


Abstract
Proton pump inhibitors (PPIs) are widely used in the treatment of acid-related diseases. However, several unmet medical needs, such as suppression of night-time acid secretion and rapid symptom relief, remain. In this study, we investigated the pharmacological effects of 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine monofumarate (TAK-438), a novel potassium-competitive acid blocker (P-CAB), on gastric acid secretion in comparison with lansoprazole, a typical PPI, and SCH28080 [3-(cyanomethyl)-2-methyl,8-(phenylmethoxy)imidazo(1,2-a)pyridine], a prototype of P-CAB. TAK-438, SCH28080, and lansoprazole inhibited H+,K+-ATPase activity in porcine gastric microsomes with IC50 values of 0.019, 0.14, and 7.6 μM, respectively, at pH 6.5. The inhibitory activity of TAK-438 was unaffected by ambient pH, whereas the inhibitory activities of SCH28080 and lansoprazole were weaker at pH 7.5. The inhibition by TAK-438 and SCH28080 was reversible and achieved in a K+-competitive manner, quite different from that by lansoprazole. TAK-438, at a dose of 4 mg/kg (as the free base) orally, completely inhibited basal and 2-deoxy-d-glucose-stimulated gastric acid secretion in rats, and its effect on both was stronger than that of lansoprazole. TAK-438 increased the pH of gastric perfusate to a higher value than did lansoprazole or SCH28080, and the effect of TAK-438 was sustained longer than that of lansoprazole or SCH28080. These results indicate that TAK-438 exerts a more potent and longer-lasting inhibitory action on gastric acid secretion than either lansoprazole or SCH28080. TAK-438 is a novel antisecretory drug that may provide a new option for the patients with acid-related disease that is refractory to, or inadequately controlled by, treatment with PPIs.
Introduction
Acid-related diseases of the upper gastrointestinal tract, especially gastroesophageal reflux disease (GERD), remain a widespread problem worldwide. A recent systematic review reported a prevalence of GERD, based on the occurrence of typical reflux symptoms, ranging from less than 5% in Asia to 10 to 20% in the Western world (Dent et al., 2005). Reflux disease is becoming increasingly more common, and the chronic nature and high prevalence of reflux disease necessitates a long-term treatment strategy. Proton pump inhibitors (PPIs) are widely used in the treatment of acid-related diseases, such as GERD and peptic ulcer disease (Graham et al., 2002; Frazzoni et al., 2003; Robinson, 2005), and also in combination with antibiotics to eradicate Helicobacter pylori (Malfertheiner et al., 2003). PPIs inhibit gastric H+,K+-ATPase activity by covalently binding to its sulfhydryl group, which results in inhibition of gastric acid secretion (Sachs et al., 1988; Nagaya et al., 1990; Wolfe and Sachs, 2000). Despite their potent inhibitory activity against acid secretion and their worldwide clinical application, several areas where treatment could be further refined or enhanced have been suggested, including the management of patients with gastroesophageal symptoms who do not respond adequately to PPI therapy (Fass et al., 2005). PPIs exert their maximal activity after treatment with daily doses on 4 to 5 consecutive days (Dammann and Burkhardt, 1999). Because PPI therapy frequently does not control acid inhibition throughout a 24-h period, even when taken twice daily (Katz et al., 2000), night-time acid secretion may cause symptoms in some patients (Ang and Fock, 2006). A further limitation of PPIs is the degree of interpatient variability observed in their effect on acid secretion that results from the polymorphism of the hepatic CYP2C19 responsible for their metabolism (Furuta et al., 2005).
A new class of acid suppressants, known as potassium-competitive acid blockers (P-CABs) or acid pump antagonists, inhibits gastric H+,K+-ATPase in a K+-competitive and reversible manner in clear contrast to PPIs (Vakil, 2004; Andersson and Carlsson, 2005; Geibel, 2005). P-CABs have higher pKa values than PPIs, and they are stable at low pH. These properties allow P-CABs to become highly concentrated in the strongly acidic compartment of the gastric parietal cell at the luminal surface of the H+,K+-ATPase and to exert a less variable onset of their effect because, unlike PPIs, they do not require a gastroprotective formulation (Wurst and Hartmann, 1996; Parsons and Keeling, 2005). SCH28080 [3-(cyanomethyl)-2-methyl,8-(phenylmethoxy)imidazo(1,2-a)pyridine], a prototype P-CAB, has been shown to bind to the phosphoenzyme with an extracytosolic conformation of the monovalent cation site (E2P) form of the H+,K+-ATPase and to be strictly K+-competitive (Mendlein and Sachs, 1990). This mechanism allows rapid inhibition of the pump without the need for acidity at its luminal surface because the pump is blocked in mid-cycle. Several structural derivatives, imidazopyridines such as SCH28080 (Wallmark et al., 1987) and AZD0865 [8-[(2,6-dimethylbenzyl)amino]-N-[2-hydroxyethyl]-2,3-dimethylimidazo[1,2-a]pyridine-6-carboxyamide] (Gedda et al., 2007), pyrimidines (Yu et al., 2004), imidazonaphthyridines (Simon et al., 2007), and pyrrolopyridazines (Ito et al., 2007), have been evaluated as P-CABs. These compounds were found to exhibit a fast onset of inhibition of acid secretion on the basis of rapid achievement of their peak plasma concentration. Moreover, the full effect was observed on the 1st day of administration. However, P-CABs are not being used clinically worldwide because of their insufficient efficacy or hepatic toxicity (Parsons and Keeling, 2005; Kahrilas et al., 2007).
We have discovered a novel P-CAB, 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine monofumarate (TAK-438, Fig. 1), a pyrrole derivative with a chemical structure that is completely different from the P-CABs developed to date. In this study, we evaluated the mechanism of inhibition of TAK-438 on porcine gastric H+,K+-ATPase, as well as its effect on gastric acid secretion, and the pH of gastric perfusate in rats in comparison with lansoprazole [2-[[[3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridyl]methyl]sulfinyl]-1H-benzimidazole], a typical PPI. In some experiments, we also compared TAK-438 with SCH28080, a prototype P-CAB.

Chemical structure of TAK-438.
Materials and Methods
Chemicals
TAK-438 and lansoprazole were synthesized by Takeda Pharmaceutical Company Limited. TAK-438, lansoprazole, and SCH28080 (purchased from Sigma-Aldrich, St. Louis, MO) were dissolved in dimethyl sulfoxide to assess their effect on H+,K+-ATPase activity in vitro. In the in vivo experiments, TAK-438 was administered orally as a suspension in 0.5% methylcellulose solution. Lansoprazole was administered orally as a suspension in 0.5% methylcellulose solution containing 1% NaHCO3. Each compound was administered intravenously as a solution in a mixture of N, N- dimethylacetamide and polyethylene glycol 400 in a volume ratio of 1:1.
Histamine 2HCl and 2-deoxy-d-glucose (2DG) were obtained from Wako Pure Chemicals (Osaka, Japan). All of the other reagents and solvents used were of the best grade available.
Animals
All animal experiments were carried out in accordance with ethical guidelines established by the Animal Care and Use Committee of Takeda Pharmaceutical Company Limited. Male 7- or 8-week-old Sprague-Dawley rats (CLEA Japan, Inc., Tokyo, Japan) were used. The animals were fasted for 24 h before the experiment but with free access to water.
Preparation of Porcine Gastric H+,K+-ATPase
Porcine stomachs were obtained from Katayama Chemical, Ltd. (Osaka, Japan). The fundus of the stomach was isolated, rinsed with tap water, and washed with 3 M NaCl to remove the superficial cells, cell debris, and mucus. The oxyntic cell-rich mucosa was scraped off and homogenized in a homogenizing buffer (0.25 M sucrose, 1 mM EDTA, and 10 mM Tris HCl, pH 6.8) in a Polytron homogenizer. The homogenate was centrifuged at 20,000g for 30 min, and the supernatant was collected and centrifuged at 100,000g for 90 min. The pellets were collected and suspended in a homogenizing buffer. The preparation was layered over 7.5% (w/w) Ficoll solution in the homogenizing buffer and centrifuged at 100,000g for 60 min. The microsomal fraction concentrated in the middle layer was collected, diluted with homogenizing buffer, and centrifuged at 100,000g for 90 min. The pellet was collected and suspended in a homogenizing buffer to yield a final protein concentration of 0.5 mg/ml. The suspension obtained was stored at −80°C until used.
Measurement of H+,K+-ATPase Activity
The gastric vesicles (0.124 μg) were incubated for 30 min at 37°C in 45 μl of 50 mM Tris-HEPES buffer (pH 6.5 or 7.5) containing 5 mM MgCl2, 10 μM valinomycin, 0 or 10 mM KCl and various concentrations of TAK-438, SCH28080, or lansoprazole. The enzyme reaction was initiated by adding 5 μl of 2 mM ATP, and the mixture was incubated at 37°C for 40 min. The reaction was stopped by adding 15 μl of malachite green reagent (0.12% malachite green, 7.5% hexa-ammonium heptamolybdate, and 11% Tween 20 in a volume ratio of 100:25:2). The ATPase activity was determined by measuring the inorganic phosphate released by the hydrolysis of ATP (Fiske and Subbarow, 1925). H+,K+-ATPase activity was calculated after subtraction of the corresponding enzyme activity in the absence of KCl. In some experiments, H+,K+-ATPase activity was measured in the presence of 100 μM dithiothreitol (DTT).
To investigate the reversibility of the inhibition of H+,K+-ATPase activity by the three compounds tested, a dilution procedure was conducted after incubation with each compound. A 2-μl volume of the compound solution was preincubated with 12 μl of 0.002% HCl solution for 5 min and incubated with 4 μl (0.5 mg/ml) of pig gastric vesicles for 5 min at room temperature. After neutralizing the enzyme-compound mixture with 2 μl of 500 mM HEPES-Tris buffer (pH 6.5), a 1.1-μl volume of the mixture was diluted with 39 μl of 50 mM HEPES-Tris buffer containing 12.5 μM valinomycin and 5 mM MgCl2. As an undiluted control, 4.9 μl of the compound was added to dilution mixtures at the same concentration as that of the original incubation; for the dilution treatment group, 4.9 μl of vehicle was added to the dilution mixtures. After 60 min, the reaction was initiated by adding 5 μl of 2 mM ATP with or without 10 mM KCl, and the reaction mixture was incubated for 60 min at 37°C. The reaction was then stopped, and the ATPase activity was determined as described previously.
To measure H+,K+-ATPase activity in the kinetic study, 5 μl of solutions containing various concentrations of TAK-438 or SCH28080 were added to the enzyme mixture with various concentration of KCl, and the mixtures were incubated at 37°C for 30 min. The ATPase activity was measured as described previously.
Measurement of Na+,K+-ATPase Activity
The activity of Na+,K+-ATPase from porcine cerebral cortex (Sigma-Aldrich) was measured as described for H+,K+-ATPase, with the exception that the composition of 40 μl of the enzyme mixture was 4 μg of Na+,K+-ATPase, 50 mM Tris-HEPES, pH 7.5, and 2 mM MgCl2, with or without 100 mM NaCl or 10 mM KCl.
Measurement of Gastric Acid Secretion
Gastric Acid Secretion in Pylorus-Ligated Rats.
Drugs and the vehicle were given orally (2 ml/kg) to rats in a blind manner. The pylorus was ligated under light anesthesia 1 h after drug and vehicle administration, and the abdomen was closed by suturing. At 3 h after the pyloric ligation, the rats were sacrificed by CO2 asphyxiation, and their stomachs were removed. The gastric contents were collected and centrifuged at 3000 rpm for 10 min. The volume of each sample was measured, and the acid concentration was determined by automatic titrator (COM-555SC; Hiranuma Sangyo Co., Ltd., Ibaragi, Japan), and total acid output during the 3-h period (in microequivalents) was calculated.
2DG-Stimulated Acid Secretion in Anesthetized Rats.
Drugs and the vehicle were given orally (2 ml/kg) to rats in a blind manner. The pylorus was ligated after anesthetization with urethane (1.2 g/kg i.p.), and the abdomen was closed; 2DG (200 mg/kg/10 ml) then was injected subcutaneously 1 h after drug and vehicle administration. At 3 h after 2DG administration, the rats were sacrificed by CO2 asphyxiation, and their stomachs were removed. The gastric contents were collected and centrifuged at 3000 rpm for 10 min. Total acid output during the 3-h period (in microequivalents) was calculated as described previously.
Measurement of the pH of Gastric Perfusate under Histamine Stimulation in Anesthetized Rats
The animals were anesthetized with urethane (1.2 g/kg i.p.). The abdomen was opened, and the stomach was exposed. Cannulas were introduced into the stomach from the duodenum and also from the forestomach, and the esophagus was ligated. The stomach was perfused with saline at a rate of 0.5 ml/min, and the pH of the perfusate was continuously measured with a glass electrode (6961-15C and 2461A-15T; Horiba, Kyoto, Japan). Histamine 2HCl (8 mg/kg/h) was infused intravenously via the cervical vein. When the pH had stabilized, drugs and the vehicle were administered intravenously. The pH of the perfusate was measured for 5 h after administration of the drug or vehicle.
Statistics
Data were expressed as the means ± S.E., calculated with the Microsoft Excel software program (Microsoft, Redmond, WA). IC50 or ID50 values and their 95% confidence intervals were calculated by logistic regression analysis. In the gastric acid secretion experiment, the differences between each drug treatment group and the vehicle group were tested for statistical significance by a one-tailed Shirley-Williams test. p Values <0.025 were considered significant.
Results
Effect of TAK-438, SCH28080, and Lansoprazole on Gastric H+,K+-ATPase Activity.
The inhibitory effect of TAK-438, SCH28080, and lansoprazole on gastric H+,K+-ATPase activity is shown in Fig. 2. Under weakly acidic conditions (pH 6.5), all three compounds inhibited gastric H+,K+-ATPase activity in a concentration-dependent manner. The inhibitory activity of TAK-438 was the most potent. The IC50 values of TAK-438, SCH28080, and lansoprazole were 0.019, 0.14, and 7.6 μM, respectively (Table 1). Under neutral conditions (pH 7.5), the inhibitory activity of TAK-438 was almost the same as that under weakly acidic conditions. On the other hand, the enzyme inhibitory activities of SCH28080 and lansoprazole were weaker under the neutral condition (Table 1).

Inhibitory activities of TAK-438 (A), SCH28080 (B), and lansoprazole (C) on porcine gastric H+,K+-ATPase activity. The enzyme was preincubated for 30 min with various concentrations of TAK-438, SCH28080, and lansoprazole at pH 6.5 (closed circles) and pH 7.5 (open triangles). Each point represents the mean ± S.E. of three different experiments.
IC50 values of TAK-438, SCH28080, and lansoprazole against porcine H+, K+-ATPase
Effect of TAK-438, SCH28080, and lansoprazole on Na+,K+-ATPase Activity.
With regard to selectivity of inhibitory activity, the effects of the three compounds on Na+,K+-ATPase activity also were examined. These studies showed that neither TAK-438 nor SCH28080 inhibited Na+,K+-ATPase activity, even at concentrations 500 times higher than their IC50 values against gastric H+,K+-ATPase activity (Table 2). Lansoprazole slightly inhibited Na+,K+-ATPase activity at 100 μM.
Effect of TAK-438, SCH28080, and lansoprazole on porcine Na+,K+-ATPase
Effect of DTT on the Inhibitory Activities of TAK-438, SCH28080, and Lansoprazole on H+,K+-ATPase.
PPIs exert their activity by binding to sulfhydryl groups of H+,K+-ATPase, and the thiol reagent DTT inhibits the binding of PPIs. In this study, the influence of DTT on the enzyme inhibitory action of TAK-438, SCH28080, and lansoprazole was investigated at a submaximal inhibitory concentration of each of the three compounds. The inhibitory activities of TAK-438 (0.1 μM) and SCH28080 (1 μM) were unaffected by the presence of a 100 μM concentration of DTT, but the activity of lansoprazole (100 μM) was markedly diminished by DTT (Fig. 3).

Effect of DTT on the inhibitory activities of TAK-438, SCH28080, and lansoprazole against H+,K+-ATPase. The inhibitory effect of TAK-438, SCH28080, and lansoprazole on H+,K+-ATPase activity was measured in the presence (closed columns) and absence (open columns) of 100 μM DTT. Each column represents the mean ± S.E. of three different experiments.
Effect of Dilution on the Inhibitory Activities of TAK-438, SCH28080, and Lansoprazole on H+,K+-ATPase.
The effect of dilution on the inhibitory effect of TAK-438, SCH28080, and lansoprazole on gastric H+,K+-ATPase activity is shown in Fig. 4. The percentage inhibition of gastric H+,K+-ATPase activity by TAK-438 and SCH28080 at the submaximal concentration was 70.2 and 47.7%, respectively. The enzyme inhibitory activities of TAK-438 and SCH28080 were markedly diminished by dilution of the reaction mixture. The percentage inhibition after dilution was 12.3% by TAK-438 and 7.2% by SCH28080. By contrast, the percentage inhibition by lansoprazole at the submaximal concentration was 85.6%, and it was unaffected by dilution.

Effect of dilution on the inhibitory activities of TAK-438, SCH28080, and lansoprazole against H+,K+-ATPase. H+,K+-ATPase was preincubated for 5 min with each compound, and the inhibitory activity was measured 60 min after dilution of the mixture (closed columns) or without diluting the mixture (open columns). Each column represents the mean ± S.E. of five different experiments.
Kinetics of the Inhibitory Activities of TAK-438 and SCH28080 on Gastric H+,K+-ATPase.
To clarify the mechanism of enzyme inhibition, Lineweaver-Burk analyses of TAK-438 and SCH28080 were conducted. Figure 5 shows the Lineweaver-Burk (double-reciprocal) plots between the H+,K+-ATPase activity and K+ concentration, indicating that both TAK-438 and SCH28080 inhibited gastric H+,K+-ATPase in a K+-competitive manner. The calculated Ki values of TAK-438 and SCH28080 were 0.003 and 0.193 μM, respectively. The inhibitory activity of TAK-438 was 64 times more potent than that of SCH28080.

Lineweaver-Burk plots of K+ concentration versus H+,K+-ATPase activity in the presence of TAK-438 (A) and SCH28080 (B). The concentrations of TAK-438 were 0 (open squares), 25 (closed triangles), and 50 nM (closed circles). The concentrations of SCH28080 were 0 (open squares), 100 (closed squares), 300 (closed triangles), and 1000 nM (closed circles). Each point represents the mean ± S.E. of three different experiments.
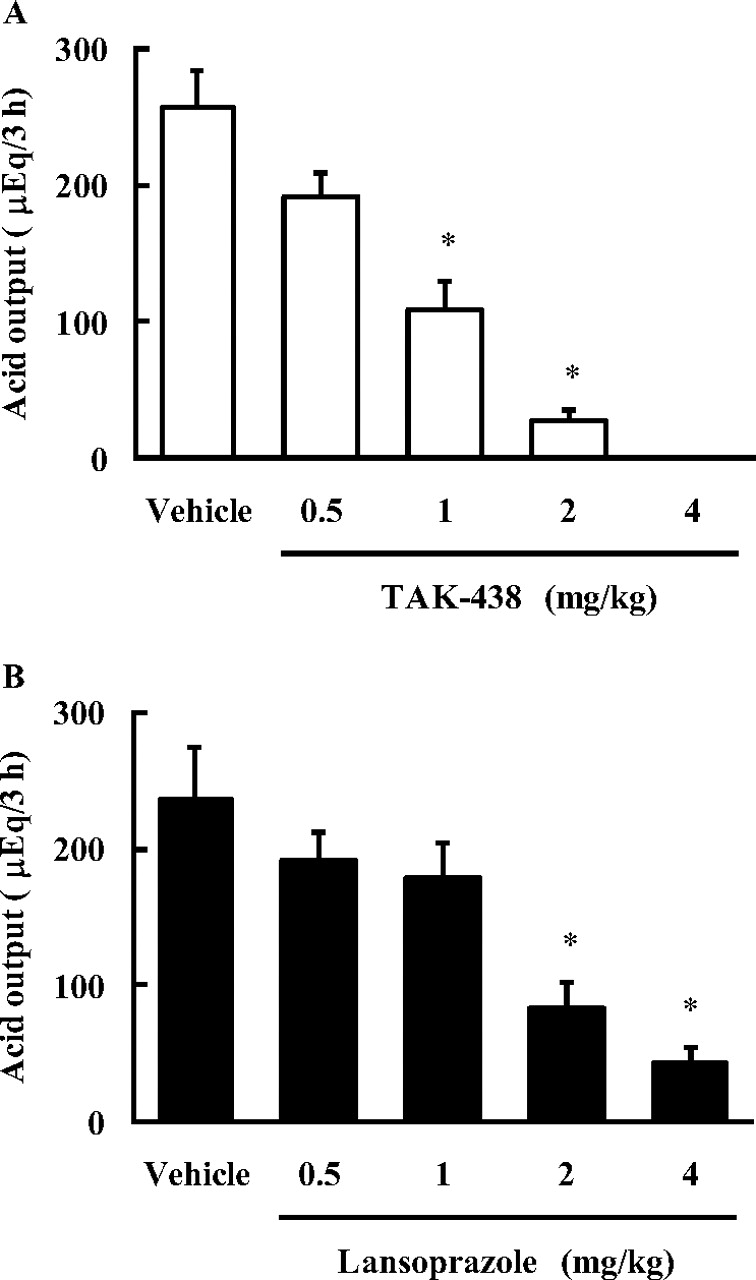
Effect of TAK-438 and Lansoprazole on Basal Gastric Acid Secretion in Pylorus-Ligated Rats.
Basal gastric acid secretion was determined by pylorus ligation in conscious rats, and the control basal gastric acid output was approximately 250 to 320 μEq/3 h. TAK-438 at doses of 0.5, 1, 2, and 4 mg/kg (all doses are shown as the free base) inhibited basal gastric acid secretion in a dose-dependent manner (Fig. 6 A), and the ID50 value was 1.26 mg/kg (Table 3). Lansoprazole also inhibited basal gastric acid secretion (Fig. 6B), and the ID50 value was 1.47 mg/kg (Table 3). TAK-438 completely inhibited gastric acid secretion at the 4 mg/kg dose, whereas inhibition by lansoprazole was incomplete, even at the 8 mg/kg dose.

Effect of TAK-438 (A) and lansoprazole (B) on gastric acid secretion in pylorus-ligated rats. TAK-438, lansoprazole, and the vehicle were administered orally 1 h before pylorus ligation. Gastric contents were collected 3 h after the ligation, and total acid output was calculated. Each column represents the mean ± S.E. from six to eight rats. The statistical significance of the difference was determined by a one-tailed Shirley-Williams test. *, p < 0.025 versus vehicle.
ID50 values of TAK-438 and lansoprazole against gastric acid secretion in rats
Effect of TAK-438 and Lansoprazole on 2DG-Stimulated Gastric Acid Secretion in Anesthetized Rats.
In a preliminary experiment, gastric acid secretion in urethane-anesthetized rats was negligible in the absence of a secretagogue. When gastric acid secretion was stimulated with 2DG, the acid output was approximately 250 μEq/3 h. Both TAK-438 and lansoprazole inhibited 2DG-stimulated gastric acid secretion in a dose-dependent manner (Fig. 7), and the ID50 values of TAK-438 and lansoprazole were 0.83 and 1.57 mg/kg, respectively (Table 3). TAK-438 completely inhibited gastric acid secretion at the 4 mg/kg dose, whereas inhibition by lansoprazole was incomplete at that dose.

Effect of TAK-438 (A) and lansoprazole (B) on 2DG-stimulated acid secretion in anesthetized rats. TAK-438, lansoprazole, and the vehicle were administered orally 1 h before pylorus ligation and 2DG (200 mg/kg s.c.) administration. Gastric contents were collected 3 h after 2DG administration, and total acid output was calculated. Each column represents the mean ± S.E. from seven or eight rats. The statistical significance of the difference was determined by a one-tailed Shirley-Williams test. *, p < 0.025 versus vehicle.
Effect of TAK-438, SCH28080, and Lansoprazole on the pH of Gastric Perfusate under Histamine Stimulation in Anesthetized Rats.
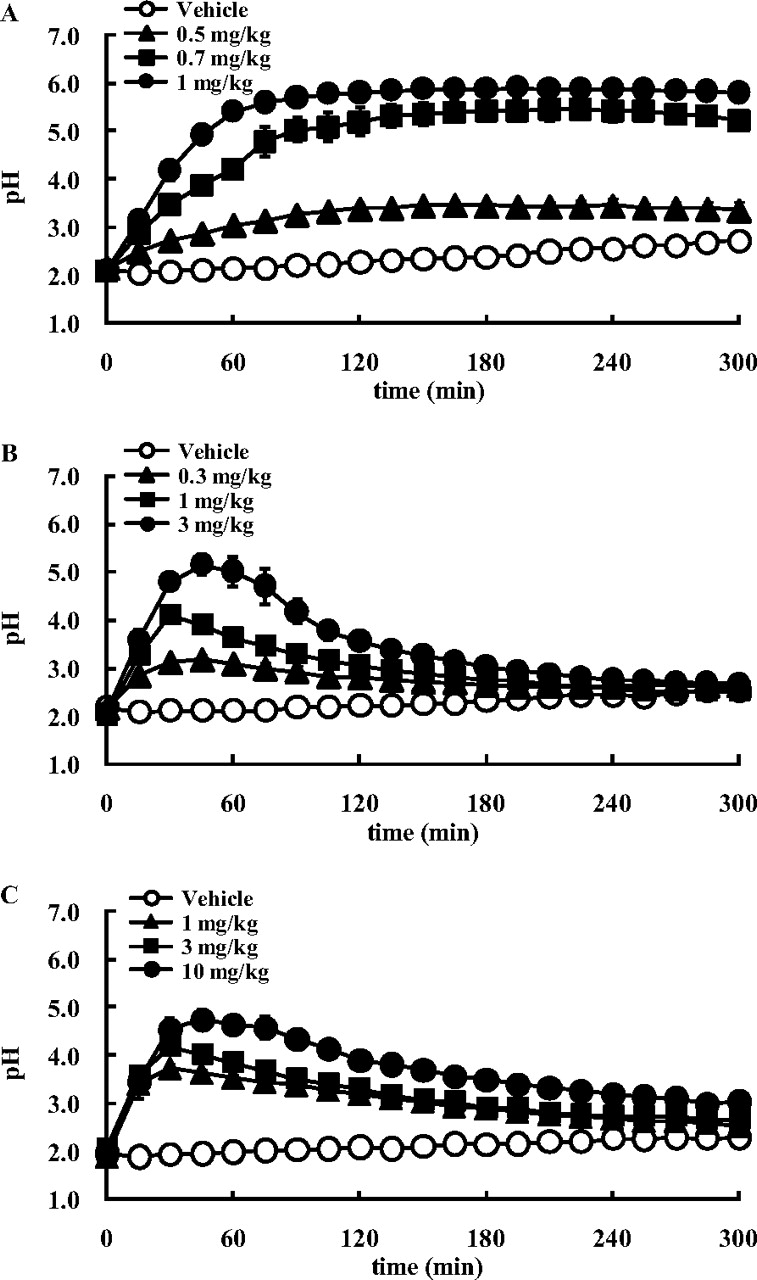
The effect of TAK-438, SCH28080, and lansoprazole on the pH of gastric perfusate is shown in Fig. 8. The pH value of saline in this experimental condition was 6.0 to 6.3. Intravenous infusion of histamine 2HCl at 8 mg/kg/h stimulated gastric acid secretion and decreased the pH of the gastric perfusate to around 2. Intravenous administration of TAK-438 dose-dependently increased the pH of the gastric perfusate, and the increase in pH was sustained for 5 h after administration. At the 1 mg/kg dose, the pH plateaued 90 min after administration, and the highest pH value reached was 5.9. SCH28080 also dose-dependently increased the pH of the gastric perfusate, and the pH reached 5.2 at the 3 mg/kg dose. Lansoprazole increased the pH of the perfusate, but even at the 10 mg/kg dose, the highest pH reached was only 4.7. It took 30 to 60 min for the pH of the perfusate to reach its peak value after administration of SCH28080 and lansoprazole. The effect of SCH28080 and lansoprazole on the pH of the gastric perfusate had diminished 2 h after administration, and it had almost disappeared at 5 h after administration.
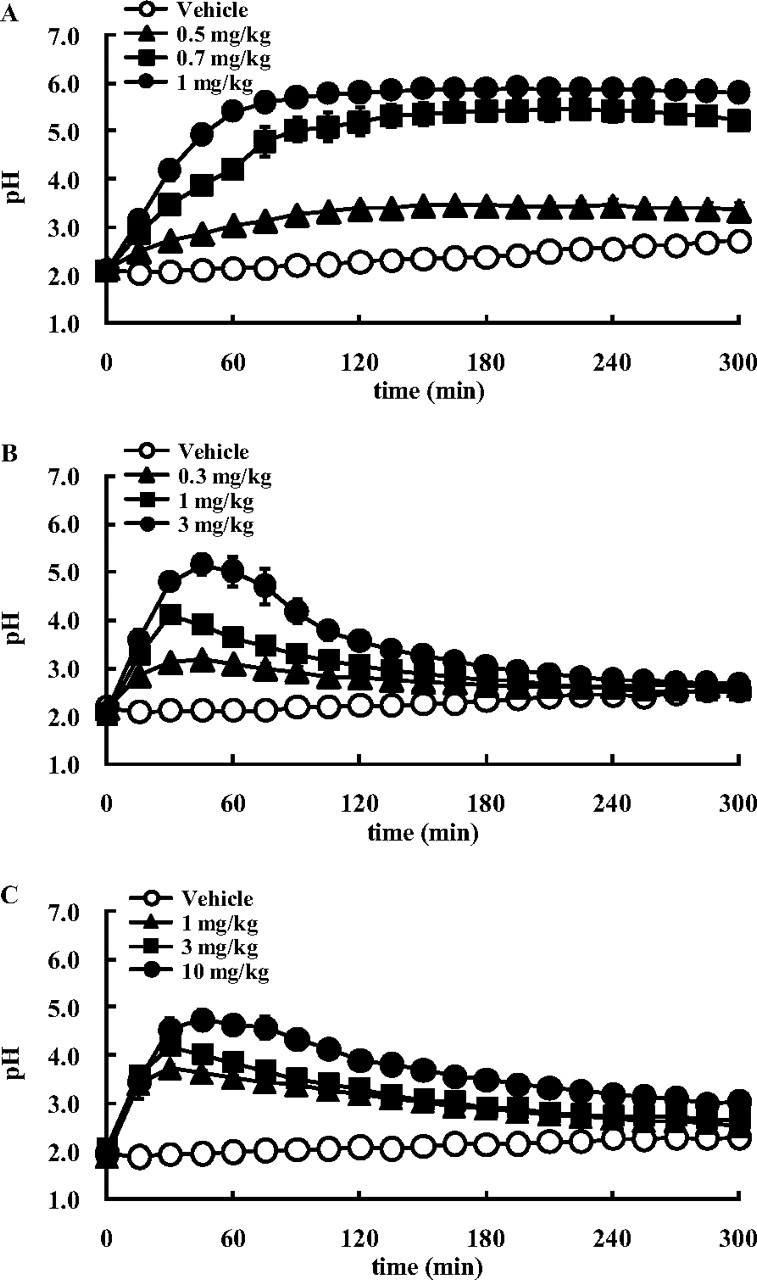
Effect of TAK-438 (A), SCH28080 (B), and lansoprazole (C) on the pH of gastric perfusate under histamine stimulation in anesthetized rats. The stomach was perfused with saline at a rate of 0.5 ml/min, and the pH was measured continuously. Histamine 2HCl (8 mg/kg/h) was infused intravenously, and the drug or the vehicle (open circles) was administered intravenously after the pH had stabilized. Each point represents the mean ± S.E. from four rats.
Discussion
The results of this study showed that TAK-438 had a more potent inhibitory effect on porcine gastric H+,K+-ATPase activity than did lansoprazole, a typical PPI, or SCH28080, a prototype P-CAB. In addition, the inhibitory effect of TAK-438 was unaffected by ambient pH, whereas the inhibitory effect of lansoprazole and SCH28080 was attenuated at pH 7.5. PPIs exert their inhibitory activity after undergoing molecular rearrangement under the acidic condition in parietal cells that allows them to covalently bind to key cysteine residues in the transmembrane domains of H+,K+-ATPase (Sachs et al., 1995). Thus, their activity decreases under neutral conditions. P-CABs are weak bases; e.g., SCH28080 has a pKa value of 5.6 (Bell et al., 1992). P-CABs are instantly protonated in an acidic environment, and in the protonated form, they are thought to bind to and inhibit H+,K+-ATPase (Andersson and Carlsson, 2005). The inhibitory activity of previous P-CABs, including SCH28080, has also been reported to be weaker under neutral conditions (Pope et al., 1995; Tsukimi et al., 2000). Given that AZD0865 has a pKa value of 6.1, the theoretical protonated compound is approximately 33% at pH 6.4 and only below 5% at pH 7.4; therefore, AZD0865 shows a more potent inhibitory effect at pH 6.4 than at pH 7.4 (Gedda et al., 2007). In comparison, the pKa value of TAK-438 is 9.37, higher than that of other P-CABs; thus, TAK-438 should be protonated instantly and exert a potent inhibitory activity even in a neutral environment.
The inhibitory effect of PPIs on H+,K+-ATPase activity is known to be blocked by a thiol reagent such as DTT, which interferes with the covalent binding between PPIs and H+,K+-ATPase (Nagaya et al., 1989). The inhibitory effect of lansoprazole on H+,K+-ATPase activity was found to be blocked by treatment with DTT, whereas the inhibitory activities of TAK-438 and SCH28080 were unaffected. In another experiment, the inhibitory activity of lansoprazole was found to be the same after dilution, which reflects the covalent nature of PPI binding to gastric H+,K+-ATPase. By contrast, the inhibitory effect of TAK-438 and SCH28080 was reversed by dilution. In the kinetic experiments, Lineweaver-Burk plot analysis showed that TAK-438 and SCH28080 inhibited gastric H+,K+-ATPase by competing with potassium ions. These findings confirm that TAK-438 is a P-CAB that does not form any covalent bonds and whose mechanism of H+,K+-ATPase inhibition is reversible and potassium-competitive, in contrast to that of PPIs. Furthermore, TAK-438 is quite different from the other P-CABs because the effect of TAK-438 was found to be unaffected by ambient pH. Given that PPIs are only able to bind to activated acid pumps, it takes 4 to 5 consecutive days to achieve maximal acid suppression at therapeutic doses (Tytgat, 2001). The results of this study show that TAK-438 is capable of inhibiting H+,K+-ATPase under both acidic and neutral conditions, indicating that TAK-438 can bind to both activated and resting acid pumps; thus, TAK-438 may be able to achieve maximal acid suppression from the 1st day, much faster than PPIs.
The amino acid sequences of H+,K+-ATPase and Na+,K+-ATPase are highly homologous, but TAK-438 did not inhibit Na+,K+-ATPase, even at 10 μM. This result indicates that the binding of TAK-438 to gastric H+,K+-ATPase is highly selective. Moreover, it has recently been reported that the only organ in humans expressing significant levels of the P-CAB target gastric H+,K+-ATPase is the stomach (Herrmann et al., 2007).
The in vivo antisecretory effect of TAK-438 was investigated in comparison with lansoprazole in pylorus-ligated rats. The ID50 value of TAK-438 was lower than that of lansoprazole (Table 3). TAK-438 at 4 mg/kg completely inhibited basal and 2DG-stimulated acid secretion, whereas lansoprazole showed potent but incomplete inhibition of the acid secretion at 4 or 8 mg/kg. It has been reported that intraduodenal administration of lansoprazole does not completely inhibit basal and 2DG-stimulated acid secretion, even at a 30 mg/kg dose (Satoh et al., 1989; Nagaya et al., 1991). It is noteworthy that TAK-438 completely inhibits acid secretion, probably because acid activation is not required for TAK-438 to exert its acid secretion inhibiting effect, in contrast to lansoprazole, which requires acid secretion for its activity. The inhibitory effect of TAK-438 on H+,K+-ATPase at pH 6.5 in vitro was approximately 400 times more potent than that of lansoprazole (Table 1), but based on the ID50 values, the antisecretory effect of TAK-438 in vivo was only approximately1.2 to 2.0 times more potent than that of lansoprazole (Table 3). This discrepancy between the findings in vitro and in vivo can be explained by the difference in pH conditions. The pH under the experimental conditions in vitro was 7.5 or 6.5, almost neutral, whereas the pH of the secretory canaliculi of the parietal cell in vivo was approximately 1 to 2. Because lansoprazole is fully transformed into its active form under acidic conditions, it exhibited more potent antisecretory activity in vivo than in vitro. The same discrepancy between Ki value in vitro and the intragastric pH-increasing effect in vivo of TAK-438 and SCH28080 was observed and can also be explained by the difference in pH conditions. As mentioned previously, the efficacy of TAK-438 was unaffected by ambient pH, but that of SCH28080 was affected.
In clinical studies, an intragastric pH of >4 holding time is measured as an important parameter for the treatment of GERD (Yu et al., 2004; Kahrilas et al., 2007), because the rate of healing of reflux esophagitis and of symptom relief is associated with intragastric pH of >4 holding time (Bell et al., 1992; Hunt, 1999). PPI cannot inhibit night-time acid secretion completely even when administered before dinner, because of its time-limited duration of acid suppression (Katz et al., 2006). As a result of covalent binding, the duration of the inhibitory effect of PPIs is much longer than their plasma half-life. However, the short half-life of PPIs in plasma, their requirement for acid activation, and their instability in acidic environments impair their efficacy as acid inhibitors, particularly against night-time acid secretion. It would seem that faster healing of esophagitis and better symptom relief would be achieved with a therapeutic agent that had both a longer duration of gastric acid suppression and that increased intragastric pH to a higher level. We investigated the effect of TAK-438 on the pH of gastric perfusate in anesthetized rats to evaluate its intragastric pH-increasing effect and duration of action, and the results indicated that TAK-438 had both a more potent and a longer duration of action than that of lansoprazole or SCH28080. In this study, 1 mg/kg TAK-438 increased the pH of the gastric perfusate to 5.9, indicating almost complete inhibition, because the pH of saline was approximately 6 and the effect was maintained for more than 5 h. Complete inhibition was probably observed because TAK-438 did not require acid secretion to exert its effect and the higher gastric pH may show great therapeutic efficacy not only for GERD but also for H. pylori eradication and gastric bleeding. Neither lansoprazole nor SCH28080 exhibited such strong or long-lasting inhibition, even at 10 or 3 mg/kg. These results suggest that TAK-438 has the potential to increase intragastric pH in humans to a higher level and for a longer time than the PPIs or P-CABs that have been developed to date.
It took approximately 90 to 120 min for the effect of TAK-438 to reach a plateau, whereas it took 30 to 60 min for SCH28080 and lansoprazole to reach their peak pH. The longer duration, but slightly slower onset of action, of TAK-438 cannot be explained pharmacokinetically because we monitored the plasma concentrations of TAK-438 after intravenous administration to rats and the t1/2 was 1.2 h (data not shown). Because TAK-438 is protonated under neutral pH conditions, it may take slightly longer for TAK-438 to reach its target site, i.e., gastric H+,K+-ATPase in parietal cell canaliculi. Therefore, TAK-438 might show a slightly slower onset of action. However, once TAK-438 is bound to gastric H+,K+-ATPase, its binding is tight and the dissociation from gastric H+,K+-ATPase is very slow because TAK-438 has sulfonyl and methylamine moieties in its structure (Shin et al., 2010). Therefore, it may exert a more potent and longer-lasting effect than PPIs and other P-CABs. Further studies to clarify the mechanism of the long duration of action of TAK-438 are in progress.
In conclusion, TAK-438 demonstrated a potent inhibitory effect on gastric H+,K+-ATPase in a reversible and K+-competitive manner, and the inhibitory effect was observed under both acidic and neutral conditions in vitro. In vivo, TAK-438 exerted a more potent inhibitory effect on gastric acid secretion and a stronger and longer-lasting effect on increase in the pH of gastric perfusate than did lansoprazole and SCH28080. These findings demonstrate that TAK-438 is a novel antisecretory drug that may provide an improvement over current PPI-based treatment and may also be a new option for the treatment of patients, with acid-related disease that is refractory to or inadequately controlled by treatment with PPIs.
Acknowledgments
We thank Dr. Hideaki Nagaya for helpful suggestions. We also thank Toshimasa Kyutoku and Kozo Matsushita for technical assistance.
Footnotes
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.170274.
-
ABBREVIATIONS:
- GERD
- gastroesophageal reflux disease
- PPI
- proton pump inhibitor
- P-CAB
- potassium-competitive acid blocker
- TAK-438
- 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine monofumarate
- SCH28080
- 3-(cyanomethyl)-2-methyl,8-(phenylmethoxy)imidazo(1,2-a)pyridine
- AZD0865
- 8-[(2,6-dimethylbenzyl)amino]-N-[2-hydroxyethyl]-2,3-dimethylimidazo[1,2-a]pyridine-6-carboxyamide
- 2DG
- 2-deoxy-d-glucose
- DTT
- dithiothreitol.
- Received May 16, 2010.
- Accepted July 6, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}