


Abstract
Inhibition of H+,K+-ATPase is accepted as the most effective way of controlling gastric acid secretion. However, current acid suppressant therapy for gastroesophageal reflux disease, using histamine H2 receptor antagonists and proton pump inhibitors, does not fully meet the needs of all patients because of their mechanism of action. This study sought to characterize the in vitro and in vivo pharmacology of a novel acid pump antagonist, N-(2-Hydroxyethyl)-N,2-dimethyl-8-{[(4R)-5-methyl-3,4-dihydro-2H-chromen-4-yl]amino}imidazo[1,2-a]pyridine-6-carboxamide (PF-03716556), and to compare it with other acid suppressants. Porcine, canine, and human recombinant gastric H+,K+-ATPase activities were measured by ion-leaky and ion-tight assay. The affinities for a range of receptors, ion channels, and enzymes were determined to analyze selectivity profile. Acid secretion in Ghosh-Schild rats and Heidenhain pouch dogs were measured by titrating perfusate and gastric juice samples. PF-03716556 demonstrated 3-fold greater inhibitory activity than 5,6-dimethyl-2-(4-fluorophenylamino)-4-(1-methyl-1,2,3,4-tetrahydroisoquinoline-2-yl)pyrimidine (revaprazan), the only acid pump antagonist that has been available on the market, in ion-tight assay. The compound did not display any species differences, exhibiting highly selective profile including the canine kidney Na+,K+-ATPase. Kinetics experiments revealed that PF-03716556 has a competitive and reversible mode of action. More rapid onset of action than 5-methoxy-2-{[(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]-sulfinyl}-benzimidazole (omeprazole) and 3-fold greater potency than revaprazan were observed in Ghosh-Schild rats and Heidenhain pouch dogs. PF-03716556, a novel acid pump antagonist, could improve upon or even replace current pharmacological treatment for gastroesophageal reflux disease.
Gastroesophageal reflux disease (GERD), an acid-related disease of the upper gastrointestinal tract, is extremely common worldwide. Gastric acid may reflux into the esophagus or oral cavity following antireflux barrier dysfunction; the reflux results in a variety of symptoms and mucosal lesions of the esophagus (Katz et al., 2000). In patients with GERD, esophageal adenocarcinoma and other severe complications can develop (Zhang et al., 2008), significantly affecting their quality of life. As the likelihood of esophageal mucosal lesion development and symptom severity are statistically correlated with the extent of acid exposure, control and maintenance of gastric acidity is important for the treatment of GERD (Bell et al., 1992).
The gastric H+,K+-ATPase, which is responsible for gastric acid secretion, is a P2-type ATPase located in the apical membrane of parietal cells. The enzyme is an α/β heterodimer. The α subunit, which has cation-binding and ATP-binding sites, plays a crucial role in catalytic activity. The β subunit is responsible for functional expression. Acid secretion is electroneutral as the H+,K+-ATPase transports H+ into the secretory canaliculus of the parietal cell in exchange for K+ (Ganser and Forte, 1973; Sachs et al., 1976; Shin et al., 2008). Inhibition of the H+,K+-ATPase is currently the most effective way to control gastric acid secretion and remains an attractive target for the medical treatment of acid-related diseases.
The first effective medical treatments for acid-related diseases were histamine H2 receptor antagonists (H2RAs), which have been prescribed for decades to promote lesion healing and symptom relief in patients with peptic ulcer disease and GERD (Chiba et al., 1997). The gastric acid secretion stimulated by the cholinergic pathway, however, is not inhibited by H2RAs because of its mechanism of action. In addition, pharmacological tolerance to H2RAs develops during 14 days of continuous administration in subjects without Helicobacter pylori infection (Lachman and Howden, 2000; Komazawa et al., 2003).
The introduction of proton pump inhibitors (PPIs) has significantly improved acid-suppressive therapy. The superior efficacy of PPIs over H2RAs is attributed to the fact that they directly inhibit gastric H+,K+-ATPase independently of the nature of the stimulus and display a longer duration of action and an antisecretory activity (Andersson and Carlsson, 2005). With the irreversible nature of binding, PPIs undergo a conformational change under the acidic conditions in parietal cells that allows them to bind covalently to key cysteine residues in the transmembrane domains of the H+,K+-ATPase (Sachs et al., 1995). PPIs are extensively used as effective acid suppressants for the treatment of moderate-to-severe GERD and other acid-related diseases (Robinson and Horn, 2003). Because PPIs can only bind covalently to activated acid pumps, not resting acid pumps, it takes several days of continuous daily administration to achieve maximal acid suppression at therapeutic doses (Tytgat, 2001). Thus, patients may suffer from continuing GERD symptoms for several days after initiating PPI therapy. Because this slow onset of action probably results from the mechanism of action of all PPIs, it will be difficult to improve upon the current treatment profile of PPIs (Vakil, 2004). Therefore, GERD therapy could be significantly improved with the advent of effective acid suppressant therapy with a rapid onset of action.
A new class of acid suppressants, known as potassium-competitive acid blockers or acid pump antagonists (APAs), demonstrate reversible inhibition of gastric acid secretion. These agents compete with K+ for ionic binding to the H+,K+-ATPase near the K+-binding site (Pope and Sachs, 1992; Wurst and Hartmann, 1996; Andersson and Carlsson, 2005). The prototypic agent, SCH28080, has been used extensively to investigate the mechanism of action of the class (Beil et al., 1986; Wallmark et al., 1987; Keeling et al., 1988). Current agents, including soraprazan, AZD0865, and 5,6-dimethyl-2-(4-fluorophenylamino)-4-(1-methyl-1,2,3,4-tetrahydroisoquinoline-2-yl)pyrimidine (revaprazan), have been developed as APAs that exhibit reversible inhibition in a K+-competitive manner (Han et al., 1998; Gedda et al., 2007; Simon et al., 2007). This mechanism of action is expected to have a rapid onset of action; initial research demonstrated that gastric acid secretion is nearly completely inhibited within 30 min of administration (Wurst and Hartmann, 1996).
In this study, we characterized a novel, potent, and selective acid pump antagonist, PF-03716556 (Fig. 1), a chromane-substituted 2-alkyl imidazopyridine derivative, in vitro and in vivo. PF-03716556 inhibits the porcine, canine, and human recombinant gastric H+,K+-ATPase in a competitive manner without any biologically relevant activity against other tested receptors, ion channels, and enzymes, including the Na+,K+-ATPase. PF-03716556 inhibits gastric acid secretion in a dose-dependent manner in Ghosh-Schild rats and Heidenhain pouch dogs, producing full efficacy on treatment day 1.
Materials and Methods
Ethics Approvals. Experimental procedures in this study were performed according to the Pfizer guidelines and policies concerning laboratory animal care and use. All protocols were approved by the Animal Ethics Committee of Pfizer Global Research and Development in Nagoya Laboratories (Pfizer Global Research and Development, Pfizer Japan Inc., Taketoyo, Aichi, Japan).
Preparation of Porcine and Canine Gastric H+,K+-ATPase. The fresh porcine or canine fundic mucosa was scraped from the underlying muscular layer of stomach; these samples were then minced and homogenized with a Potter-Elvehjem homogenizer in 250 mM sucrose; or with a Polytron homogenizer (Brinkmann Instruments, Westbury, NY) in buffer containing 1 mM EGTA, 250 mM sucrose, and 5 mM Tris, pH 7.4, at 4°C for porcine and canine samples, respectively. The homogenate was first filtered through gauze, then centrifuged at 20,000g for 30 min at 4°C. Supernatants were recentrifuged at 115,000g for 30 min at 4°C. Pellets were resuspended in 250 mM sucrose and then separated by differential zonal-density gradient centrifugation in a vertical rotor at 130,000g for 60 min at 4°C for porcine samples or at 132,000g for 90 min at 4°C with a swing rotor for canine samples. The density gradient was prepared with 250 mM sucrose as the upper layer and 7% (w/v) Ficoll in 250 mM sucrose as the lower layer. We collected the fraction just above the Ficoll interface, which we designated the ion-tight vesicle. These samples were diluted 10-fold in pure water; ion-leaky vesicles were generated by permeabilization using freeze dry processing. These vesicles were stored at -80°C until use. Freeze-dried vesicles were reconstituted with the original volume of pure water, and protein concentration was determined using a bicinchoninic acid protein assay kit (Pierce Chemical. Rockford, IL) according to the manufacturer's protocol.

Chemical structure of PF-03716556.

Inhibitory activity of PF-03716556 against porcine (A; circles), canine (A; triangles), and human recombinant gastric H+,K+-ATPase (B) (ion-leaky assay). Each value represents mean ± S.E.M. of three independent experiments. B, inset, Western blot analysis: anti-H+,K+-ATPase α subunit (C-terminal) polyclonal antibody, rabbit, 95-kDa. HEK293 cell membranes from nontransfected (N) and transfected (T) are shown. Individual pIC50 values are shown in Table 1.
Preparation of Human Recombinant Gastric H+,K+-ATPase. H+,K+-ATPase cDNA clones (α and β subunits) were obtained by reverse transcription and polymerase chain reaction using total RNA from human stomach as a template. The amplified cDNA fragments encoding the α and β subunits were subcloned into the appropriate sites of the mammalian expression vectors pcDNA3.1/Zeo(-), which carries the eukaryotic selection marker Zeocin (phleomycin; Invitrogen, Carlsbad, CA), and pcDNA3.1(+), which bears the eukaryotic selection marker Geneticin (G418; Invitrogen). To generate stable transfectants, we transfected vectors into human embryonic kidney (HEK) 293 cells using FuGENE 6 (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instructions. Stable transfectants were selected by culture in 0.5 mg/ml G418 and 0.1 mg/ml phleomycin for 4 weeks. HEK293 cells stably expressing human gastric H+,K+-ATPase were seeded in T-225 cell culture flasks in culture medium containing Dulbecco's modified Eagle's medium, 10% heat-inactivated fetal bovine serum, 0.5 mg/ml G418, and 0.1 mg/ml phleomycin. Medium was changed after 9 days of subculture in humidified incubator at 37°C with 5% CO2. Cells were harvested with 1 mM EDTA/phosphate-buffered saline(-) after an additional 2-day culture. After centrifugation at 1000 rpm for 5 min at 4°C, packed cells were resuspended in buffer containing 0.5 mM MgSO4, protease inhibitors, and 50 mM Tris-HCl, pH 7.4, and homogenized with a Polytron homogenizer for 40 s. Homogenates were centrifuged at 1000 rpm for 5 min at 4°C; the resulting supernatants were recentrifuged at 40,000g for 30 min at 4°C. Pellets were resuspended in 250 mM sucrose using a Polytron homogenizer. The resulting samples were aliquoted and stored at -80°C until use. Protein concentrations of the membrane fraction were determined by bicinchoninic acid protein assay (Pierce Chemical).
Preparation of Canine Kidney Gastric Na+,K+-ATPase. Purified canine kidney Na+,K+-ATPase was purchased from Sigma-Aldrich (St. Louis, MO). Forty-two milligrams of enzyme was reconstituted in 250 mM sucrose to a final concentration of 7 mg/ml protein. This solution was aliquoted and stored at -80°C until use.
H+,K+-ATPase Activity in Ion-Leaky/Ion-Tight Gastric Vesicles (Ion-Leaky/Ion-Tight Assay). As described previously (Keeling et al., 1988), gastric H+,K+-ATPase activity was measured in a 60-μl reaction mixture containing either the test compound with 0.3 μg of freeze-dried vesicles, 5 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, and 40 mM Bis-Tris, pH 6.4, at 37°C for ion-leaky assays or the test compound, 2 μg of vesicles, 150 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, 17 μM valinomycin, and 5 mM Tris, pH 7.4, at 37°C for ion-tight assays, in 96-well clear polystyrene plates (nontissue culture-treated). For 0% inhibition and 100% inhibition controls, enzymatic reactions were performed in the presence of 1% dimethyl sulfoxide (DMSO) and 100 μM SCH28080, respectively. Reaction mixtures were incubated at 37°C for 30 min in the presence of Na2ATP; reactions were aborted by the addition of 30 μl of 10% SDS containing antifoam A. A colorimetric reagent was prepared by mixing 10% l-ascorbic acid, pH 5.0, and 35 mM ammonium molybdate in 15 mM zinc acetate, pH 5.0, at a ratio of 4:1. We then added 200 μl of colorimetric reagent to each well. After incubation at 37°C for 30 min (ion-leaky assays) or at 37°C for 20 min (ion-tight assays), the optical density of each well was measured at 750 nm using a plate reader. Inorganic phosphate solution containing KH2PO4 and K2HPO4 was used as a standard.
Na+,K+-ATPase Activity. Canine kidney Na+,K+-ATPase activity was measured in 60-μl reaction mixtures containing the test compound, 11 μg of protein, 100 mM NaCl, 2 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, and 40 mM Tris, pH 7.4, at 37°C in a 96-well clear polystyrene plates. For 0% inhibition and 100% inhibition controls, we performed enzymatic reactions in the presence of 1% DMSO and 100 μM ouabain, respectively. Reaction mixtures were incubated at 37°C for 30 min after addition of Na2ATP. Reactions were terminated by the addition of 30 μl of 10% SDS containing antifoam A. Samples were then incubated with 200 μl of colorimetric reagent for 15 min at 37°C. The optical density of each well was measured at 750 nm using a plate reader. The inorganic phosphate solution described above was used as a standard.
Enzyme Kinetics. Porcine gastric H+,K+-ATPase activity was measured in 60-μl reaction mixtures containing the test compound; 1 μg of vesicles; 2, 2.5, 3.5, 5, and 10 mM KCl; 3 mM MgSO4; 3 mM Na2ATP; and 40 mM Bis-Tris, pH 6.4, at 37°C in 96-well clear polystyrene plates. Reaction mixtures were incubated at 37°C for 30 min after addition of Na2ATP; reactions were terminated with 30 μl of 10% SDS containing antifoam A. After additional incubation with 200 μl of colorimetric reagent per well at 37°C for 10 min, we measured the optical density of each well at 750 nm using a plate reader. The inorganic phosphate solution containing KH2PO4 and K2HPO4 was used as a standard.

Inhibitory activity of PF-03716556 against the porcine gastric H+,K+-ATPase in acidic condition (ion-tight assay). Each value represents mean ± S.E.M. of three independent experiments. pIC50 values are shown in Table 1.
Receptor, Ion Channel, and Enzyme Selectivity Profile. Selectivity profile analysis was performed by Cerep (Celle l'Evescault, France). The affinities of PF-03716556 for a range of receptors, ion channels, and enzymes were determined in duplicate at 10 or 30 μM using standard radioligand binding techniques. The respective reference compounds were tested at several concentrations to obtain concentration-response curves to validate the experiment.
Measurement of Gastric Acid Secretion in Ghosh-Schild Rats. Acid secretion in gastric lumen-perfused rats was measured according to the method described previously (Watanabe et al., 2000). Male Sprague-Dawley rats (250–300 g; Charles River Laboratories Japan, Inc., Yokohama, Japan) were singly housed in cages under standard conditions of 21–22°C, controlled humidity, and light from 7:00 AM to 7:00 PM daily. Rats were allowed to acclimatize to the animal facility for 2 weeks and fasted for 18 h with free access to water before experiments. Rats were anesthetized with urethane (1.4 g/kg; 2 ml/kg i.p.), maintained at 35°C, and tracheotomized. After a middle abdominal incision, a dual polyethylene cannula was inserted into the forestomach and perfused the stomach with saline, pH 5.0, at 37°C at a rate of 1 ml/min. We determined the acid output in the perfusate at 5-min intervals by titration with 0.02 N NaOH to pH 5.0. After determining basal acid secretion for 30 min, we stimulated acid secretion by continuous intravenous infusion of pentagastrin (16 μg/kg/h; 1 ml/h). Revaprazan (3–30 mg/kg; 1 ml/kg) dissolved in 0.5% methylcellulose, PF-03716556 (1–10 mg/kg; 1 ml/kg) dissolved in 5% DMSO and 15% Cremophor EL (BASF Wyandotte, Wyandotte, MI), or vehicle alone was administered intraduodenally after acid secretion reached a plateau phase, in which stable acid secretion lasted at least 15 min.
Measurement of Gastric Acid Secretion in Heidenhain Pouch Dogs. In male beagles (7–15 kg; Marshall Farms USA, Inc., Oak Park, IL), we constructed a gastric pouch according to the Heidenhain method (Heidenhain, 1879). In brief, dogs were anesthetized with isoflurane, and the abdominal cavity was opened under aseptic conditions. After exposing the stomach in the surgical field, a portion of the greater curvature opposite the splenic hilum was converted into a pouch with adequate blood supply from the intact gastroepiploic artery. The main body of the stomach was reconstituted, while the pouch drained into an implanted metal cannula. After closing the pouch, the cannula was brought out of the abdominal cavity through the left lateral abdominal wall. Animals were allowed to recover from surgery for at least 3 weeks in single housing under standard conditions. They received standard food once daily at 11:00 AM and water ad libitum. Animals were fasted overnight before the experiment with free access to water. Gastric juice samples were collected by gravity drainage every 15 min throughout the experiment. Acidity in the gastric juice was measured by titration to an endpoint of pH 7.0. Acid secretion was stimulated by continuous intravenous infusion of histamine (80 μg/kg/h; 5 ml/h). Revaprazan (1 and 3 mg/kg; 5 ml/body), 5-methoxy-2-{[(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]-sulfinyl}benzimidazole (omeprazole) (0.3 and 0.6 mg/kg; 5 ml/body), PF-03716556 (0.3–3 mg/kg; 5 ml/body), dissolved in 0.5% methylcellulose, or vehicle alone was administered orally 60 or 90 min after beginning the histamine infusion.
Data Analysis. Ion-leaky, ion-tight, and Na+,K+-ATPase assays were performed in triplicate. The averages of three independent experiments were used for analysis. For the ion-leaky and ion-tight assays, curve fitting used nonlinear regression with Prism version 4.02 (GraphPad Software Inc., San Diego, CA) to determine a pIC50 value. Each value is shown as a mean ± S.E.M. The mode of inhibition of the porcine gastric H+,K+-ATPase by PF-03716556 was determined by goodness-of-fit and a graphic method using a Lineweaver-Burk plot. Average data were calculated with Enzyme Kinetics Module 1.1 SigmaPlot version 7.101 (Systat Software, Inc., San Jose, CA).
Results
Inhibition of H+,K+-ATPase Activity in Vitro. PF-03716556 inhibited H+,K+-ATPase activity of porcine ion-leaky membrane vesicles in a concentration-dependent manner, with a pIC50 value of 6.026 ± 0.112 at pH 6.4 (Fig. 2A). In the ion-leaky membranes of canine vesicles and human recombinant cells, the pIC50 values at pH 6.4 were 6.038 ± 0.039 (Fig. 2A) and 6.009 ± 0.209 (Fig. 2B), respectively. In porcine ion-leaky membrane vesicles, revaprazan and omeprazole inhibited H+,K+-ATPase activity in a concentration-dependent manner, with pIC50 values of 6.203 ± 0.005 and 5.412 ± 0.005, respectively, at pH 6.4 (Table 1).
In vitro activities of PF-03716556, revaprazan, and omeprazole Data are mean ± S.E.M.
In porcine ion-tight membrane vesicles, PF-03716556 inhibited H+,K+-ATPase activity in a concentration-dependent manner, with a pIC50 value of 7.095 ± 0.077 at pH 7.4 (Fig. 3). Revaprazan and omeprazole inhibited H+,K+-ATPase activity in porcine ion-tight membrane vesicles in a concentration-dependent manner, with pIC50 values of 6.323 ± 0.015 and 5.763 ± 0.196, respectively, at pH 7.4 (Table 1). PF-03716556 did not inhibit canine kidney Na+,K+-ATPase at concentrations as high as 100 μM, which are 100-fold greater than the concentrations needed to inhibit the gastric H+,K+-ATPase. This result suggests that PF-03716556 exhibited high selectivity for the H+,K+-ATPase over the Na+,K+-ATPase (Table 1). Kinetic experiments for PF-03716556 were performed to confirm mode of inhibition of the porcine gastric H+,K+-ATPase. Statistical analysis for goodness-of-fit revealed that PF-03716556 displayed competitive inhibition against potassium ions. Lineweaver-Burk analysis demonstrated that inhibition of the H+,K+-ATPase was reversible (Fig. 4).

Lineweaver-Burk plot for PF-03716556 (competitive inhibition). Kinetics experiments were performed using the porcine gastric H+,K+-ATPase. Enzyme reaction was measured at pH 6.4. Averaged data were calculated with the Enzyme Kinetics Module 1.1 SigmaPlot version 7.101. Graph presented is from a representative experiment of three independent experiments.
We determined the selectivity profile for PF-03716556 by evaluating the effect of PF-03716556 on the specific binding of radioligands to receptors, ion channels, and enzymes. This analysis, performed by Cerep, determined that PF-03716556 (10 or 30 μM) did not exhibit any biologically relevant activity against any of the tested receptors, ion channels, or enzymes expressed in native tissues, cell lines, and transfectants (Tables 2 and 3).
Receptor and ion channel selectivity profile for 10 μM PF-03716556
Enzyme selectivity profile for PF-03716556
Inhibition of Gastric Acid Secretion in Ghosh-Schild Rats. Basal acid secretion in the anesthetized rats achieved a steady state within 60 min after surgery and lasted for at least 30 min before pentagastrin infusion. Gastric acid secretion was increased by intravenous infusion of pentagastrin, achieving a plateau within 60 to 90 min. Stable stimulated acid secretion lasted for more than 210 min. Although we did not observe a dose-dependent inhibition of gastric acid secretion after intraduodenal administration of revaprazan at doses tested 3 to 30 mg/kg, maximal efficacy was apparent at 10 mg/kg (Fig. 5A). Intraduodenal administration of PF-03716556 inhibited gastric acid secretion in a dose-dependent manner over concentrations ranging from 1 to 10 mg/kg. Complete inhibition was observed at 10 mg/kg PF-03716556, with faster onset of action than revaprazan (Fig. 5B) and was sustained for more than 210 min after administration.
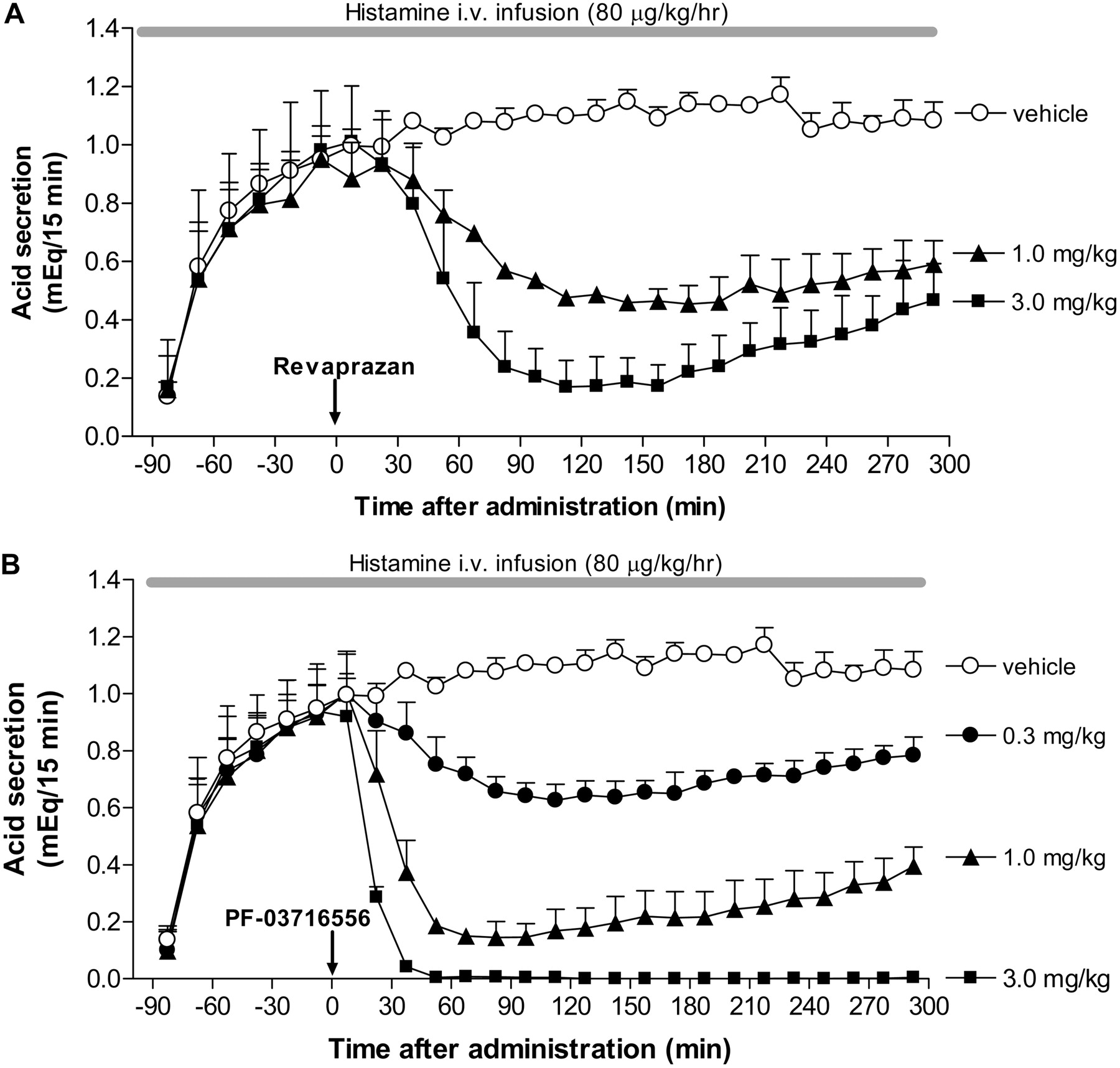
Inhibition of Gastric Acid Secretion in Heidenhain Pouch Dogs. Gastric acid secretion increased after intravenous infusion of histamine, achieving a plateau level within 90 min that lasted for more than 300 min. Oral revaprazan inhibited gastric acid secretion in a dose-dependent manner at 1 and 3 mg/kg (Fig. 6A). Dose-dependent inhibition of gastric acid secretion was observed following oral administration of PF-03716556 at doses ranging from 0.3 to 3 mg/kg; complete inhibition was observed at 3 mg/kg (Fig. 6B). This inhibitory effect was sustained for more than 300 min after dose.
We administered PF-03716556 or omeprazole repeatedly over 5 days to observe the inhibitory effects on histamine-stimulated acid secretion. Oral administration of omeprazole inhibited gastric acid secretion in a dose-dependent manner at doses of 0.3 and 0.6 mg/kg on treatment days 1 and 5 (Fig. 7A). Administration of 0.6 mg/kg omeprazole, which is a clinically relevant dose, for 5 days completely suppressed gastric acid secretion. The peak level of acid inhibition following omeprazole administration, however, was less than 40% on day 1. PF-03716556 inhibited gastric acid secretion on treatment day 5 after repeated administration; the inhibitory effect of PF-03716556 was maintained at a high level from day 1 to day 5 (Fig. 7B).
Discussion
The gastric H+,K+-ATPase, a transmembrane enzyme present in parietal cells, is the target molecule for APAs. In this study, we assessed the inhibitory effect of PF-03716556 on the gastric H+,K+-ATPase in the enzymatic fraction (ion-leaky assay) or on the enzyme within ion-tight vesicles (ion-tight assay). Isolated ion-tight vesicles have a low ion-permeability; because the binding site for both K+ and APAs resides inside ion-tight vesicles, enzymatic action of the gastric H+,K+-ATPase exchanges of K+ for H+, resulting in a pH gradient across the membrane of ion-tight vesicles (Reenstra and Forte, 1990). Because ion-leaky vesicles, generated by freeze dry processing, do not have a pH gradient, ion-tight vesicles are more similar to the conditions present at the luminal face of parietal cells in vivo. This study demonstrated that PF-03716556 inhibited porcine, canine, and human recombinant gastric H+,K+-ATPase, with similar pIC50 values, implying that there was no species selectivity in the ion-leaky assay. In the ion-tight assay, PF-03716556 demonstrated greater inhibitory activity than revaprazan, the only APA that has been available on the market. PF-03716556 was more potent in the ion-tight assay than the ion-leaky assay, suggesting that PF-03716556 concentrates within acidic regions, as was observed for another APA, AZD0865 (Gedda et al., 2007). Kinetic experiments revealed that PF-03716556 competed with potassium ions for porcine H+,K+-ATPase, suggesting that the inhibitory effect of PF-03716556 is reversible. An intrinsic property of this class is the reversibility of their binding to H+,K+-ATPase. Although the reversibility of H+,K+-ATPase inhibition by PF-03716556 was not specifically investigated, in in vitro assays other structurally related imidazopyridine derivatives demonstrated reversibility of H+,K+-ATPase inhibition on washout (data not shown).
The in vivo efficacy of PF-03716556 was assessed in both Ghosh-Schild rats and Heidenhain pouch dogs. These are well established animal models that have been used previously to characterize PPIs and H2RAs. Consistent with the observed potency in vitro, PF-03716556 displayed approximately 3-fold greater potency than revaprazan in Ghosh-Schild rats. Intraduodenal administration of revaprazan at doses of 10 and 30 mg/kg displayed equivalent inhibitory efficacies. The lack of dose dependence is probably secondary to a low absorption profile in rats, as reported previously (Han et al., 1998). In Heidenhain pouch dogs, PF-03716556 also displayed 3-fold greater efficacy than revaprazan. This drug also exhibited dose-dependent inhibition with rapid onset after oral dosing. Oral administration of 3 mg/kg PF-03716556 demonstrated gastric acid suppression lasting more than 300 min after dose, indicating an improved duration-of-action profile over revaprazan.
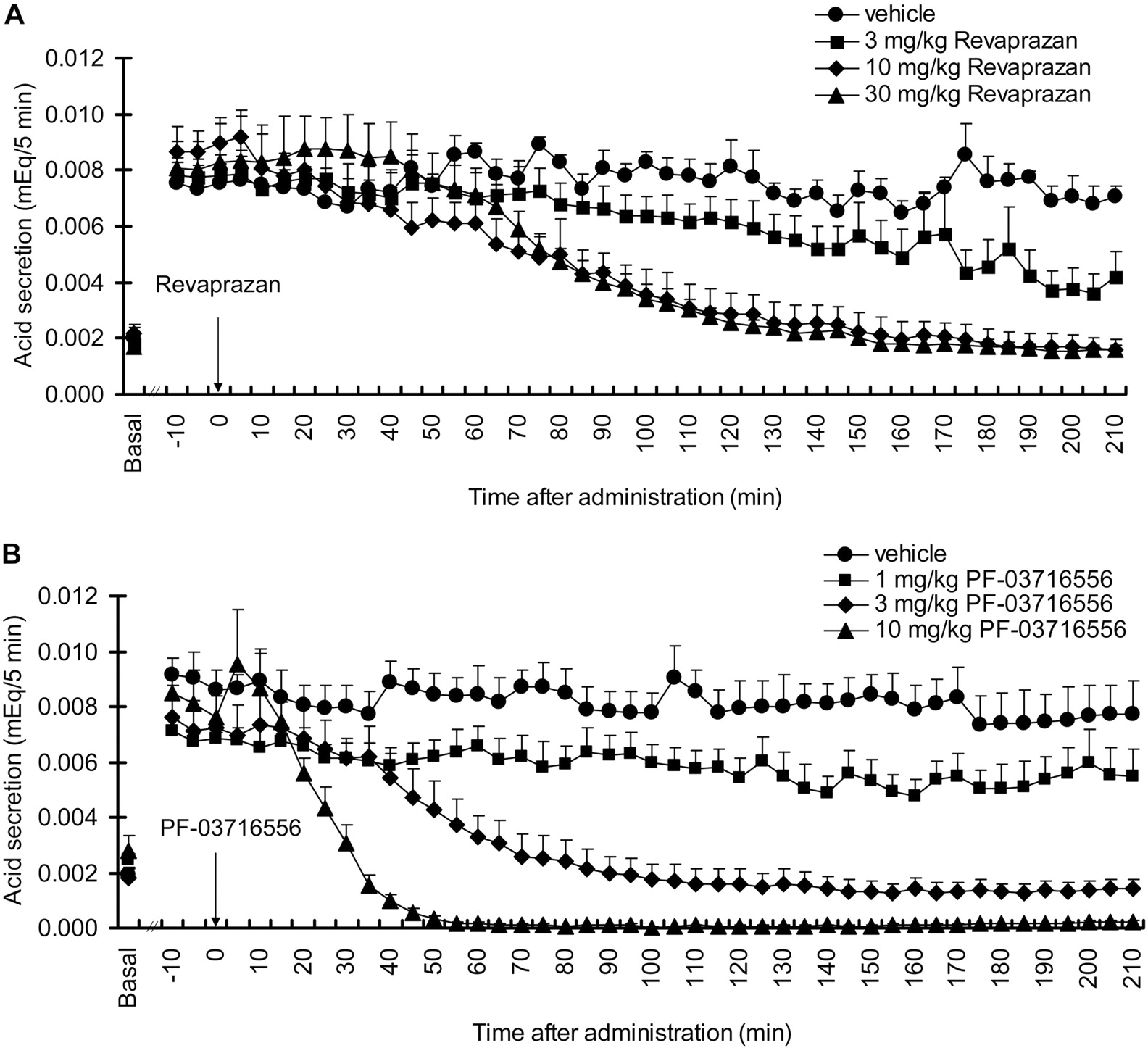
Effect of revaprazan (A) and PF-03716556 (B) on acid secretion stimulated by pentagastrin infusion in Ghosh-Schild rats. Revaprazan (3–30 mg/kg), PF-03716556 (1–10 mg/kg), or vehicle was administered intraduodenally at the time point 0. The average acid secretion for 30 min before the pentagastrin infusion was shown as Basal. Each value represents mean + S.E.M. from four rats.

Effect of revaprazan (A) and PF-03716556 (B) on gastric acid secretion in Heidenhain pouch dogs. Gastric acid secretion was stimulated by histamine infusion after overnight fasting. Revaprazan (1.0 and 3.0 mg/kg), PF-03716556 (0.3–3.0 mg/kg), or vehicle was administered orally 90 min after commencement of the histamine infusion. Each value represents mean + S.E.M. from three dogs.

Effect of omeprazole (A) and PF-03716556 (B) on gastric acid secretion before and after single or repeated administration in Heidenhain pouch dogs. Omeprazole (0.3 and 0.6 mg/kg) or PF-03716556 (1.0 mg/kg) was administered orally once daily for 5 days. At treatment day 1 and day 5, the effect on gastric acid secretion stimulated by histamine infusion was examined. Each value represents mean + S.E.M. from two to three dogs.
PPIs require repeated dosing over several days to reach a steady, maximal effect at therapeutic doses in humans. We investigated the pharmacological profile of PF-03716556 using Heidenhain pouch dogs to differentiate it from omeprazole with a focus on the onset of action. The efficacy of omeprazole increased during repeated administration; at 0.6 mg/kg, the clinically relevant dose, omeprazole displayed complete gastric acid suppression by treatment day 5, consistent with previous reports (Larsson et al., 1985; Wallmark, 1989; Uchiyama et al., 1999). In contrast, PF-03716556 inhibited gastric acid secretion by 70% within 1 h of the first 1 mg/kg dose, and the efficacy did not change after 5-day dosing, indicating that PF-03716556 suppresses gastric acid secretion at full efficacy after a single dosing without the development of tolerance. These results obtained using Heidenhain pouch dogs indicate the advantages in the rapidity of onset of action over PPIs and in superior potency in comparison with another APA that is currently clinically available.
In summary, PF-03716556 demonstrated K+-competitive and reversible inhibition of gastric H+,K+-ATPase with no species differences among the porcine, canine, and human enzymes. This compound is more potent in acidic conditions (ion-tight assay) and demonstrated high selectivity for the H+,K+-ATPase in vitro, without any detectable activity against the Na+,K+-ATPase. PF-03716556 produced greater inhibition than revaprazan in both the in vitro (ion-tight assay) and in vivo (Ghosh-Schild rats and Heidenhain pouch dogs) conditions. PF-03716556 offers long-lasting and maximal efficacy within 30 min of a single dosing with responses that are maintained for at least 5 days of repeated dosing with no signs of tolerance. There are still some unmet medical needs in managing patients with GERD despite the success of PPIs. Compounds in this class may achieve complete relief from heartburn by raising intragastric pH with fast onset and long duration of action and may provide significant benefit to the patients who do not adequately respond to PPIs. Consequently, PF-03716556, a novel acid pump antagonist, could improve upon or even replace current pharmacological treatment for GERD.
Acknowledgments
We thank Drs. Garry J. Douglas (Pfizer) and Masaomi Tajimi (RaQualia Pharma Inc., Taketoyo, Aichi, Japan) for helpful suggestions. We also thank Tomomi Oida for technical assistance.
Footnotes
-
This study was funded by Pfizer.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.108.146415.
-
ABBREVIATIONS: GERD, gastroesophageal reflux disease; H2RA, histamine H2 receptor antagonist; PPI, proton pump inhibitor; APA, acid pump antagonist; SCH28080, 3-(cyanomethyl)-2-methyl,8-(phenylmethoxy)imidazo(1,2-a)pyridine; AZD0865, 8-[(2,6-dimethylbenzyl)amino]-N-[2-hydroxyethyl]-2,3-dimethylimidazo[1,2-a]pyridine-6-carboxyamide; PF-03716556, N-(2-hydroxyethyl)-N,2-dimethyl-8-{[(4R)-5-methyl-3,4-dihydro-2H-chromen-4-yl]amino}imidazo[1,2-a]pyridine-6-carboxamide; HEK, human embryonic kidney; DMSO, dimethyl sulfoxide; ICI 118551, (±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol; CP 55940, 3-(2-hydroxy-4-(1,1-dimethylheptyl)phenyl)-4-(3-hydroxypropyl)-cyclohexanol; WIN551212-2, -(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3,-de]-1,4-benzoxazinyl]-(1-naphtalenyl)methanone; CGP 54626, [S-(R*,R*)]-3-[[1-(3,4-dichlorophenyl)ethyl]amino]-2-hydroxypropyl](cyclohexylmethyl) phosphinic acid; CGS 19755, cis-4-phosphonomethyl-2-piperidine carboxylic acid; U 50488, 3,4-dichloro-N-methyl-N-(2-(1-pyrrolidinyl)-cyclohexyl)-benzeneacetamide; (±)-DOI, (±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane; MDL 72222, 1 αH,3 αH,5 αH-tropan-3-yl-3,5-dichlorobenzoate; D600, benzeneacetonitrile; NS398, N-[2-(cyclohexyloxyl)-4-nitrophenyl]-methane sulfonamide; SB202190, 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole.
- Received September 22, 2008.
- Accepted October 30, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}