


Abstract
Tegoprazan, a novel potassium-competitive acid blocker (P-CAB), is a next-generation therapeutics developed for the treatment of acid-related gastrointestinal diseases such as gastroesophageal reflux disease (GERD) and peptic ulcers. In the present study, the in vitro and in vivo pharmacological properties of tegoprazan were compared with those of esomeprazole, a representative proton pump inhibitor. In vitro enzyme assays were performed using ion-leaky vesicles containing gastric H+/K+-ATPases isolated from pigs. The in vivo efficacies of tegoprazan were evaluated in rat models of GERD and peptic ulcer. Tegoprazan inhibited the activity of porcine H+/K+-ATPase with an IC50 value of 0.53 μM in a reversible manner, whereas esomeprazole showed weak and irreversible inhibition with an IC50 value of 42.52 μM. In a GERD model, tegoprazan showed dose-dependent efficacy in inhibiting esophageal injury and gastric acid secretion with an ED50 of 2.0 mg/kg, which was 15-fold more potent than that of esomeprazole. In peptic ulcer models, tegoprazan exhibited superior antiulcer activity compared with esomeprazole. The ED50 of tegoprazan in the naproxen-, ethanol-, and water-immersion restraint stress-induced peptic ulcer models were 0.1, 1.4, and 0.1 mg/kg, respectively. In the acetic acid–induced peptic ulcer model, the curative ratio of tegoprazan at 10 mg/kg was higher than that of esomeprazole at 30 mg/kg (44.2% vs. 32.7%, respectively), after 5 days of repeated oral administration. Thus, tegoprazan is a novel P-CAB that shows potent and reversible inhibition of gastric H+/K+-ATPase and may provide stronger efficacy compared with previous proton pump inhibitors.
Introduction
Acid-related diseases such as gastroesophageal reflux disease (GERD) and peptic ulcer disease (PUD) are common disorders in the upper digestive system associated with gastric acid secretion (Sachs et al., 2010). Drugs based on different mechanisms have been developed to control gastric acid secretion for the treatment of acid-related diseases or to alleviate the related symptoms (Burget et al., 1990). Treatments for acid-related diseases have been revolutionized by drugs that exhibit improved efficacy and pharmacokinetic properties based on a new mechanism of acid suppression (Inatomi et al., 2016). Histamine-2 receptor antagonists (H2RA) such as ranitidine are first-generation acid suppressants that have been widely used for more than 40 years (Feldman and Burton, 1990). However, the healing rates of H2RA in GERD and PUD are only 50% and 74%, respectively, mainly because of insufficient suppression of gastric acid secretion (Robinson, 2004). Proton pump inhibitors (PPIs), such as esomeprazole, are second-generation acid suppressants targeting gastric H+/K+-ATPase, the enzyme responsible for the final step of gastric acid secretion on the luminal side of gastric parietal cells (Rabon and Reuben, 1990). Because of its enhanced acid suppression effects, esomeprazole significantly increased the healing rate in GERD or PUD compared with H2RA (Edwards et al., 2006). Thus, PPIs became a standard therapy for various acid-related gastrointestinal disorders (Frazzoni et al., 2003; Robinson, 2005). However, PPI therapy exhibits some limitations because of its mechanism of action and drug properties (Fass et al., 2005). PPIs as prodrugs require the acid activation process to convert into active sulfenamide forms, which are responsible for irreversible inhibition of H+/K+-ATPase via covalent binding (Sachs et al., 1988; Nagaya et al., 1989; Shin and Sachs, 2002). Therefore, it is recommended that PPIs be taken before meals for the acid activation (Sachs et al., 1995). Moreover, because of the short plasma half-life of PPIs and the de novo cell surface insertion of H+/K+-ATPase, approximately 3–5 days are required to reach the maximum efficacy in gastric pH elevation (Katz et al., 2000; Ang and Fock, 2006). PPI efficacy is also affected by CYP2C19 polymorphism because most PPIs are substrates of CYP2C19 (Dammann and Burkhardt, 1999; Furuta et al., 2005). Indeed, a field study suggests that 40% of patients with GERD responded poorly to standard PPI therapy, possibly because of insufficient suppression of gastric acid secretion (Cicala et al., 2013).
To overcome these limitations of PPI therapy, a new class of acid suppressants known as potassium-competitive acid blockers (P-CABs) has been developed (Andersson and Carlsson, 2005). Unlike PPIs, P-CABs directly inhibit gastric H+/K+-ATPase in a K+-competitive and reversible manner, and thus do not require the acid activation process for inhibition (Vakil, 2004). Prototype P-CABs such as SCH28080 or AZD0865 are based on the imidazolopyridine structure and exhibit characteristics typical to P-CABs, including potent and reversible K+ competitive inhibition of H+/K+-ATPase with rapid onset of action (Beil et al., 1986). However, the development of SCH28080 and AZD0865 was suspended because of hepatotoxicity concerns (Parsons and Keeling, 2005). Revaprazan, based on the pyrimidine-2 amine structure, is the first P-CAB marketed in South Korea. However, the acid suppression ability and gastric pH >4.0 holding time of revaprazan are not superior to conventional PPIs (Kim et al., 2010). Vonoprazan, a pyrrole derivative recently marketed in Japan, exhibits superior acid suppression compared with PPIs (Garnock-Jones, 2015). However, safety concerns about the long-term use of vonoprazan have been raised because of significant elevation of serum gastrin levels compared with that of conventional PPI therapy, possibly because of its high pKa value (>9) and strong inhibition of acid secretion (Ashida et al., 2016; Murakami et al., 2016; Miwa et al., 2017). The elevated serum gastrin level is closely correlated with the increased incidence of gastric endocrine cell tumors in a nonclinical carcinogenicity study (Cappiello and Layton, 1965). However, the safety risk of elevated gastrin levels due to vonoprazan in humans remains unclear.
Tegoprazan (Fig. 1), a novel P-CAB based on the benzimidazole structure, is a potent and highly selective inhibitor of H+/K+-ATPase with characteristics typical to P-CAB. Tegoprazan exhibited potent inhibition of acid secretion in the Heidenhain pouch dog model with rapid onset of maximal efficacy (Takahashi and Take, 2018).

Structure of tegoprazan.
In the present study, we demonstrate that tegoprazan effectively controls gastric acid–related diseases using animal models of GERD and PUD. The in vitro and in vivo pharmacological characteristics were compared with those of esomeprazole, a representative PPI.
Materials and Methods
Chemicals
Tegoprazan (CJ HealthCare, Seoul, Republic of Korea), esomeprazole (Hetero, Shouguang, People’s Republic of China) and SCH28080 (S4443; Sigma-Aldrich, St. Louis, MO) were dissolved in DMSO to assess the in vitro activity of H+/K+-ATPase. For in vivo experiments, tegoprazan and esomeprazole were dissolved in 0.5% methyl cellulose solution. Drugs were administered to animals via the oral route or intraduodenal injection. All the reagents and solvents used in this study were of analytical grade or higher.
Animals
All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals from the CJ HealthCare animal ethics committee (No. CJ-0196-00, CJ-0234-00, CJ-0310-00, CJ-0311-00). Porcine stomachs were obtained from a local abattoir (Daesung Corporation, Daejeon, Republic of Korea). Male Sprague-Dawley rats were purchased from Han Lym Laboratory (Hwaseong, Republic of Korea).
Preparation of Porcine Gastric H+/K+-ATPase
Vesicles containing H+/K+-ATPases were isolated from fresh porcine stomachs as previously described (Takahashi and Take, 2018). Porcine stomachs were dissected alongside the greater curvature, followed by washing with ice-cold PBS. The gastric mucosal layer was scraped off from the underlying muscular layer and then added to lysis buffer containing 250 mM sucrose and 40 mM HEPES, followed by homogenization on ice for 20 minutes. The homogenate was centrifuged at 20,000g, 4°C for 30 minutes, and the supernatant was further centrifuged at 115,000g, 4°C for 30 minutes. The pellets were resuspended in lysis buffer and centrifuged at 130,000g, 4°C for 1 hour. The pellets were resuspended in distilled H2O and immediately frozen in liquid nitrogen. To produce lyophilized vesicles, the resuspended vesicles were permeabilized by freeze drying processing and then stored at −70°C. Prior to use, gastric vesicles were thawed by adding an equal volume of distilled H2O. The final concentration of gastric vesicles was 3 μg/μl.
Measurement of H+/K+-ATPase Activity
The H+/K+-ATPase activities were determined by colorimetric measurement of inorganic phosphate production in a reaction mixture containing isolated porcine gastric vesicles as described previously (Keeling et al., 1989; Mori et al., 2009). The porcine gastric H+/K+-ATPase activities were measured in a 60 μl reaction mixture containing 4.5 μg of porcine ion-leaky vesicles in a reaction buffer (20 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, 40 mM bis-tris, pH 6.4) containing various concentrations of tegoprazan or esomeprazole at 37°C in a 96-well polystyrene plate. For 0% or 100% inhibition controls, the enzyme reaction was performed in the presence of 1% DMSO (a vehicle control) or 100 μM SCH28080, a selective inhibitor of gastric H+/K+-ATPase. The reaction was started by adding Na2ATP, and the mixture was incubated at 37°C for 30 minutes, and then 30 μl of 10% sodium dodecyl sulfate containing antifoam A was added to stop the reaction. Inorganic phosphates were measured in 200 μl of colorimetric reagent consisting of four parts of 10% l-ascorbic acid, pH 5, and one part of 35 mM ammonium molybdate in 15 mM zinc acetate, pH 5. After incubation with the colorimetric reagent at 37°C, the optical density was measured at 750 nm with a plate reader (Power wave XS; Bio-TEK, Winooski, VT). The standard inorganic phosphate solution was prepared using KH2PO4. Tegoprazan and esomeprazole were dissolved in DMSO to a concentration of 10 mM, and then diluted in the reaction mixture to the appropriate final concentrations.
Reversibility of H+/K+-ATPase Activity
To examine the reversibility of the tegoprazan- or esomeprazole-induced H+/K+-ATPase inhibition, the enzyme activities before and after the washout of drugs were compared. Porcine gastric H+/K+-ATPase activities were measured in a 600 μl reaction mixture containing 60 μg of porcine ion-leaky vesicles and reaction buffer (5 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, 40 mM bis-tris, pH 6.4) at 37°C in a 96-well polystyrene plate. The reaction was started by adding Na2ATP, and the mixture was incubated at 37°C for 40 minutes. After incubation, 60 μl of the solution was collected, followed by the measurement of H+/K+-ATPase activity as described above. The remaining 540 μl was centrifuged at 27,800g, 4°C for 60 minutes. After removing the supernatant, the pellet was resuspended in distilled water prior to further centrifugation at 27,800g, 4°C for 20 minutes. After removing the supernatant, 600 μl of the reaction mixture, not containing gastric vesicles, was added to the pellet. The final mixture was incubated at 37°C for 80 minutes. After the incubation, 60 μl of the solution was collected for each compound, and H+/K+-ATPase activity was measured.
Measurement of Gastric Acid Secretion in Pylorus-Ligated Rats
Drugs were orally administered to five to six rats after fasting for 24 hours. A vehicle control group was also used at 0.5% methylcellulose. After 30 minutes of drug administration, the pylorus was ligated under weak anesthesia (3% isoflurane inhalation), and the abdomen was closed by surgical suturing. After 5 hours of pyloric ligation, the rats were sacrificed and the stomachs were removed. The gastric contents were collected and centrifuged at 3000 rpm for 5 minutes. The pH of the gastric contents was measured using an automatic titrator (Orion 3 star; Thermo Fisher Scientific, Waltham, MA). For multiple-dosing experiments, drugs were orally administered daily for 4 days, and the fifth dosing was administered after fasting for 24 hours. The pylorus-ligated model was prepared using a single-administration design.
Induction of Reflux Esophagitis in Rats
The rat reflux esophagitis model was induced as described previously (Rao and Vijayakumar, 2008). Rats were fasted for 24 hours and anesthetized by inhaling 3% isoflurane (Model 100 vaporizer, Harvard Apparatus, Holliston, MA), and the stomach was exposed after a 1-cm incision was made in the left upper quadrant of the abdomen. The duodenal pyloric ring and the border between the forestomach and the glandular area of the exposed stomach were ligated by surgical suture. After ligation of the pyloric ring, each drug was intraduodenally administered at 5 ml/kg using 3-ml syringes. The stomach was returned to its original position, and the abdomen was closed using autoclips. After surgery, rats were not allowed to eat or drink for 4 hours. Rats were then sacrificed by CO2 asphyxiation, and their abdomens were incised. The esophagus was exposed up to the neck region, and the portions of esophagus near the neck and just below the ligated pylorus near the duodenum were cut to excise both the esophagus and stomach. The gastric contents were collected and centrifuged at 3000 rpm for 5 minutes. Using the supernatant, the amount and pH of the gastric juice were measured, and total acid output was calculated. The stomach was incised along the greater curvature extending to the esophagus, and a photo was acquired with a digital camera. The esophagus epithelial tissues were scraped using a slide glass on ice. After adding lysis buffer (200 mM NaCl, 10 mM Tris, 5 EDTA [5 mM ethylenediaminetetraacetic acid], 1 mM phenylmethylsulfonyl fluoride, 10% glycerin, and 1% protease inhibitor cocktail), the tissue was lysed for 1 hour on ice. The lysate was centrifuged for 1500g at 4°C for 15 minutes, and the supernatant was collected for the protein assay. The amount of myeloperoxidase (MPO) in 100 μg of protein was measured according to the MPO Kit instructions (RAT MPO ELISA Kit; Hycult Biotech, Plymouth Meeting, PA).
Induction of Peptic Ulcer in Rats
Nonsteroidal Anti-Inflammatory Drug–Induced Peptic Ulcer Model.
Rats were fasted for 24 hours before oral administration of tegoprazan and esomeprazole. One hour after drug administration, naproxen (30 mg/kg) was orally administered to each rat three times at 2-hour intervals. The rats were sacrificed at 4 hours after the final naproxen administration. The stomach was collected and fixed in 2% formalin on ice for 1 hour. The great curvature of each stomach was incised and photographed using a digital camera. The ulcer area was analyzed with an image analyzer (ImageJ; National Institutes of Health), the area of the lesion was summed for each rat, and the ulcer index (%) was calculated per total area of the stomach. For multiple-dosing experiments, tegoprazan and esomeprazole were orally administered daily for 4 days, and the fifth dosing was administered after fasting for 24 hours. The naproxen-induced peptic ulcer model was prepared in a single-administration design.
Ethanol-Induced Peptic Ulcer Model.
Rats were fasted for 48 hours before oral administration of tegoprazan and esomeprazole. One hour after drug administration, absolute ethanol (5 ml/kg) was orally administered to each rat. The rats were then sacrificed at 1 hour after ethanol administration, and the area of gastric ulcers was calculated as described above.
Water Immersion–Restraint Stress–Induced Peptic Ulcer Model.
Water immersion–restraint stress (WIRS) is commonly used as an experimental model of stress-mediated acute gastric ulcer lesions because of its clinical relevance and reproducibility. Rats were fasted for 48 hours before oral administration of tegoprazan and esomeprazole. Ten minutes after drug administration, the rats were placed in a restraint cage and immersed in water at 21°C for 4 hours. The rats were sacrificed 4 hours later, and the area of gastric ulcers was calculated as described above.
Acetic Acid–Induced Peptic Ulcer Model.
Gastric ulcers (namely, kissing ulcers) were induced by intraluminal exposure to acetic acid. Before surgery, the rats were fasted overnight with free access to water. On the next day, rats were anesthetized with xylazine/ketamine (7.5 and 60 mg/kg) by intraperitoneal administration, and the stomach was exposed via midline epigastric incision. The antrum was clamped using a sponge forceps (inner diameter, 6 mm) and 100 μl of 60% acetic acid was then injected into the clamped area for 40 seconds. The incision was then closed by suturing. Each animal was allowed to recover from anesthesia. Two days after the surgical procedure, tegoprazan and esomeprazole were orally administered daily for 5 days. Animals were fasted for 24 hours before the sacrifice. After anesthesia, the stomach was collected from each animal, and then opened along the greater curvature, rinsed with saline, pinned out on a cork board, and photographed. The total ulcerated area (in square millimeters) was measured using Leica Application Suite V4 Software (Leica Microsystems, Wetzlar, Germany). The curative ratio was calculated as follows: curative ratio (%) = {(ulcer area of control group − ulcer area of test group)/ulcer area of control group} × 100.
Stomach Distribution Study in Rats
Dosing and Sample Collection.
Tegoprazan was orally administered at a dose of 10 mg/kg to fasted Sprague-Dawley male rats. The blood and stomach were collected at 0.25, 0.5, 1, 6, 12, and 24 hours after dosing. Blood was immediately centrifuged to obtain the plasma, and stomach tissue was weighed and rinsed with physiologic saline to remove the blood or contents. All plasma and tissue samples were stored at −20°C until analysis.
Measurement of Tegoprazan.
A 100-μl plasma sample added with 20 μl of internal standard (donepezil, 1 μg/ml in 50% methanol) and 1 ml of acetonitrile to induce protein precipitation. The mixture was vigorously mixed and centrifuged, and then 100 μl of the supernatant was added with 700 μl of 0.1% formic acid in water-acetonitrile (50:50, v/v) and mixed by vortexing. After centrifugation, 5 μl of the supernatants of each sample was directly injected into a liquid chromatography-tandem mass spectrometry system. Each stomach sample was thawed and mixed with ice-cold distilled water at 1:1 (in grams per milliliter). After processing in a homogenizer, 100 μl of each tissue homogenate was added to 20 μl of internal standard and 1 ml of acetonitrile. Next, the tissue samples were treated in the same manner as the plasma sample.
Chromatography was performed on a reversed-phase column (2.0 × 75 mm, 3 μm; Cadenza CW-C18 column; Imtakt, Portland, OR) using isocratic elution with a mobile phase of 0.1% formic acid in water-acetonitrile (70:30, v/v) at a flow rate of 0.2 ml/min. The column and autosampler tray temperature were maintained at 35°C and 4°C, respectively. Mass spectrometry was operated in positive turbo-ion spray model (API5000; AB Sciex, Framingham, MA) at the transition mass/charge ratio 338.3–220.3 for tegoprazan and 380.4–91.2 for internal standard. The limit of quantification was 10 ng/ml for tegoprazan with acceptable precision (coefficient of variation, <15%) and accuracy (relative error, <15%).
Pharmacokinetics.
Non-compartmental pharmacokinetic analysis of all data were performed using WinNonlin 5.2 (Pharsight, Cary, NC).
Data Analysis
Data are presented as the mean ± S.D. or S.E. IC50 or ED50 values were obtained from the logistic fit of dose-response data (Prism version 5; GraphPad Software, San Diego, CA). In the reversibility test and GERD model, the differences between each drug treatment group and control group were tested for statistical significance by Student’s t tests. P values less than 0.05 were considered statistically significant. In the gastric acid secretion experiment and peptic ulcer models, the differences between each drug treatment group and vehicle group were tested for statistical significance by the nonparametric Mann-Whitney U tests. P values less than 0.05 were considered statistically significant.
Results
Effects of Tegoprazan and Esomeprazole on Porcine Gastric H+/K+-ATPase Activity.
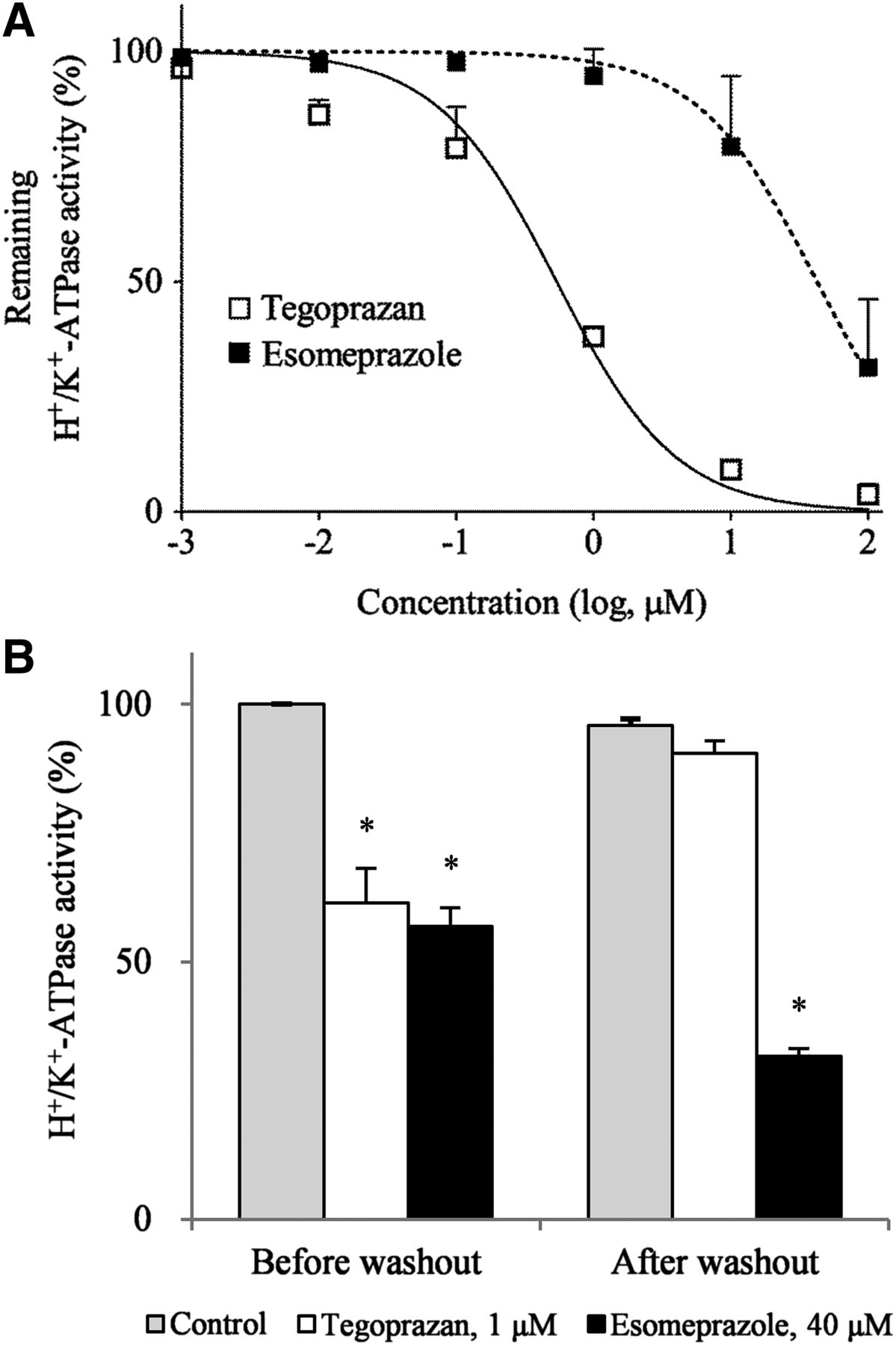
Because direct and reversible inhibition of H+/K+-ATPase are critical characteristics of P-CABs, the inhibition potency and recovery of H+/K+-ATPase activity after washout of the test drugs were evaluated using porcine H+/K+-ATPase containing vesicles under ion-leaky conditions. Because the potassium-binding site of the H+/K+-ATPase is inside the vesicle in this preparation, potassium ion outside the vesicle cannot access the ATPase in the absence of an ionophore. In the ion-leaky vesicle, potassium can freely access the potassium-binding site in the assay solution (Takahashi and Take, 2018). Under the ion-leaky condition, both compounds inhibited H+/K+-ATPase activity in a dose-dependent manner. The IC50 values of tegoprazan and esomeprazole were 0.53 and 42.52 μM, respectively. The inhibitory activity of tegoprazan was 80-fold higher than that of esomeprazole (Fig. 2A). We also tested the reversibility of the inhibitory potency of both compounds on the H+/K+-ATPase activity. The assay of dose-dependent inhibition by tegoprazan and esomeprazole on the H+/K+-ATPase activity showed that the concentrations causing approximately 50% of equal inhibitory effects were 1 and 40 μM, respectively. When the H+/K+-ATPase activity of the control group was set as 100% before the washout, the activity of the control group after the washout was 95.9%. Before washout, tegoprazan (1 μM) showed a percentage of control activity of 61.5% compared with the control. After washout, the H+/K+-ATPase activity level returned to that of the control, demonstrating the reversibility of the inhibitory mechanism. In contrast, esomeprazole (40 μM) showed a percentage of control activity of 56.9% compared with the control before the washout and exerted 32.0% activity even after washout (Fig. 2B). These results are in agreement with those of previous studies showing that the inhibitory effects of PPIs are irreversible.

In vitro inhibitory activities and reversibility of tegoprazan and esomeprazole on porcine gastric H+/K+-ATPase. (A) The reaction was initiated by adding tegoprazan and esomeprazole to the reaction buffer followed by incubation at 21°C for 30 minutes. Data are presented as the mean ± S.E. of two independent experiments. (B) The washout method assay was employed to confirm the reversible mechanism of action of tegoprazan. Assays were performed using porcine gastric H+/K+-ATPase in freeze-dried vesicles. Data are presented as the mean ± S.D. of three independent experiments. The significance of differences between means was determined by the Student’s t test. *P < 0.05 vs. control.
Effects of Tegoprazan and Esomeprazole on Gastric Acid Secretion in Pylorus-Ligated Rats.
This study was conducted to test the inhibitory potency of tegoprazan and esomeprazole on gastric acid secretion through pylorus ligation in conscious rats. Because esomeprazole may require several days of dosing to reach full efficacy, the drugs were evaluated against each other in single and 5-day repeated dosing designs. The basal gastric pH values of the vehicle control for 5 hours after the surgical procedure were pH 1.39 and 1.51 at single dosing and on 5 consecutive days, respectively. For the single-day and multiday dosing regimens, the gastric pH values of 3 mg/kg tegoprazan were 4.13 and 4.34, and for 10 mg/kg tegoprazan were 6.86 and 6.63, respectively. Thus, tegoprazan inhibited basal gastric acid secretion in a dose-dependent manner and was effective at a single dose (Fig. 3A). For the single and multiday dosing regimen, the gastric pH values of 10 mg/kg esomeprazole were 1.95 and 2.98, and for 30 mg/kg esomeprazole were 4.86 and 6.02, respectively. Esomeprazole also inhibited basal gastric acid secretion in a dose-dependent manner and then showed full efficacy in a multiday dosing regimen (Fig. 3B). Tegoprazan completely inhibited gastric acid secretion at the 10 mg/kg single dosing, whereas inhibition by esomeprazole was incomplete, even for the 30 mg/kg multiday dosing regimen.

Effect of tegoprazan and esomeprazole on gastric acid secretion in pylorus-ligated rats. Tegoprazan (A) and esomeprazole (B) were administered orally 30 minutes before pylorus ligation. Five hours later, the rats were sacrificed, and the gastric contents were collected and pH was measured. Each column represents the mean ± S.D. from five to six rats. The significance of differences between the means was determined by the Mann-Whitney U test. *P < 0.05 vs. vehicle.
Effects of Tegoprazan and Esomeprazole on Rats with Experimental Reflux Esophagitis.
By dual ligation at the duodenal pyloric ring and the forestomach, we prepared a rat model to induce the reflux of gastric contents into the esophagus. We observed esophageal mucous membrane damage from the low pH of the refluxed gastric contents. In this study, the inhibitory effects of tegoprazan on esophageal mucosa injury and gastric acid secretion were evaluated and compared with those of esomeprazole. Severe longitudinal hemorrhagic lesions with blackish exudates were observed in the esophagus of the reflux esophagitis vehicle group. However, tegoprazan at doses of 0.3 and 1 mg/kg showed reduced esophagitis area with mild hemorrhagic lesions; tegoprazan at doses of 3–30 mg/kg showed no lesions in reflux esophagitis. Esomeprazole at doses of 0.3 and 1 mg/kg were not effective for decreasing reflux esophagitis, and higher doses of 3–30 mg/kg showed no lesions in reflux esophagitis (Fig. 4A). Gastric total acid output in the vehicle group was 114 mEq/l per 5 hours, and the 0.3–30 mg/kg tegoprazan inhibited gastric acid output in a dose-dependent manner (Fig. 4B) with an ED50 of 2.0 mg/kg (Table 1). Esomeprazole exerted an inhibitory effect only at the high dose of 30 mg/kg (Fig. 4B), with an ED50 value of 32.5 mg/kg (Table 1). Damage to the esophageal mucous tissue caused by gastric acid reflux was measured from the amount of MPO in the esophageal mucous tissue. The vehicle group contained 820 ng of MPO per 100 μg of protein, whereas both the tegoprazan and esomeprazole groups showed reduced MPO levels in a dose-dependent manner (Fig. 4C), with ED50 values of 5.9 and 29.2 mg/kg, respectively (Table 1).

Effect of tegoprazan and esomeprazole in a rat model of reflux esophagitis. Reflux esophagitis was surgically induced by ligating both the pylorus and foregut of rat stomach. The rats were laparotomized under anesthesia, and the transitional region between the forestomach and glandular portion, and the duodenum was ligated with a silk thread. Prior to duodenum ligation, tegoprazan and esomeprazole were intraduodenally injected. Four hours after surgery, the gastric contents and esophageal mucosa tissues were collected. (A) Macroscopic appearance of the esophageal mucosa in rats. (B) Gastric contents were collected 4 hours after surgery, and total acid output was calculated. (C) Esophageal mucosal proteins were extracted from mucosa tissues using lysis buffer, and MPO concentration was measured. Each column represents the mean ± S.D. from three rats. The significance of the difference between means was determined by the Student’s t test. *P < 0.05 vs. vehicle.
ED50 values of tegoprazan and esomeprazole in reflux esophagitis model
Effects of Tegoprazan and Esomeprazole on Rats with Naproxen-Induced Gastric Ulcer.
This study was conducted to evaluate the antiulcer efficacy of tegoprazan and esomeprazole in a naproxen-induced acute gastric ulcer rat model. Because esomeprazole may require several days of dosing to reach full efficacy, the drugs were evaluated against each other in single and 5-day repeated dosing designs. The ulcer lesions (%) were calculated by dividing the area of the ulcer lesion per total area of the stomach. For the single-day and multiday dosing regimens, the ED50 value of tegoprazan at 0.1, 1, and 10 mg/kg were 0.1 and 0.1 mg/kg, respectively. Thus, tegoprazan exerted a antiulcer effect in a dose-dependent manner and was effective at a single dose. The ED50 values of esomeprazole for the single and multidosing regimens were 3.9 and 0.9 mg/kg, respectively (Fig. 5A; Table 2). This model also confirmed that the efficacy of esomeprazole was increased by repeated administration rather than single-dose administration. Despite the multidosing regimen, tegoprazan was 9-fold more potent than esomeprazole. In terms of the gross appearance of gastric ulcers, the vehicle group produced a punctuate type ulcer that was clean and deep. However, tegoprazan at a dose of 1 mg/kg resulted in significantly reduced ulcer lesions with a shallower depth. Esomeprazole at a dose of 3 mg/kg also reduced ulcer lesions, but not significantly (Fig. 5A).

Effect of tegoprazan and esomeprazole in rat models of gastric ulcer. Acute gastric ulcers were induced by three different inducers. (A) After 30 minutes of tegoprazan and esomeprazole administration on a single day (closed columns) and daily for 5 days (open columns), naproxen was orally administered to each animal three times at 2-hour intervals. Four hours after the last naproxen administration, the rats were sacrificed. (B) After 1 hour of tegoprazan and esomeprazole administration, absolute ethanol was orally administered for 1 hour. (C) Ten minutes after tegoprazan and esomeprazole administration, rats were placed in a restraint cage and immersed in water at 21°C for 4 hours. The ulcer area was analyzed by an image analyzer and the ulcer lesion was calculated per total lesion of the stomach. Each column represents the mean ± S.E. from 6 to 10 rats. The significance of the difference between means was determined by the Mann-Whitney U test. *P < 0.05 vs. vehicle.
ED50 values of tegoprazan and esomeprazole in peptic ulcer models
Effects of Tegoprazan and Esomeprazole on Rats with Ethanol-Induced Gastric Ulcer.
We assessed the effects of tegoprazan and esomeprazole based on acute damage to the gastric mucous due to administration of absolute ethanol. The calculation of ulcer lesions (percentage) followed the naproxen ulcer model. Tegoprazan at doses of 0.1, 1, and 10 mg/kg exerted a antiulcer effect in a dose-dependent manner with an ED50 of 1.4 mg/kg. Esomeprazole also suppressed gastric ulcer in a dose-dependent manner at 0.3, 3, and 30 mg/kg, with an ED50 of 7.3 mg/kg (Fig. 5B; Table 2). In terms of macroscopic appearance of gastric ulcers, the vehicle group produced elongated bands of severe hemorrhagic lesions parallel to the long axis of the stomach. Tegoprazan at a dose of 1 mg/kg significantly reduced the ulcer area and severity of hemorrhagic bands. However, esomeprazole at a dose of 3 mg/kg did not reduce ulcer lesions (Fig. 5B).
Effects of Tegoprazan and Esomeprazole on Rats with WIRS-Induced Gastric Ulcer.
We assessed the effects of tegoprazan and esomeprazole against gastric ulcer induced by WIRS for 4 hours. The ulcer lesions (%) were calculated as described for the naproxen ulcer model. Tegoprazan at doses of 0.1, 1, and 10 mg/kg exerted an antiulcer effect in a dose-dependent manner with an ED50 of 0.1 mg/kg. Esomeprazole suppressed gastric ulcer at only 30 mg/kg with an ED50 of 9.7 mg/kg (Fig. 5C; Table 2). For the macroscopic appearance of gastric ulcers, the vehicle group showed serious gastric bleeding erosions, as indicated by mucosal hemorrhage and mucosal erosive lesions. Hemorrhage was observed mainly in the gastric body and antrum. However, tegoprazan at dose of 1 mg/kg showed a significantly reduced hemorrhage area, whereas esomeprazole at a dose of 3 mg/kg did not reduce hemorrhage lesions (Fig. 5C).
Effects of Tegoprazan and Esomeprazole on Rats with Acetic Acid–Induced Gastric Ulcer.
To evaluate the effects of tegoprazan and esomeprazole on acetic acid–induced gastric ulcer models, the ulcer lesions and curative ratio (percentage) were evaluated. Intraluminal application of 60% acetic acid produced two round, deep gastric ulcers in the stomachs of mice in the vehicle group, with the ulcer area calculated as 14.46 mm2. Oral administration of tegoprazan (0.1, 1, and 10 mg/kg/d for 5 consecutive days) significantly reduced the gastric ulcer areas to 9.5, 8.9, and 8.1 mm2, respectively. The curative ratio of tegoprazan was dose dependently increased by 34.2%, 38.0%, and 44.2%. Esomeprazole (30 mg/kg) also reduced the gastric ulcer area to 9.7 mm2, and the curative ratio of esomeprazole was 32.7% (Fig. 6A). The total ulcerated area (in square millimeters) was measured using Leica application Suite V4 Software (Fig. 6B).

Effect of tegoprazan and esomeprazole in a rat model of acetic acid–induced gastric ulcer. The rats were anesthetized with xylazine/ketamine, and the stomach was incised via midline epigastric incision. The antrum was clamped using a sponge forceps, and 100 μl of 60% acetic acid was then injected into the clamped area for 40 seconds. After 2 days, tegoprazan and esomeprazole were orally administered for 5 days. (A) Gastric ulcer area data from 10 rats are presented as the mean ± S.D. The significance of the difference between means was determined by the Mann-Whitney U test. *P < 0.05 vs. vehicle. (B) Gross appearance of gastric mucosa in rats and quantitative analysis.
Stomach Distribution of Tegoprazan in Rats.
The concentration-time profiles of tegoprazan in the plasma and stomach after oral administration of tegoprazan at a dose of 10 mg/kg are shown in Fig. 7. Tegoprazan reached a Cmax at 0.25 hour in both the blood and stomach, and the concentration of tegoprazan in the stomach was higher than that in the plasma at all sampling times; additionally, tegoprazan was present in the gastric tissue 24 hours after administration to rats. This indicated that tegoprazan is very rapidly absorbed and distributed into the stomach.

Concentration-time profiles of tegoprazan in plasma and the stomach after oral administration of tegoprazan at a dose of 10 mg/kg in rats. Plasma and stomach samples were obtained 0.25, 0.5, 1, 6, 12, and 24 hours after dosing. Plasma and gastric concentration of tegoprazan are shown in nanograms per milliliter and nanograms per gram, respectively. Each point represents the mean ± S.D. of five rats.
Discussion
Recently, the basic pharmacological properties of tegoprazan, such as its potent and selective inhibition of gastric H+/K+-ATPase and in vivo inhibitory efficacy for gastric acid secretion in a Heidenhain pouch dog model, were reported (Takahashi and Take, 2018). In this study, we further demonstrated that tegoprazan inhibits H+/K+-ATPase with 80-fold higher potency than esomeprazole under ion-leaky conditions at pH 6.4. Under these conditions, the direct inhibitory effect on H+/K+-ATPase can be compared without the potential for drug accumulation in the acidic compartment or an acid activation process. In a previous report, tegoprazan was 3-fold more potent in inhibiting H+/K+-ATPase activity under ion-tight conditions than under ion-leaky conditions, suggesting that tegoprazan, a weakly basic compound with a pKa of 5.1, can be concentrated in acidic compartments (Takahashi and Take, 2018). Moreover, the concentration of tegoprazan in the stomach was higher than that in the plasma, and a concentration of 152 ng/g remained in the stomach at 12 hours after oral administration at a dose of 10 mg/kg (Fig. 7). The stomach is the target organ of tegoprazan, and its rapid distribution into the stomach may be advantageous for achieving the pharmacodynamic effect of tegoprazan. Therefore, these pharmacological properties of tegoprazan suggest that it may be a more potent and effective agent for inhibiting gastric acid secretion in vivo compared with esomeprazole. Another important characteristic of tegoprazan that differentiates it from esomeprazole is the reversibility of its inhibition of H+/K+-ATPase activity. The inhibitory activity of esomeprazole remained the same after washout, reflecting the irreversible covalent nature of PPI binding to gastric H+/K+-ATPase. In contrast, the inhibitory effect of tegoprazan was reversed by washout. This ensures that gastric acid suppression was not unnecessarily prolonged when stopping the administration of tegoprazan, thereby decreasing the risk of side effects such as acid rebounding. Previous Linweaver-Burk plot analysis show that P-CABs such as tegoprazan and SCH28080 inhibit gastric H+/K+-ATPase by competing with potassium ions (Hori et al., 2010; Takahashi and Take, 2018). These findings confirm that tegoprazan is a P-CAB that does not form covalent bonds and whose mechanism of H+/K+-ATPase inhibition, in contrast to that of PPIs, is reversible and based on competition with potassium.
The in vivo antisecretory effect of tegoprazan was investigated and compared with that of esomeprazole in pylorus-ligated rats. Tegoprazan at a single dose of 10 mg/kg completely inhibited basal acid secretion, whereas esomeprazole only achieved a pH of 6 even with the multiday dosing regimen of 30 mg/kg. Notably, the inhibition of acid secretion by tegoprazan was complete, likely because acid activation was not required for tegoprazan to exert its inhibitory effect, in contrast to esomeprazole, which requires acid activation. The inhibitory effect of tegoprazan on H+/K+-ATPase at pH 6.4 in vitro was approximately 80-fold greater than that of esomeprazole (Fig. 2A), but the antisecretory effect of tegoprazan in vivo was only approximately 5-fold to 10-fold more potent than that of esomeprazole (Fig. 3). This discrepancy between the in vitro and in vivo finding can be explained by the difference in pH conditions. The pH under the in vitro conditions was 6.4 (almost neutral), whereas that of secretory canaliculi of the parietal cells in vivo was 1 to 2. Because esomeprazole is fully transformed into its active form under acidic conditions, its antisecretory activity is more potent in vivo than in vitro. However, tegoprazan showed more potent inhibitory activity than esomeprazole.
For the treatment of GERD, attaining and maintaining an intragastric pH of >4 is important (Kahrilas et al., 2007), as the rate of healing of reflux esophagitis and onset of rapid symptom relief are associated with an intragastric pH of >4 (Bell and Hunt, 1992). We prepared an experimental model of reflux esophagitis for only 4 hours to compare the effects of rapid symptom relief after dual ligation at the duodenal pyloric ring and forestomach. The ED50 value of the total acid output of tegoprazan was 16-fold more potent than that of esomeprazole. This potent inhibition of acid secretion by tegoprazan can be explained by its ability to maintain its activity unaffected by the ambient pH, combined with the potent activity of tegoprazan itself. P-CABs are instantly protonated in an acidic environment and bind to and inhibit H+/K+-ATPase in this protonated form (Andersson and Carlsson, 2005). The pKa value of tegoprazan is 5.1, whereas that of esomeprazole is 4.0. Therefore, tegoprazan can achieve a higher concentration than esomeprazole in acidic environments such as the secretory canaliculi of the gastric parietal cells. Furthermore, PPIs are activated under acidic conditions, and the active compound can inhibit only the H+/K+-ATPase that has already been transferred to the apical membrane and activated (Forte and Yao, 1996). The resting proton pumps are internalized in a tubulovesicle that is not transferred to the apical membrane. Therefore, PPIs cannot inhibit these resting proton pumps because the active form of PPIs cannot penetrate the membrane (Nagaya et al., 1989). Moreover, the inhibition of acid secretion by PPIs was decreased because of the short plasma half-life and its rapid degradation in the secretory canaliculi. However, P-CABs are unaffected by ambient pH and can inhibit H+/K+-ATPase under both acidic and neutral conditions, indicating that P-CABs can bind to both activated and resting acid pumps (Scott et al., 2015). Thus, tegoprazan may be able to achieve maximal acid suppression from the first dosing, and its onset time is much more rapid than those of PPIs such as esomeprazole. A previous study showed that vonoprazan, another P-CAB, exerts a more potent and longer-lasting inhibitory effect on gastric acid secretion but is unaffected by ambient pH in rats and dogs. However, because the pKa value of vonoprazan is 9.3, it exists in many protonated forms under neutral pH conditions; thus, compared with other P-CABs such as tegoprazan, it may take slightly longer for vonoprazan to reach its target site, such as the gastric H+/K+-ATPase in parietal cell canaliculi (Hori et al., 2010). Therefore, tegoprazan may be advantageous for rapid symptom relief in GERD treatment.
To determine whether the indications for tegoprazan usage can be expanded to PUD, we tested the efficacy of tegoprazan in various animal models of PUD. The ED50 values of tegoprazan in NSAID-, ethanol-, and WIRS-induced acute gastric ulcer models were 5-fold to 97-fold more potent than those of esomeprazole. The concentration of tegoprazan was much higher in the stomach than in the plasma and was present in the gastric tissue even 24 hours after administration in rats. The potent antisecretory effect of tegoprazan appears to be associated with its high concentration in the stomach and its rapid onset time. In a previous study (Takahashi and Take, 2018), the effect of tegoprazan on the pH of the gastric perfusate under subcutaneous infusion of 6 μg/kg per hour pentagastrin to the gastric fistula of dogs was reported. Oral administration of tegoprazan at 3 mg/kg increased the pH from 2 to 4 after 30 minutes, and then to >6 1 hour after tegoprazan administration. The increase in pH by tegoprazan was unaffected by subsequent pentagastrin infusion, and the pH was maintained at approximately 7 for 5 hours. In contrast, lansoprazole was previously shown to have a significant antisecretory effect without cimetidine pretreatment, but this effect disappeared when cimetidine was administered before lansoprazole (Hori et al., 2011). PPIs could not be transformed to their active form under neutral conditions resulting from the pretreatment with cimetidine. In our rat model of subchronic acetic acid–induced peptic ulcer, tegoprazan also exhibited superior antiulcer activity compared with esomeprazole after 5 days of repeated oral administration. However, PPIs can exhibit maximal activity after treatment with daily doses for 4 to 5 consecutive days. Combined with the natural healing process, PPI in the subchronic model was slightly more potent than in the single-administration model. In our PUD model studies, we obtained similar efficacies to that of vonoprazan, which has been used for the treatment of PUD in Japan (data not shown). The potent antiulcer activity of tegoprazan shows that it may be effective for patients with peptic ulcer.
In conclusion, tegoprazan is a novel P-CAB that potently and reversibly inhibits gastric H+/K+-ATPase. It exhibits superior efficacy in rat models of gastric acid–related diseases compared with esomeprazole. Therefore, tegoprazan is a new therapeutic option that may have enhanced efficacy compared with existing PPI therapies for the treatment of acid-related diseases.
Acknowledgments
The authors thank all researchers involved in the tegoprazan program at CJ HealthCare Corporation. This work was supported by grants of the New Drug Development project, Korea Drug Development Fund (KDDF-201404-10).
Authorship Contributions
Participated in research design: D. K. Kim, S. J. Lee, Park, B.-T. Kim, Song, Moon, and Ryu.
Conducted experiments: D. K. Kim, K.-H. Lee, and S.-j. Kim.
Contributed new reagents or analytic tools: D. K. Kim, and K.-H. Lee.
Performed data analysis: D. K. Kim, K.-H. Lee, and S.-J. Kim.
Wrote or contributed to the writing of the manuscript: D. K. Kim and Ryu.
Footnotes
- Received November 12, 2018.
- Accepted March 11, 2019.
This research study was supported by CJ HealthCare Corporation and no external funding was received.
Abbreviations
- AZD0865
- 8-[(2,6-dimethylbenzyl)amino]-N-[2-hydroxyethyl]-2,3-dimethylimidazo[1,2-a]pyridine-6-carboxyamide
- GERD
- gastroesophageal reflux disease
- H2RA
- histamine-2 receptor antagonist
- MPO
- myeloperoxidase
- P-CAB
- potassium-competitive acid blocker
- PPI
- proton pump inhibitor
- PUD
- peptic ulcer disease
- revaprazan
- 5,6-dimethyl-2-(4-fluorophenylamino)-4-(1-methyl-1,2,3,4-tetrahydroisoquinolin-2-yl)pyrimidine
- SCH28080
- 3-(cyanomethyl)-2-methyl,8-(phenylmethoxy)imidazo(1,2-a)pyridine
- tegoprazan
- 7-[[(4S)-5,7-difluoro-3,4-dihydro-2H-chromen-4-yl]oxy]-N,N,2-trimethyl-3H-benzimidazole-5-carboxamide
- vonoprazan
- 1-[5-(2-fluorophenyl)-1-(pyridine-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine monofumarate
- WIRS
- water immersion–restraint stress
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}