly active in each of the tests predictive of potential antipositive activity (Brunelloet al., 1995; Kahn and Davis, 1995). Similarly in line with previous work, clozapine was active in each paradigm, although only at substantially higher doses, in correspondence with its lower affinity at D2 receptors (Brunello et al., 1995; Meltzer, 1995). The activity of haloperidol and clozapine in these models is consistent with their clinical efficacy over, respectively, low and high dose ranges in controlling the positive symptoms of schizophrenia (Kane and Freeman, 1994; Meltzer, 1995; Pickar, 1995). In accordance with its intermediate affinity for D2 receptors, S 16924 also manifested robust and dose-dependent activity in each of these models over a dose-range lying between those of haloperidol and clozapine (Maurel-Remy et al., 1995a and b). These data support, then, the hypothesis that S 16924 should be effective in controlling the positive symptoms of schizophrenia via antagonist actions at D2 receptors. The robust inhibition by S 16924 of hyperlocomotion induced by the NMDA receptor channel blocker, dizocilpine, is of particular interest inasmuch as a reduction in glutamatergic transmission may be involved in the pathogenesis of schizophrenia (Bartha et al., 1997; Evins et al.,1997). Further, clozapine reduces the psychosis elicited by a further open channel blockade at NMDA receptors, ketamine, in man (Malhotraet al., 1997). Notwithstanding the importance of D2 receptor blockade, selective antagonists at D1 receptors, such as SCH 39166, are also potently active in the models used herein. Further, D1 receptor antagonism may enhance the antipsychotic effects of D2 receptor blockade although, clinically, selective blockade of D1receptors does not appear to be sufficient for antipsychotic activity (Josselin et al., 1997; Martin et al., 1994;Pickar, 1995). Indeed, as indicated by the rotation model employed herein (table 1), in contrast to haloperidol, S 16924 and clozapine both exert antagonist properties at D1 receptors at doses close to those blocking D2 receptors. Further, the selective α1-AR antagonist, prazosin, similarly attenuates the hyperlocomotion provoked by dizocilpine and other psychostimulants (Blanc et al., 1994; Mathé et al., 1996). These observations suggest that the antagonist properties of S 16924 and clozapine at D1 and α1-AR receptors may also contribute to their potential antipositive actions. Indeed, it might be conjectured that combined D2, D1 and α1-AR receptor blockade, a property shared by clozapine and S 16924, may improve efficacy in patients who respond poorly to selective D2receptor blockade with haloperidol (Prinssen et al., 1994). Finally, it has been suggested that stimulation of 5-HT1Areceptors, although poorly effective alone, may enhance the antipositive effects of D2 receptor blockade (Evenden, 1992). This observation is evidently of pertinence to S 16924, a potent 5-HT1A partial agonist, although we have not, as yet, acquired evidence that this property contributes to its actions in the models of antipositive activity summarized in table 1.
Preferential blockade of PCP-induced hyperlocomotion and DOI-induced HTWs by S 16924: importance of 5-HT2Aantagonist properties.
In contrast to amphetamine, administration of PCP to normal subjects mimics the negative-cognitive as well as the positive symptoms of schizophrenia (Gorelick and Balster, 1995;Steinpreis, 1996). We have hypothesized that preferential blockade of PCP- vs. amphetamine-induced locomotion may suggest a clozapine-like ability to control negative symptoms (Maurel-Remyet al., 1995a and b). Indeed, our study confirms the greater sensitivity of PCP- as compared to amphetamine-induced (and spontaneous) locomotion to clozapine, but not haloperidol, and a similar profile was presented by S 16924. This differential responsiveness of PCP- as compared to amphetamine-, cocaine- and dizocilpine-induced locomotion to clozapine and S 16924 may reflect the contrasting mechanisms underlying their induction of locomotion. Thus, amphetamine-, cocaine- and dizocilpine-induced locomotion is mediated (independently of 5-HT2A receptors) by nucleus accumbens-localized D2 receptors (Jackson et al., 1994; Maurel-Remy et al., 1995a and b; Moore and Kenyon, 1994). In distinction, although dependent on dopaminergic mechanisms for its expression, PCP-evoked hyperlocomotion involves serotonergic mechanisms. Indeed, it may involve the activation of nucleus accumbens-localized 5-HT2A receptors by an increase in extracellular 5-HT levels: this results from an interference by PCP of 5-HT reuptake processes (Jackson et al., 1994; Kehneet al., 1996; Maurel-Remy et al. 1995a and b;Moore and Kenyon, 1994). Correspondingly, the more potent 5-HT2Avs. D2 antagonist properties of S 16924 (table 8) and clozapine likely underlies their preferential blockade of the actions of PCP as compared to amphetamine (Maurel-Remyet al., 1995a). S 16924 and clozapine were also highly potent in blocking induction of HTWs by the hallucinogen, DOI and, in analogy to PCP-induced locomotion, 5-HT2A receptors mediate the induction of HTWs by DOI, although this population of 5-HT2A sites is localized in the FCX (Schreiber et al., 1995; Willins and Meltzer, 1997). Thus, a common feature of PCP-induced hyperlocomotion and DOI-induced HTWs is their mediation by 5-HT2A receptors and sensitivity to the potent 5-HT2A antagonist properties of S 16924 and clozapine. These data are, thus, consistent with the suggestion that, like clozapine, and by virtue of its potent 5-HT2A antagonist properties, S 16924 may control the negative symptoms of schizophrenia in which a dysfunctionment of serotonergic as well as dopaminergic transmission has been implicated (Martin et al., 1997;Meltzer, 1995; Svensson et al., 1995).
DS properties of S 16924.
As mentioned above, DOI behaves as an agonist at 5-HT2A receptors, stimulation of which underlies its DS properties in rats (Schreiber et al.,1994). Correspondingly, in line with their potent 5-HT2Aantagonist properties, S 16924 and clozapine, but not haloperidol, abolished the DS properties of DOI. A direct comparison of the DS properties of S 16924 and clozapine revealed that, in clozapine-trained animals, S 16924 showed complete (≥80%) generalization suggesting that animals share DS properties with those of clozapine. This interpretation is reinforced by the complete (≥80%) generalization of clozapine in animals trained to recognize S 16924 itself. In distinction, in line with previous studies, haloperidol achieved no more than partial (≤50%) generalization to clozapine or to S 16924 (Carey and Bergman, 1997; Nielsen, 1988). Thus, S 16924 and clozapine clearly share similar DS properties, whereas those of haloperidol are different. It has proven difficult to identify the receptorial interactions underlying the DS properties of clozapine, although a role of muscarinic receptors (in rats) and 5-HT2C receptors (in pigeons) has been proposed (Hoenicke et al., 1992; Nielsen, 1988). The present data are consistent with the latter possibility inasmuch as S 16924 is a potent ligand at 5-HT2C—but not muscarinic receptors (companion paper and tables 7 and 8). In addition, complementary studies (Dekeyne A and Millan MJ, unpublished observations) have indicated that antagonist properties at 5-HT2A and 5-HT2C receptors may be involved in the DS properties of S 16924 and clozapine in rats. Nevertheless, multiple receptorial interactions likely underlie the complex DS properties of clozapine (Carey and Bergman, 1997) and S 16924. Notably, the actions of S 16924 and clozapine in these DS paradigms were expressed at doses lower than those disrupting response rates, in line with their lack of cataleptic activity (see below).
EEG profile of S 16924.
Although it is not possible to specify which cerebral structures and neuronal circuits are implicated in the DS properties of S 16924 and clozapine, it is not unreasonable to assume that both limbic and cortical regions are involved in view of their rich monoaminergic innervation and importance in the actions of antipsychotic agents. Thus, it is of interest that S 16924 and clozapine exerted a similar influence on EEG power spectra derived from the FCX, whereas the action of haloperidol was clearly different. It is difficult to attribute particular EEG complexes to interactions at specific receptor types. However, in complementary studies (Sebban C,et al., unpublished observations) the D2/D3 antagonist, raclopride, reproduced the profile of haloperidol, consistent with a role of antagonist properties at D2 sites, whereas the profile of S 16924 and clozapine was mimicked by 5-HT2A antagonists, consistent with a role of antagonist actions at 5-HT2A receptors. The similarity in the EEG power spectra of S 16924 and clozapine, as compared to haloperidol, obtained from the FCX is of particular significance inasmuch as a functional perturbation (“hypofrontality”) of this region is implicated in the negative and cognitive-attentional symptoms of schizophrenia (Andreasen et al., 1992; Knable and Weinberger, 1997).
Modulation of cognitive-attentional function: induction of LI by S 16924.
To more directly explore the possibility that S 16924 may improve cognitive-attentional performance, and increase the “filtering” of irrelevant sensory stimuli, a function compromised in schizophrenic patients, we examined its influence in a paradigm of LI: that is, the decrease in the conditioning power of a stimulus after its previous presentation (Weiner and Feldon, 1997). In the CER model used, SRs were slightly higher for animals preexposed (10 times) to an auditory stimulus during a drinking session as compared to those which had not been preexposed (Weiner and Feldon, 1997). In line with recent studies (Moran et al., 1996), clozapine enhanced the difference between these groups via a selective action on SRs in the latter, an action mimicked by S 16924. That is, in their presence, there was a significant LI, indicating that both S 16924 and clozapine may enhance the ability to ignore irrelevant sensory stimuli. In distinction, haloperidol was ineffective herein, although under certain conditions LI can also be induced by haloperidol (Moran et al., 1996; see Ruob et al., 1997 and Weiner and Feldon, 1997). The mechanisms underlying the potential ability of S 16924 and clozapine to improve cognitive-attentional function are not, as yet, clear. However, activation and blockade of 5-HT2A receptors can disrupt and induce LI, respectively (Hitchcock et al.,1997; Moser and Moran, 1994). Thus, 5-HT2A antagonist properties of S 16924 and clozapine may contribute, although 5-HT1A agonist properties of S 16924 are unlikely to be involved because 8-OH-DPAT does not induce LI (Gray et al.,1995; Millan MJ and Schreiber R, unpublished observations). Thalamic populations of α1-AR receptors inhibit the filtering of information en route to the cortex and prazosin blocks PCP-induced disruption in a pre-pulse inhibition model (Bakshi and Geyer, 1997;McCormick and Pape, 1990). Thus, α1-AR receptor blockade might also be involved in the actions of S 16924 and clozapine. Several arguments also favor a implication of D4 receptors blockade in the LI-inducing actions of S 16924 and clozapine. Thus, the hippocampus is implicated in mechanisms underlying LI (Gray et al., 1995) and D4 receptors are concentrated both therein as well as in the FCX and other structures controlling cognition (Primus et al., 1997). Further, selective D4 receptor antagonists induce LI in rats (Millan et al., in preparation). Finally, the ability of S 16924 and clozapine to reinforce mesocortical vs. mesolimbic dopaminergic transmission (companion paper) may be of significance inasmuch as a disruption of LI is associated with a decrease and increase, respectively, in the activity of these pathways (Broersenet al., 1996; Gray et al., 1995). Indeed, disruption of LI by amphetamine likely reflects an enhancement of mesolimbic dopaminergic transmission thereby activating D2receptors in the nucleus accumbens (Gray et al., 1995;Weiner and Feldon, 1997). Correspondingly, the ability of haloperidol and higher doses of S 16924 and clozapine to prevent disruption of LI by amphetamine presumably reflects antagonism of mesolimbic D2 receptors (Gray et al., 1995; Moran et al., 1996; Weiner and Feldon, 1997). It would be of interest to further explore the, potentially multiple, mechanisms involved in the control of LI and other cognitive functions by S 16924 and to determine its potential influence on LI and cognition in schizophrenic patients (Gray et al., 1995).
Low extrapyramidal potential of S 16924: importance of 5-HT1A agonist properties.
Haloperidol potently elicited catalepsy in rats, a response reflecting interruption of activity at D2 receptors in the striatum and predictive of an extrapyramidal motor syndrome in man (Cunningham-Owens, 1996;Hoffman and Donovan, 1995). In contrast, S 16924 did not elicit catalepsy suggesting that, in analogy to clozapine (Cunningham-Owens, 1996; Keks, 1996), its extrapyramidal potential is low. Indeed, clozapine and, more potently, S 16924 inhibited the induction of catalepsy by haloperidol and the D1 receptor antagonist, SCH 39166 (Bartoszyk et al., 1996; Cunningham-Owens, 1996;Kalkman et al., 1997). There are several mechanisms that may be relevant to this low extrapyramidal potential of S 16924 and clozapine. First, selective D1 or D2antagonists may elicit catalepsy by provoking a dysequilibrium in activity at striatal D1 and D2 sites (Brunelloet al., 1995; Josselin et al., 1997) and both S 16924 and clozapine, in contrast to haloperidol and SCH 39166, show balanced antagonist activity at D1 and D2receptors (table 1 and companion paper). Second, the anticholinergic properties of clozapine have been implicated in its low extrapyramidal potential although, in view of its low affinity for muscarinic receptors, this mechanism is not relevant to S 16924 (Brunello et al., 1995; Olianas et al., 1997; Zorn et al., 1994) (table 7). Third, a marked preference of antipsychotics for 5-HT2Avs. D2 receptors has been convincingly associated with a low extrapyramidal potential, a property share by both S 16924 and clozapine (Kehne et al., 1996;Roth and Meltzer, 1995; although see Wadenberg, 1996). Fourth, 5-HT1A agonists may counter the extrapyramidal properties of neuroleptic agents, likely reflecting activation of inhibitory 5-HT1A autoreceptors in raphe nuclei (Invernizzi et al., 1988). Indeed, S 16924 is a 5-HT1A receptor partial agonist and, in contrast to haloperidol, inhibits striatal turnover of 5-HT although only weakly augmenting striatal DA synthesis (companion paper). Such a pattern of actions has been correlated with a low extrapyramidal potential, although it is controversial as to whether a reduction in striatal serotonergic transmission plays a causal role in limiting the influence of D2 receptor antagonism upon induction of catalepsy (Andersen and Kilpatrick, 1996;Lucas et al., 1997). In any case, the selective 5-HT1A antagonist, WAY 100,635, dose-dependently abolished the anticataleptic actions of S 16924 against haloperidol demonstrating that this is its principal mechanism of action. This assertion was supported by a study of methylphenidate-induced gnawing, a behavior also involving activation of striatal population of D2receptors. The ability of S 16924 to block this response was also abolished by WAY 100,635, in line with data obtained with other 5-HT1A agonists (Kleven et al., 1996). Interestingly, WAY 100,635 was less effective in blocking the anticataleptic actions of clozapine and did not modify its antagonism of methylphenidate. This suggests that agonist actions at 5-HT1A receptors are less important to the low extrapyramidal potential of clozapine than that of S 16924 (Bartoszyket al., 1996; Kalkman et al., 1997; Klevenet al., 1996). Clozapine moderates extrapyramidal movement disorders (tardive dyskinesias) elicited by prior treatment with neuroleptic drugs (Cunningham-Owens, 1996; Lieberman et al.,1991). This clinical, antidyskinetic action of clozapine may correspond to its ability to block haloperidol-induced catalepsy. Thus, S 16924 might also be of utility in the control of dyskinesias due to antecedent neuroleptic treatment. Notably, even after chronic treatment, S 16924 (and clozapine) were still capable of blocking the cataleptic actions of haloperidol suggesting that their ability to reduce extrapyramidal effects may not adapt.
Limited impact of S 16924 upon PRL secretion.
In contrast to the increase in PRL levels provoked by haloperidol, S 16924 and clozapine elicited only a modest augmentation in PRL levels, even at high doses. This is of significance since hyperprolactinaemia is associated with ovulatory dysfunction, ammenorrhea and galactorrhea (Cunningham-Owens, 1996). The differential induction of PRL secretion by S 16924 and clozapine vs. haloperidol may likely be explained by the relatively weak affinity of the former at lactotrophic D2 receptors tonically controlling PRL release (Cunningham-Owens, 1996; Millan et al., 1995a). Interestingly, at least in a transfected cell line, D2receptors controlling PRL release may be “constitutively active” (Nilsson et al., 1996). That is, the induction of PRL secretion by haloperidol may reflect negative intrinsic activity at D2 sites rather than interruption of a tonic inhibitory control exerted by DA. Although this is an intriguing hypothesis, it is unlikely to fully account for the influence of drugs upon PRL secretionin vivo inasmuch as both haloperidol and clozapine behave as inverse agonists at cloned hD2 receptors (Hall and Strange, 1997). Currently, it has not been established whether S 16924 possesses negative efficacy at D2 sites, an issue justifying further investigation inasmuch as inverse agonist actions at various populations of D2 receptors might, in theory, also explain the induction of catalepsy and DA synthesis. Irrespective of the underlying mechanisms, these data collectively suggest that, as with clozapine, S 16924 may be distinguished from haloperidol as concerns its low potential for extrapyramidal motor and endocrine side-effects.
Absence of a serotonergic syndrome.
8-OH-DPAT and other high efficacy agonists at postsynaptic 5-HT1A receptors provoke a constellation of behaviors termed the “serotonin syndrome,” and including FBP (Tricklebank, 1985). They also elicit STFs, a behavior reflecting interference with sensory transmission in the dorsal horn of the spinal cord (Millan et al., 1994). S 16924 did not, however, elicit either FBP or STFs consistent with its partial agonist properties in a cellular model of coupling at postsynaptic h5-HT1A receptors (companion paper) and other in vivo models, such as induction of hypothermia, where the magnitude of the actions of S 16924 are less pronounced than those of full agonists (Millan MJ and Dekeyne, unpublished observations). Indeed, S 16924 fully and potently blocked the induction of STFs and FBP by 8-OH-DPAT, in analogy to other ligands possessing partial agonist properties at postsynaptic 5-HT1A receptors (Millanet al., 1994). However, as described previously (Millanet al., 1994, 1995b; Tricklebank, 1985), these behaviors are dependent on dopaminergic and adrenergic mechanisms for their expression, and the actions of S 16924 at dopamine D2 and α1-AR receptors may, thus, also contribute to its inhibition of STFs. This mechanism also likely accounts for the blockade of this behavior by clozapine and haloperidol. Indeed, the mutual involvement of 5-HT1A receptors, dopaminergic and adrenergic receptors in the modulation of various motor, endocrine and other functional parameters renders the “isolation” and definition of in vivo actions of S 16924 and other multireceptorial agents at 5-HT1A, D2 and other receptor types difficult (Millan et al., 1992, 1995b). A complementary and more direct approach to estimating relative potency at 5-HT1Avs. D2 receptors was undertaken in the companion paper. Thus, in line with itsca. 10-fold higher affinity at native, rat 5-HT1Avs. D2 receptors, S 16924 (WAY 100,635-reversibly) inhibited the electrical activity of (raphe-localized) serotonergic neurones at doses (ID50 = 0.02 mg/kg, i.v.) ca. 10-fold lower than those preventing the inhibition of the electrical activity of (ventrotegmental area) dopaminergic neurones by apomorphine (0.2 mg/kg, i.v.). This question of the relative occupation and expression of activity at 5-HT2A, D2 and other receptor types is, thus, a complex issue, which is currently being clinically addressed for S 16924 using the technique of Position Emission Tomography.
Side-effect profile of S 16924 vs. clozapine: low muscarinic and histaminic affinity.
S 16924 exhibited only modest affinity for H1 receptors, a finding of significance inasmuch as the potent interaction of clozapine at H1 sites underlies several, undesirable actions affecting patient compliance. That is, sedation, drowsiness, fatigue and weight gain (Cunningham-Owens, 1996). S 16924 displayed negligible affinity for multiple, muscarinic (M1–M4) receptors, an observation of importance inasmuch as the potent interaction of clozapine with specific muscarinic receptor types is associated with a disturbance of vision, gastrointestinal transit, urinary flow and cardiovascular function, including tachycardia due to vagal (M2) blockade (Brunello et al., 1995; Cunningham-Owens, 1996). Interestingly, in contrast to other antipsychotics possessing high muscarinic affinity, clozapine elicits hypersalivation (sialorrhoea) rather than a dry mouth, possibly due to an agonist action at M4 receptors (Zorn et al., 1994; but see Olianas et al., 1997). As concerns other side-effects of clozapine, seizure activity occurs in around 5% of patients, probably due to an interference with inhibitory or excitatory amino acid transmission in the hippocampus, temporal cortex and other regions (Geaney, 1995). In this light, it is of interest that S 16924 is devoid of affinity for GABAA, GABAB, benzodiazepine, NMDA and AMPA receptors. The most disquieting, adverse effect of clozapine is, nevertheless, agranulocytosis, which occurs inca. 1 to 2% of patients and which can be fatal (Cunningham-Owens, 1996). The particular chemical structure of clozapine, allowing for the generation of a nitrenium ion, has been implicated (Liu and Uetrecht, 1995). The contrasting structure of S 16924 as compared to clozapine should be underlined in this context.
A comparison to other antipsychotic agents.
Several other, novel antipsychotics have been described that possess more marked activity at serotonergic (notably, 5-HT2A) and adrenergic (α1-AR) as compared to dopaminergic receptors, including olanzapine and quetiapine, which are structurally related to clozapine, the benzoizoxazole, risperidone, and the indole derivative, sertindole (Pickar, 1995). Although these agents do not possess significant activity at 5-HT1A receptors, the benzothiazole, ziprasidone, displays high affinity for 5-HT1A receptors, at which it behaves as a partial agonistin vitro (Seeger et al., 1995). However, in contrast to S 16924, its affinity for 5-HT1A sites is not superior to that at D2 receptors, and agonist actions at 5-HT1A receptors are not a pronounced feature of itsin vivo profile of activity (Seeger et al., 1995; Millan MJ and Brocco M, unpublished observations). More recently, several novel antipsychotics have been documented that show significant activity at 5-HT1A receptors in vitro andin vivo. Of these, both 1192U90 and SM 9018 are of note inasmuch as, as with S 16924, they also display antagonist activity at 5-HT2A receptors (Jones-Humble et al., 1996;Kato et al., 1990). Nevertheless, their chemical structures and precise functional profiles clearly differ to those of S 16924. It will be of interest to establish to what extent activity at 5-HT1A receptors contributes to the clinical actions of S 16924 and other drugs in the management of schizophrenia.
Summary and Conclusions
In summary, S 16924 displays marked similarities to clozapine and differences to haloperidol as concerns its antipsychotic profile. The parallels between S 16924 and clozapine as regards both their receptorial and functional profiles, together with the distinctive and pronounced 5-HT1A agonist properties of S 16924, suggest that it may be able to control both the positive and the negative symptoms of schizophrenia in the relative absence of extrapyramidal symptoms. Further, schizophrenic patients frequently experience marked anxiety which exacerbates negative symptoms such as social withdrawal and which also worsens compliance. In this light, it is of note that S 16924, reflecting its 5-HT1A agonist properties, expresses pronounced anxiolytic properties in several experimental models (Dekeyne et al., 1997). In addition to the importance of its marked agonist actions at 5-HT1Areceptors, the overall “clozapine-like” profile of action of S 16924 may, in particular, be attributed to its potent antagonist activity at 5-HT2A and, possibly, α1-AR and D4, as compared to D2 receptors. However, S 16924 may be differentiated from clozapine by its weak interaction with muscarinic and histaminic receptors suggesting a low potential for autonomic side-effects. The potential antipsychotic properties of S 16924 are currently undergoing clinical evaluation.
Acknowledgments
The authors thank B. Denorme, S. Girardon, H. Gressier, S. Monneyron and S. Veiga for technical assistance and C. Langaney-Le Roy and M. Soubeyran for secretarial assistance.
Footnotes
-
Send reprint requests to: Dr Mark J. Millan, Institut de Recherches Servier, Centre de Recherches de Croissy, Psychopharmacology Department, 125 Chemin de Ronde, 78290 - Croissy-sur-Seine, Paris, France.
-
↵1 Current address: Hôpital Charles Foix, 7, avenue de la République, 94205-Ivry-Sur-Seine Cedex, France.
- Abbreviations:
- 5-HT
- serotonin
- CAR
- conditioned avoidance responses
- CER
- conditional emotional response
- DA
- dopamine
- DS
- discriminative stimulus
- EEG
- electroencephalographic
- FBP
- flat body posture
- FCX
- frontal cortex
- HTW
- head-twitches
- LI
- latent inhibition
- NPE
- non preexposed
- PCP
- phencyclidine
- PE
- preexposed
- PRL
- prolactin
- SR
- suppression ratio
- STF
- spontaneous tail-flicks
- PCP
- phencyclidine
- ID50
- inhibitory dose
- CL
- confidence limit
- ANOVA
- analysis of variance
- DOI
- 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane
- IC50
- inhibitory concentration
- Received February 4, 1998.
- Accepted May 2, 1998.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}