


Abstract
Tegoprazan [(S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo[d]imidazole-6-carboxamide], a potassium-competitive acid blocker (P-CAB), is a novel potent and highly selective inhibitor of gastric H+/K+-ATPase. Tegoprazan inhibited porcine, canine, and human H+/K+-ATPases in vitro with IC50 values ranging from 0.29 to 0.52 μM, while that for canine kidney Na+/K+-ATPase was more than 100 μM. A kinetic analysis revealed that tegoprazan inhibited H+/K+-ATPase in a potassium-competitive manner and the binding was reversible. Oral single administrations of tegoprazan ranging from 0.3 to 30 mg/kg in dogs were well absorbed into the blood stream and distributed in gastric tissue/fluid higher than in plasma. Tegoprazan potently inhibited histamine-induced gastric acid secretion in dogs, and a complete inhibition was observed at 1.0 mg/kg starting from 1 hour after administration. Moreover, an oral administration of tegoprazan at 1 and 3 mg/kg reversed the pentagastrin-induced acidified gastric pH to the neutral range. Interestingly, 3 mg/kg tegoprazan immediately evoked a gastric phase III contraction of the migrating motor complex in pentagastrin-treated dogs and similar effects was observed with the other P-CAB, vonoprazan. Tegoprazan is the novel P-CAB that may provide a new option for the therapy of gastric acid–related and motility-impaired diseases.
Introduction
Acid-related gastrointestinal diseases such as gastroesophageal reflux disease, nonerosive reflux disease, gastric ulcers, and nonsteroidal anti-inflammatory drug–associated ulcers are the most common diseases among gastrointestinal disorder. Drug therapies starting from histamine receptor 2 blockers and following proton pump inhibitors (PPIs) demonstrate the efficacy of the inhibition of gastric acid secretion for the treatment of acid-related diseases, and those new drugs dramatically improved the quality of life of patients. However, the level of satisfaction through therapy using the currently available drugs is still inadequate. For example, the control of heartburn and esophageal reflux symptoms during nighttime are difficult under current PPI therapies, and the symptoms of patients during the initial 3 days after PPI therapy are not controlled (Fass and Sifrim, 2009; Chey et al., 2010; Strand et al., 2017). The common properties of the currently available PPIs (e.g., PPIs require chemical transformation to active form and simultaneous activation of H+/K+-ATPase on the parietal cell membrane for inhibition of acid secretion, bind to the target molecule irreversibly, and eliminate from plasma rapidly) often cause insufficient gastric pH control that may result in therapeutic failure (Hunt, 2012; Inatomi et al., 2016).
A new class of drugs, vonoprazan, a potassium-competitive acid blocker (P-CAB) that inhibits gastric H+/K+-ATPase through a different mechanism than PPI, was recently approved in Japan and has demonstrated potent therapeutic efficacies on reflux esophagitis, gastric and duodenal ulcers, low-dose-aspirin/nonsteroidal anti-inflammatory drug–mediated ulcers, and Helicobacter pylori eradication. The efficacy of vonoprazan in patients is reported to be superior to those of PPIs in general. The pharmacological advantages of vonoprazan in human are supposed to result from better control of gastric acid secretion reaching the neutral pH range (pH > 7) and sustainable drug distribution in gastric tissue (Miwa et al., 2017). In this study, we characterized tegoprazan [(S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo[d]imidazole-6-carboxamide] (Fig. 1), a novel P-CAB currently in the late stage of clinical development in Asian countries, and demonstrated its potent and long-lasting pharmacological efficacies in animal acid-related disease models.
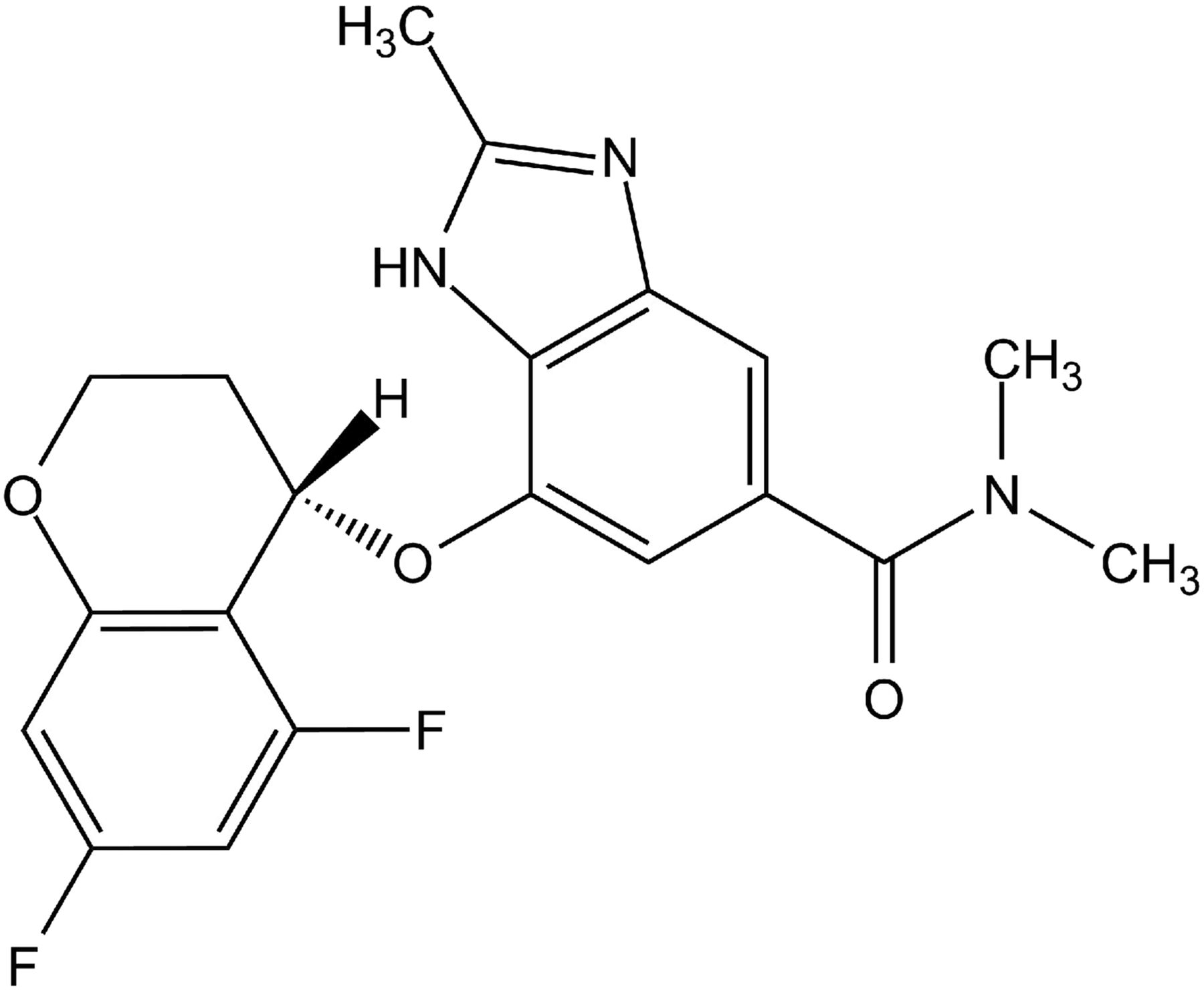
Structure of tegoprazan.
A migrating motor complex (MMC) is well characterized by the appearance of gastrointestinal contractions in the interdigestive state in dogs and humans. The MMC consists of the following four phases: phase I (quiescent period), phase II (period of intermittent and irregular low-amplitude contractions), phase III (period of short bursts of regular high-amplitude contractions), and phase IV (transition period back to phase I). The physiologic importance of gastric MMC pertains to the mechanical and chemical cleansing of the empty stomach in preparation for the next meal. Phase III contraction in the MMC is a series of gastrointestinal prokinetic contractions with slow wave frequency and high amplitude, and is observed repeatedly at 60–90-minute intervals in the empty stomach (Takahashi, 2013). Since evidence has indicated a relationship between gastric acid inhibition and disorder in phase III MMC, we evaluated the pharmacological effects of tegoprazan to evoke gastric phase III MMC contractions in the pentagastrin-treated dog model (Parkman et al., 1998).
Materials and Methods
Chemicals.
Tegoprazan, vonoprazan, omeprazole, and SCH28080 were synthesized by RaQualia Pharma Inc. (Kaminski et al., 1985; Gustavsson and Källstrӧm, 1997; Kajino et al., 2006; Hanazawa and Koike, 2007). All of the reagents and solvents used were analytical grade or higher.
Ethics Approvals and Animals.
All procedures were carried out with the approval of the Animal Ethics Committee at the institutes in which the study was conducted, RaQualia Pharma Inc. (Aichi, Japan) or Pfizer Nagoya Laboratories (Aichi, Japan), according to the Laboratory Animal Welfare guidelines. Porcine stomachs were purchased from a local abattoir. Beagle dogs were purchased from Oriental BioService Inc. (Kyoto, Japan), Marshall BioResources Japan (Tsukuba, Japan), and Beijing Marshall Biotechnology Co., Ltd (Beijing, China).
Preparation of Porcine and Canine Gastric H+/K+-ATPase Vehicles.
Porcine and canine H+/K+-ATPase vehicles were prepared as previously reported with some modifications (Mori et al., 2009). Briefly, a fresh porcine stomach was washed with saline on ice. The fundic mucosa was scraped off from the underlying muscular layer, minced and homogenized in a 250 mM sucrose solution. The homogenate was filtered with gauze, and then centrifuged at 20,000g for 30 minutes at 4°C. The supernatant was centrifuged at 115,000g for 30 minutes at 4°C. The pellet was suspended in a 250 mM sucrose solution and separated by a differential and zonal density gradient centrifugation, 250 mM sucrose (upper layer) and 7% (w/v) Ficoll in 250 mM sucrose (lower layer), at 130,000g for 60 minutes at 4°C with a vertical rotor. The fraction above the Ficoll interface was collected and diluted 10 times with water (ion-tight vehicle), permeabilized by freeze-dry processing (ion-leaky vesicle), and stored at −80°C. Before use, the freeze-dried vesicles were reconstituted with water up to the original volume. The protein concentration of reconstituted vesicles was determined using the BCA Protein Assay Kit protocol (Pierce Biotechnology, Rockford, IL).
Fresh canine stomachs were washed with saline on ice. The fundic mucosa was scraped, minced, and homogenized with a homogenizer in a solution containing 1 mM EGTA, 250 mM sucrose, and 5 mM Tris (pH 7.4 at 4°C). The homogenate was filtered with gauze, centrifuged at 20,000g for 30 minutes at 4°C, and then the supernatant was centrifuged at 115,000g for 30 minutes at 4°C. The pellet was suspended in a 250 mM sucrose solution and separated by density gradient centrifugation (the same construction as used for porcine preparation) at 132,000g for 90 minutes at 4°C with a swing rotor. The following procedures were the same as used for porcine stomach.
Preparation of Human Recombinant H+/K+-ATPase Vehicles.
Human recombinant H+/K+-ATPase was prepared as described previously with some modifications (Mori et al., 2009). Briefly, HEK293 cells stably expressing the human gastric H+/K+-ATPase were prepared by Pfizer Inc. (New York, NY). The cells were suspended in Dulbecco’s modified Eagle’s medium containing 10% heat-inactivated fetal bovine serum, 0.5 mg/ml G418, 0.1 mg/ml zeocin, 100 U/ml penicillin, and 100 µg/ml streptomycin, and were seeded in a T-225 cell culture flask. After 11-day culture in a humidified incubator at 37°C with 5% CO2, the cells were harvested in 1 mM EDTA/phosphate-buffered saline without Mg2+/Ca2+. The cells were centrifuged at 1000 rpm for 5 minutes at 4°C. The packed cells were suspended in a buffer containing 0.5 mM MgSO4, Roche Complete protease inhibitors (Mannheim, Germany), and 50 mM Tris-HCl (pH 7.4), and then homogenized with a Polytron homogenizer (Kinematica AG, Luzern, Switzerland) for 40 seconds. The homogenate was centrifuged at 1000 rpm for 5 minutes at 4°C. The supernatant was recentrifuged at 40,000g for 30 minutes at 4°C. The pellet was suspended in a 250 mM sucrose solution with the Polytron homogenizer and was aliquoted and stored at −80°C until use. The protein concentration of the membrane fraction was determined using the BCA Protein Assay Kit protocol (Pierce).
Measurement of H+/K+-ATPase Activities.
The H+/K+-ATPase activates were determined as described previously with some modifications (Keeling et al., 1988; Mori et al., 2009). The porcine and canine gastric H+/K+-ATPase activates were measured in a 60 µl reaction mixture containing 0.3 µg for porcine or 0.7 µg for canine freeze-dried vesicles (ion-leaky vesicles), test compounds, 5 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, and 40 mM Bis-tris (pH 6.4) at 37°C in a 96-well polystyrene plate. For 0% and 100% inhibition controls, the enzyme reaction was performed in the presence of 1% dimethylsulfoxide and 100 µM SCH28080, respectively. The enzyme reactions using porcine ion-tight vesicles were assayed in a 60 µl reaction mixture containing 2 µg of vehicles, test compounds, 150 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, 17 µM valinomycin (Sigma, St. Louis, MI), and 5 mM Tris (pH 7.4) at 37°C. The reaction was started by the addition of Na2ATP and the mixture was incubated at 37°C for 30 minutes, and then 30 µl of 10% SDS containing antifoam A was added to stop the reaction. Then, 200 µl of the colorimetric reagent to analyze inorganic phosphate consisting of four parts of 10% L-ascorbic acid (pH 5) and one part of 35 mM ammonium molybdate in 15 mM zinc acetate (pH 5) was added into each well. Following incubation at 37°C, the optical density was measured at 750 nm with ARVOsx (PerkinElmer, Waltham, MA). The inorganic phosphate solution prepared from KH2PO4 and K2HPO4 solutions was used as a standard for the inorganic phosphate. Test compounds were dissolved in dimethylsulfoxide at 10 mM and then diluted in the reaction mixture to give appropriate final concentrations. The human recombinant H+/K+-ATPase activity was measured in a 60 µl enzyme reaction mixture containing 10 µg of H+/K+-ATPase vehicle, the test compound, 10 µM oligomycin, 100 nM bafilomycin A1, 10 µM thapsigargin, 10 µM ouabain, 5 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, and 40 mM Bis-Tris (pH 6.4 at 37°C) in a 96-well plate. The reaction mixture was incubated for 50 minutes at 37°C after addition of Na2ATP, and 30 µl of 10% SDS containing antifoam A was dispensed to stop the reaction. The colorimetric analysis was performed in the same procedure for the porcine assay.
Kinetic Analysis of H+/K+-ATPase Inhibition.
The enzyme kinetic study was performed based on the method of porcine ion-leaky assay described previously, except for the amount of vehicle (1 µg), and was tested with various concentrations of potassium (final concentrations of 2.0, 2.5, 3.5, 5.0, and 10 mM KCl) and tegoprazan (final concentrations of 0.15, 0.30, 0.45, and 0.60 µM).
Measurement of Canine Kidney Na+/K+-ATPase Activity.
Canine kidney Na+/K+-ATPase was purchased from Sigma (A-0142). The enzyme was diluted to 7 mg/ml with 250 mM sucrose solution. The canine kidney Na+/K+-ATPase activity was measured in a 60 µl enzyme reaction mixture containing 11 µg of protein, test compound, 100 mM NaCl, 2 mM KCl, 3 mM MgSO4, 3 mM Na2ATP, and 40 mM Tris (pH 7.4 at 37°C) in a 96-well polystyrene plate. For 0% and 100% inhibition controls, the enzyme reaction was performed in the presence of 1% dimethylsulfoxide and 100 µM ouabain, respectively. The reaction was started by the addition of Na2ATP, the mixture was incubated for 30 minutes at 37°C, and then 30 µl of 10% SDS containing antifoam A was added to stop the reaction. The colorimetric analysis was performed in the same method for the porcine assay.
Data Analysis.
All of the H+/K+- ATPase and Na+/K+-ATPase assays were conducted in triplicate. The results of three independent experiments were used for analysis. The nonlinear curve fitting and IC50 determination and the kinetic study using the Lineweaver-Burk plot were analyzed using GraphPad Prism version 5 (GraphPad Software Inc., San Diego, CA). The values are shown as mean ± S.E.M.
Measurement of Plasma and Gastric Juice Concentration of Tegoprazan in Dogs.
Male beagle dogs at 8–11 months old were fasted overnight prior to dosing and dosed tegoprazan with 0.3, 3, and 30 mg/kg by mouth (n = 2) and blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, and 24 hours post dosing. Male Heidenhain pouch (HP) dogs were orally dosed tegoprazan with 1 or 3 mg/kg, and blood and gastric juice were collected at 5 or 16 hours after the drug dose. Plasma and gastric concentrations of tegoprazan were quantified using liquid chromatography–tandem mass spectrometry (API4000 Triple Quadrupole mass spectrometer; Applied Biosystems, Waltham, MA) and calculated using Analyst Program version 1.4.1 (Applied Biosystems). Pharmacokinetic parameters were determined with noncompartmental analysis using WinNonlin version 5.2.1 (Pharsight Corporation, Sunnyvale, CA).
Inhibition of Binding/Function against Pharmacologically Relevant Molecules.
Inhibitions of binding or functional activity against pharmacologically relevant molecules, receptors, ion channels, transporters, and enzymes, were tested in the presence of 10 µM tegoprazan. Regarding the Na+/K+-ATPase, tegoprazan was tested at 30 µM. All of the assays were conducted by Eurofins Cerep (Celle-Lvescault France) under their validated assay methods in the presence of standard inhibitors as an assay control (http://www.cerep.fr/cerep/users/pages/catalog/assay/catalog.asp).
Measurement of Gastric Acid Secretion in the HP Dog Model.
A gastric pouch was constructed according to the method introduced by Heidenhain (1879). Briefly, male beagle dogs (7–12 kg body weight) were anesthetized with midazolam (0.2 mg/kg, i.m.) and medetomidine (0.05 mg/kg, i.m.) and isoflurane inhalation. The abdominal cavity was opened after the injection of atropine sulfate (0.5 mg/ml, 1 ml, i.m.) and infusion of Lactec D (100 ml/h). After exposing the stomach in the surgical field, a portion of the greater curvature opposite the splenic hilum was converted into a pouch with an adequate blood supply from the intact gastroepiploic artery. The main body of the stomach was reconstituted, while the pouch drained into an implanted metal cannula. After closing the pouch, the cannula was brought out of the abdominal cavity through the left lateral abdominal wall. Animals were allowed to recover from surgery for at least at last 3 weeks. The experiment was started after overnight fasting with free access to water. Acid secretion was stimulated by continuous intravenous infusion of histamine (80 μg/mg per hour; Sigma) and maintained throughout the experimental periods. Gastric juice samples were collected by gravity drainage every 15 minutes throughout the experiment. At 90 minutes after initiation of the histamine infusion, tegoprazan, omeprazole, or vehicle (0.5% methylcellulose; Wako, Osaka, Japan) was administrated orally. In the 5-day repeated dosing study, test drug or vehicle was administered orally for 5 days and its inhibitory effect on histamine-stimulated acid secretion was examined on treatment days 1 and 5. The collected gastric juice samples were centrifuged and acidity in the supernatant was determined using an automatic titrator (AUT-501; TOA/DKK, Tokyo, Japan). Gastric acid secretion at intervals of every 15 minutes (acidity × volume of the supernatant) was expressed as milliequivalents per 15 minutes. All results were calculated by MS EXCEL software (Microsoft, Redmond, WA).
Measurement of Gastric pH in Dogs.
Gastrostomy was conducted as follows. The dogs are anesthetized with midazolam (0.2 mg/kg, i.m.) and medetomidine (0.05 mg/kg, i.m.) and isoflurane inhalation. The abdominal cavity was opened after the injection of atropine sulfate (0.5 mg/ml, 1 ml, i.m.) and infusion of Lactec D (100 ml/h). A gastrostomy tube (D12-C, 6 cm; Natsume Seisakusho Co. Ltd., Tokyo, Japan) was connected to the center of the body of the stomach and closed. The gastrostomy tube was taken out from the abdominal cavity through the left lateral abdominal wall and closed. The experiments were started at least 1 month after the surgery.
After overnight fasting, the dogs were placed in suspended-type dog slings and attached to a dip-type reference microelectrode (MI-402; Microelectrode Inc.) in the back. An esophageal pH microelectrode (MI-508; Microelectrode Inc. Bedford, NH) was introduced into the gastrostomy tube and fixed at 10 mm above the gastric lumen. Both microelectrodes were connected to a pH meter (F52; Horiba, Kyoto, Japan) and measured the gastric pH values. During the pH measurement, pentagastrin (6 μg/kg per hour, s.c. infusion; Sigma) was injected continuously. Test drug or vehicle was administered orally or intravenously 1.5 hours after starting the pentagastrin injection and the pH was measured more than 5 hours after drug administration. In the 5-day consecutive administration experiment, drugs were administered before feeding on days 1–4 and the aforementioned procedures were done on day 5. The gastric pH values were gathered at 5- or 6-second intervals and the median of each 15 minutes was used for the analysis. Fig. 6 shows mean of the median from four dogs.
Measurement of Gastric Antral Motility in Conscious Dogs.
The gastric antral motility was determined as described previously (Mikami et al., 2008). Briefly, healthy male beagles weighing 9–15 kg were anesthetized with isoflurane, and the abdominal cavity was opened under aseptic conditions. Extraluminal force transducers (IS-12S; Star Medical, Tokyo, Japan) were sutured onto the seromuscular layer of the gastric antrum 3 cm proximal to the pyloric ring, according to the method from Itoh et al. (1977). The transducer’s lead wires were taken out of the abdominal cavity through a skin incision between the scapulae. After surgery, the dogs were placed in protective jackets and housed in individual cages. Gastric motility was recorded starting at least 2 weeks after surgery. After an overnight fast, the dogs were placed in a shielded room, and gastric motility was recorded in the fasted state. Gastric motility was measured with a telemetry system (GTS-800; Star Medical), and data were acquired on a personal computer with acquisition software (Eight Star; Star Medical). After confirming the incidence of an interdigestive migrating complex (a typical motility pattern of the upper gastrointestinal tract in the fasted state) at regular intervals, test drugs or vehicle (0.1% methylcellulose) were administered orally, and gastric motility was recorded for 8 hours. To measure the gastric motility quantitatively, motor indices that represent areas of contractions were calculated. The signals from the force transducer were acquired on a personal computer and analyzed by processing software (Analyze II; Star Medical).
Results
Tegoprazan Inhibits Porcine, Canine, and Human H+/K+-ATPase Activity.
The inhibitory effects of tegoprazan on porcine, canine stomach H+/K+-ATPase, and recombinant human H+/K+-ATPase expressed in HEK293 cells were tested. The H+/K+-ATPase vehicle preparation method initially provides an ion-tight vehicle. Since the potassium binding site of the H+/K+-ATPase exists inside the vehicle in this preparation, potassium ion outside the vehicle cannot access the ATPase without the presence of ionophore. The ion-tight vehicles were then processed using the freeze-dry procedure to destroy the vehicle structure and provide the ion-leaky vehicle preparation. In the ion-leaky vehicle, potassium can freely access the potassium-binding site in the assay solution (Reenstra and Forte, 1990). We determined the inhibitory activities of tegoprazan on the H+/K+-ATPase using the ion-leaky vehicles. Inhibitions of H+/K+-ATPase purified from porcine, canine stomach, and human recombinant by tegoprazan were dose dependent and the IC50 values were 0.47, 0.29, and 0.52 μM, respectively (Fig. 2; Table 1). These results suggested that there is no species-specific inhibition of H+/K+-ATPase by tegoprazan among those tested. The inhibition of ATPase activity by tegoprazan was also tested using the porcine ion-tight vesicle preparations. The IC50 value of tegoprazan in the porcine ion-tight vesicle was 0.13 μM, which was slightly more potent than that for the porcine ion-leaky vesicle (Table 1).

Inhibition of porcine, canine, and human H+/K+-ATPase and canine Na+/K+-ATPase by tegoprazan was tested with an ion-leaky vehicle. Each enzyme preparation was incubated with tegoprazan at 37°C for 30 minutes (50 minutes for human), and the inorganic phosphate concentration in the reaction was analyzed using the colorimetric method. Each value represents the mean ± S.E.M. of three independent experiments.
The IC50 values of tegoprazan in the in vitro studies
The data for H+/K+-ATPase show the mean values (95% confidence interval) from three independent experiments. Three experiments for canine Na+/K+-ATPase resulted in IC50 > 100 μM.
Tegoprazan Is a Highly Selective Inhibitor of H+/K+-ATPase.
The H+/K+-ATPase is a member of P-type ATPase family. In this family, Na+/K+-ATPase, expressed in renal tubular cells and taking an important role in ion transport in kidney, has a similar molecular structure to H+/K+-ATPase. To identify the selectivity of tegoprazan in the family, the inhibitory effect of tegoprazan on canine kidney Na+/K+-ATPase was determined. Tegoprazan inhibited Na+/K+-ATPase less than 30% at a concentration of 100 μM. The results indicate tegoprazan shows more than 340-fold selective inhibition of canine H+/K+-ATPase over canine Na+/K+-ATPase (Table 1). The selectivity of tegoprazan was further determined against various pharmacologically relevant molecules in vitro. Inhibition of specific-ligand bindings or functional activities by 10 μM tegoprazan on receptors, enzymes, ion channels, and transporters was measured. Ten micromolars of tegoprazan did not inhibit the binding or functional activities of the molecules listed in Table 2 more than 50%, except for rat melatonin receptor ML2 (MT3). Tegoprazan at 10 μM inhibited 81% of the binding of melatonin to ML2 receptor and the IC50 value was 1.2 μM.
Percentage of inhibition of the binding/function of various molecules by tegoprazan
Data show the mean of two data points.
Tegoprazan Inhibits Gastric H+/K+-ATPase in a Potassium-Competitive and Reversible Manner.
To study the kinetics of the inhibition of H+/K+-ATPase by tegoprazan, experiments with various concentrations of potassium and tegoprazan were conducted and analyzed using the Lineweaver-Burk plot. The inhibition of H+/K+-ATPase by tegoprazan was in a potassium-competitive manner as shown in Fig. 3A. This potassium-competitive typical kinetic result indicates that the binding of tegoprazan to H+/K+-ATPase is reversible. The reversibility was also confirmed by the other experiment, which compares the inhibitory potencies of tegoprazan before and after the dilution of tegoprazan concentration in the H+/K+-ATPase enzyme reaction. Tegoprazan at 3 μM inhibited 86% of H+/K+-ATPase activity, whereas the inhibition was decreased to 34% after the dilution of tegoprazan concentration to 0.15 μM. The results suggested that tegoprazan once bound to H+/K+-ATPase at higher concentration was eliminated by decreasing tegoprazan concentration in the reaction mixture (Fig. 3B).

Mechanism of inhibition of porcine H+/K+-ATPase by tegoprazan was determined. (A) The enzyme kinetic analysis of the inhibition of porcine H+/K+-ATPase by tegoprazan was carried out with tegoprazan concentrations at 0, 0.15, 0.30, 0.45, and 0.60 μM, and KCl concentrations at 15, 21, 30, and 60 mM. The results were analyzed with the Lineweaver-Burk plot using GraphPad Prism 5 for Windows. The graph presents a mean of three independent experiments. (B) Binding reversibility of tegoprazan to porcine H+/K+-ATPase was tested. H+/K+-ATPase was incubated with 3 or 1 μM of tegoprazan, and then the tegoprazan concentration in the reaction mixture was diluted to 0.15 or 0.05 μM without any other changes to the composition of the reaction mixture. The graph shows the inhibition of H+/K+-ATPase activity before and after the tegoprazan concentration. Each value represents the mean ± S.E.M. of three independent experiments.
Tegoprazan Distributes Higher in Gastric Juice than in Plasma in Dogs.
To determine the oral exposure of tegoprazan in dogs, the plasma pharmacokinetic profile was tested. Single oral dosing of tegoprazan at 0.3, 3, and 30 mg/kg (the pharmacologically effective dose range) to Beagle dogs was well absorbed into the systemic circulation in a dose-proportional manner (Fig. 4A). The Cmax and AUC0–inf values were 2490 ng/ml and 12,731 ng·h per milliliter, respectively, at oral 3 mg/kg dosing. The elimination of tegoprazan from the blood flow was relatively rapid and the plasma half-life was in the range of 3.3–3.5 hours at the doses tested (Table 3). To understand the exposure of tegoprazan in the targeted disease organ, tegoprazan concentration in gastric juice after oral administration was then determined and compared with that in plasma. Tegoprazan was dosed in HP dogs (n = 3) at 1 or 3 mg/kg orally and gastric juice and blood samples were collected 5 or 16 hours after tegoprazan administration, respectively, and then the drug concentrations were analyzed. The mean tegoprazan concentrations in gastric juice were higher than those in plasma 5 hours after oral 1 mg/kg dosing and 16 hours after 3 mg/kg dosing (Fig. 4B).

Plasma and gastric distributions of tegoprazan were tested. (A) Plasma concentrations of tegoprazan in male dogs following single oral administration at 0.3, 3, and 30 mg/kg (n = 2) were quantified using liquid chromatography–tandem mass spectrometry. The blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, and 24 hours post dosing. (B) Concentrations of tegoprazan in plasma and gastric juice at 5 or 16 hours after oral administration at 1 or 3 mg/kg to HP dogs, respectively, were determined. Each value represents the mean ± S.E.M. from three dogs.
Pharmacokinetic parameters of tegoprazan in dog
Data represent mean (S.D.) of three animals.
Tegoprazan Inhibits Gastric Acid Secretion in the HP Dog Model.
To identify the therapeutic efficacy of tegoprazan in the animal disease model, the effects of single administration of tegoprazan on gastric acid secretion were determined in the HP dogs. The gastric acid secretion was increased by intravenous infusion of histamine treatment (80 μg/kg per hour) to a plateau level within 90 minutes, and this effect was maintained for more than 5 hours. Oral single administration of tegoprazan doses at 0.1, 0.3, and 1.0 mg/kg dose dependently inhibited the histamine-induced gastric acid secretion and complete inhibition was observed at 1 mg/kg within 1 hour after tegoprazan dosing. Tegoprazan at 1 mg/kg in dogs is a dose corresponding to the human clinical dose used in a phase II study, 50 or 100 mg once a day (Clinical Trials, NCT03006874, https://www.clinicaltrials.gov/ct2/show/NCT03006874?term=03006874&rank=1); 1 or 2 mg/kg based on a calculation with the human body weight as 50 kg. In this experiment, the efficacies of all doses of tegoprazan were maintained for more than 5 hours after tegoprazan administration (Fig. 5A). To determine the further long-term efficacy of tegoprazan on control of gastric acid secretion, histamine infusion was started 14.5 hours after tegoprazan administration and the change in gastric acid secretion was monitored for 21 hours; oral tegoprazan at 3 mg/kg inhibited histamine-induced gastric acid secretion for more than 21 hours after dosing (Fig. 5B). To test the rapidness of the pharmacological onset of tegoprazan in comparison with PPIs, the efficacies of tegoprazan and omeprazole on gastric acid secretion were measured on day 1 and on day 5 after consecutive administration. The oral tegoprazan at 0.3 mg/kg inhibited gastric acid secretion potently on day 1 and the inhibitory potency was similar to that observed on day 5 (Fig. 5C). Inhibition of gastric acid secretion by omeprazole at 0.6 mg/kg, a dose corresponding to the human clinical dose (20 or 40 mg once a day), was weak to moderate on day 1; however, on day 5 inhibition was almost complete (Fig. 5D). The experiments demonstrated that tegoprazan shows maximum inhibition of acid secretion from the day of initial treatment while the inhibition by omeprazole on day 1 is weak or moderate in dogs.

Inhibition of gastric acid secretion by tegoprazan and omeprazole was tested. (A) Intravenous histamine infusion (80 μg/kg per hour) to the HP dog was dosed from 1 or 1.5 hours before to 5 hours after the tegoprazan treatment. Single dosing of tegoprazan 0.1, 0.3, and 1.0 mg/kg or vehicle was orally administrated and the gastric acid levels were determined. Each value represents the mean + S.E.M. from four dogs. (B) Tegoprazan 3 mg/kg or vehicle was administrated orally and the histamine infusion was initiated at 14.5 hours after the tegoprazan dosing. The gastric acid levels were monitored starting from histamine dosing to 21 hours. Each value represents the mean ± S.E.M. from three dogs. (C) Effect of tegoprazan on the gastric acid secretion on the initial day of the treatment was compared with that after 5 days of consecutive treatment. Tegoprazan (0.3 mg/kg, by mouth every day) was administrated orally once daily for 5 days. Each value represents the mean ± S.E.M. from three dogs. (D) Omeprazole 0.6 mg/kg was dosed for 5 days in the same dog model and the gastric acid secretion was determined on days 1 and 5. Each value represents the mean ± S.E.M. from the results of three dogs.
Tegoprazan Reverses Pentagastrin-Induced Gastric pH Decrease in Dogs.
The therapeutic efficacy of tegoprazan was also determined in terms of intragastric pH control in dogs. After overnight fasting, subcutaneous pentagastrin infusion with 6 μg/kg per hour in dogs resulted in stable gastric pH of approximately 2. Single oral administrations of 0.3, 1, and 3 mg/kg tegoprazan 1.5 hours after initiation of pentagastrin treatment demonstrated dose-dependent gastric pH escalation. Tegoprazan at 1 mg/kg increased gastric pH by more than 7 at maximum, and the mean intragastric pH > 4 holding time ratio during the experimental period was 54%. Three milligrams per kilogram of tegoprazan achieved stable gastric pH to the neutral range within 1 hour and the effect was sustained throughout the experimental period; the mean intragastric pH > 4 holding time ratio during the experimental period was 89% (Fig. 6A). The efficacy of vonoprazan on the gastric pH control was determined and compared with that of tegoprazan in this model. A single oral dose of vonoprazan at 0.4 mg/kg, a dose corresponding to the human clinical dose (10–40 mg a day), showed a moderate and sustained increase of gastric pH ranging from 4 to 5 and the mean intragastric pH > 4 holding time ratio during experimental period was 33%. Complete pH control corresponding to tegoprazan 3 mg/kg was observed with 1 mg/kg vonoprazan (Fig. 6B). These experiments demonstrated that the efficacy of P-CABs, tegoprazan, and vonoprazan on gastric pH was potent to achieve the neutral range.

Effects of tegoprazan and vonoprazan on the intragastric pH were tested. (A) The gastric fistula dogs were treated with pentagastrin 6 μg/kg per hour subcutaneous infusion, which results in the immediate decrease of gastric pH to ∼2. Oral tegoprazan (at 0.3, 1, and 3 mg/kg) was dosed 1.5 hours after the starting of pentagastrin administration and the pH analysis was continued for 5 hours at 5-minute intervals. (B) Effects of the oral single clinical corresponding dose of vonoprazan (0.4 mg/kg) and higher (1 mg/kg) on the gastric pH were tested. Each result represents the mean of the pH from the results of four dogs.
Tegoprazan Activates Gastrointestinal Motility in Dogs.
In addition to the control of gastric pH, the ability to control the gastrointestinal motility by drugs is another index for therapy in some types of gastrointestinal diseases. The effects of tegoprazan on the gastric motilities were tested in dogs using the telemetry system. The dog gastric antrum after overnight fasting caused a phase III MMC contraction within 1 to 2 hour intervals (Fig. 7A, upper plot). Subcutaneous continuous injection of pentagastrin (6 μg/kg per hour, 1 ml/h), which stimulates gastric acid secretion, completely disappeared in the phase III MMC signals and started irregular and low-height waves, similar to the contractile pattern after feeding (Fig. 7A, middle plot). Oral administration of tegoprazan at 3 mg/kg to the dog after the pentagastrin-treatment suppressed the pentagastrin-induced signals and resumed the phase III MMC signals (Fig. 7A, bottom plot). A similar effect on the gastric phase III MMC contraction was also observed with oral vonoprazan at 1 mg/kg, the dose that increased the gastric pH to the neutral range in dogs (Fig. 7B).

Effect of tegoprazan on the dog gastric motility was studied. (A) The dogs indwelling the force transducer were fasted overnight and the gastric antral motility was determined. During the phase III period MMC contraction was observed, and vehicle treatment right after the contraction signal did not show any change in the phase III contraction pattern. Treatment with pentagastrin (PG) quickly terminated the phase III signals and initiated continuous and random frequency signals. Administration of oral tegoprazan at 3 mg/kg after the pentagastrin infusion restored the phase III contraction signals that disappeared after pentagastrin treatment. The plots show the time courses in three dogs. (B) The experiment was conducted with 1 mg/kg vonoprazan in the same dog model.
Discussion
The in vitro pharmacological studies demonstrated that tegoprazan has potent inhibitory activities in porcine, canine, and human H+/K+-ATPases in the ion-leaky vehicle preparation with IC50 values ranging from 0.29 to 0.52 μM, and there was no species-selective difference in inhibition among the species tested. The IC50 value of tegoprazan in porcine H+/K+-ATPase in the ion-tight vehicle preparation was 0.13 μM. The IC50 value of the canine kidney Na+/K+-ATPase, a highly homologous molecule to H+/K+-ATPase, by tegoprazan was more than 100 μM, and the results suggest that tegoprazan shows more than 340-fold selective inhibition to H+/K+-ATPase over Na+/K+-ATPase in dogs. The study against 115 pharmacologically relevant molecules further revealed the excellent selectivity profile of tegoprazan against a wide range of molecules except for the ML2 (MT3) receptor. Inhibition of H+/K+-ATPase by tegoprazan was in a potassium-competitive and reversible manner, and these profiles were the same as reported for vonoprazan (Kim et al., 2010; Miwa et al., 2017). This result indicates that tegoprazan has the potential to show pharmacological profiles similar to those of vonoprazan in human.
The pharmacokinetic evaluation of a single oral dose of tegoprazan in dogs demonstrated efficient and dose-proportional absorption into the blood flow. However, the plasma half-life of the tegoprazan, 3.3–3.5 hours, was not long enough to maintain a protein-unbounded plasma drug concentration above the IC50 value in H+/K+-ATPase in the in vitro assay at ∼20 hours after oral dosing. We analyzed the concentration of tegoprazan in gastric juice, the target tissue/fluid, and the mean concentration of tegoprazan in gastric juice at 16 hours after the oral 3 mg/kg administration was 603 ng/ml, which was approximately 6-fold higher than that in plasma. The protein-unbounded tegoprazan concentration in this gastric juice was calculated to be 130 nM (the protein binding ratio of tegoprazan in dog plasma was used for the calculation). This was in the range of the in vitro IC50 values of H+/K+-ATPase tested in the canine ion-leaky preparation (290 nM) as well as in the porcine ion-tight preparation (130 nM) which is supposed to maintain the in vivo environment of H+/K+-ATPase better than in the ion-leaky preparation (Reenstra and Forte, 1990). The results suggested that the sustainable existence of tegoprazan in the gastric tissue/fluid enables the long-term gastric acid control in dogs.
The oral tegoprazan administration at the doses corresponding to human use potently suppressed gastric acid secretion from the initial day of the treatment in the dog model. The potency of the inhibition of acid secretion on day 1 was almost the same as that on day 5, and the results indicate tegoprazan showed maximal efficacy from the initial day of the treatment. In contrast, the inhibition of gastric acid secretion by oral omeprazole also at the dose corresponding to human use was weak to moderate on day 1, while after 5 days of daily administration it reached maximum inhibition. In the clinical use of omeprazole and other PPIs, the maximum efficacy on acid suppression is generally obtained 3–5 days after the initiation of drug dosing (Strand et al., 2017). Since the evidence for omeprazole observed in this dog model precisely reflects the delayed onset generally observed in the clinical environment, the results observed for tegoprazan in the same model strongly suggested the opportunity of the potent inhibition of gastric acid secretion by tegoprazan from the initial day of therapy in human use.
We demonstrated that the single dosing of tegoprazan at 1 and 3 mg/kg, the doses corresponding to human use, achieved maximum gastric pH more of than 7, and the single 3 mg/kg dosing of tegoprazan maintained gastric pH of more than 6.5 for 5 hours after drug administration. In another study, single 3 mg/kg tegoprazan continuously inhibited gastric acid secretion until 21 hours after administration. Besides, similar potent and stable control of gastric pH by 1 mg/kg vonoprazan was reported in contrast to the limited potency and short term efficacy of 10 mg/kg lansoprazole, a clinically available PPI, in a rat model (Hori et al., 2010). The aforementioned results suggest that P-CABs may generally have the efficacy to control gastric pH reaching the neutral range for almost a day and the efficacy exceeds that of PPIs. These properties of tegoprazan, a potent and long-lasting acid inhibitor, may be able to contribute to better satisfaction in therapy, e.g., for PPI-refractory patients.
Several studies have shown evidence that an increase in gastric pH with inhibitors of acid secretion stimulate phase III MMC contractions; however, the results are controversial (Gielkens et al., 1998; Parkman et al., 1998). To identify whether the aforementioned evidence could be observed with P-CABs, we determined tegoprazan to be one of the most potent and quick inhibitor of acid secretion in our dog model. We demonstrated that tegoprazan potently and quickly recovered the impaired gastric phase III MMC in the dog model. A similar effect was also identified with another P-CAB, vonoprazan. Since the stable and reproducible pharmacological effects on phase III contraction were observed by tegoprazan as well as vonoprazan, we suggest that our experiments reconfirmed the evidence that gastric acid inhibition mediates phase III contraction. However, the mechanism for the stimulation of phase III contraction by gastric acid inhibition still remains to be elucidated. In the clinical setting, it has been reported that impairment of gastric phase III contraction is attenuated in functional dyspepsia patients and the dyspeptic symptoms in the postprandial state were improved once the gastric MMC was recovered (Takahashi, 2013). The property of tegoprazan to restore the phase III MMC contraction may be a new option in therapeutics to improve the symptoms in functional dyspepsia patients.
P-CAB is a new class of drugs for acid-related gastrointestinal diseases and is positioned as the next generation of PPIs. Clinical evaluation of vonoprazan has demonstrated its potent and long-acting efficacy in patients with gastroesophageal reflux disease and peptic ulcers compared with that of the currently available PPIs. In vitro and in vivo animal pharmacology studies of tegoprazan in this study have demonstrated that tegoprazan has similar pharmacological properties to those of vonoprazan. Tegoprazan is a novel P-CAB that may provide a new option for therapy in gastric acid-related diseases as well as in gastrointestinal motility-impaired diseases in clinical use.
Acknowledgments
We especially thank all of the researchers involved in the RQ-00000004/PF-03922155 program at RaQualia Pharma Inc. and Pfizer Global Research and Development for the support given.
Authorship Contributions
Participated in research design: Takahashi, Take.
Conducted experiments: Takahashi, Take.
Wrote or contributed to the writing of the manuscript: Takahashi, Take.
Footnotes
- Received July 30, 2017.
- Accepted November 14, 2017.
↵1 Current affiliation: Hamamatsu Pharma Research, Hamamatsu, Shizuoka, Japan.
This research study was supported by RaQualia Pharma Inc., and no external funding was received.
Abbreviations
- HP
- Heidenhain pouch
- MMC
- migrating motor complex
- P-CAB
- potassium-competitive acid blocker
- PPI
- proton pump inhibitor
- tegoprazan
- (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo[d]imidazole-6-carboxamide
- Copyright © 2018 The Author(s).
This is an open access article distributed under the CC BY Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}