


Visual Overview

Abstract
Glucagon-like peptide-2 (GLP-2) agonists have therapeutic potential in clinical indications in which the integrity or absorptive function of the intestinal mucosa is compromised, such as in short bowel syndrome (SBS). Native hGLP-2, a 33–amino acid peptide secreted from the small intestine, contributes to nutritional absorption but has a very short half-life because of enzymatic cleavage and renal clearance and thus is of limited therapeutic value. The GLP-2 analog teduglutide (Revestive/Gattex; Shire Inc.) has been approved for use in SBS since 2012 but has a once-daily injection regimen. Pharmacokinetic (PK) and pharmacodynamic studies confirm that apraglutide, a novel GLP-2 analog, has very low clearance, long elimination half-life, and high plasma protein binding compared with GLP-2 analogs teduglutide and glepaglutide. Apraglutide and teduglutide retain potency and selectivity at the GLP-2 receptor comparable to native hGLP-2, whereas glepaglutide was less potent and less selective. In rat intravenous PK studies, hGLP-2, teduglutide, glepaglutide, and apraglutide had clearances of 25, 9.9, 2.8, and 0.27 ml/kg per minute, respectively, and elimination half-lives of 6.4, 19, 16, and 159 minutes, respectively. The unique PK profile of apraglutide administered via intravenous and subcutaneous routes was confirmed in monkey and minipig and translated into significantly greater in vivo pharmacodynamic activity, measured as small intestinal growth in rats. Apraglutide showed greater intestinotrophic activity than the other peptides when administered at less-frequent dosing intervals because of its prolonged half-life. We postulate that apraglutide offers several advantages over existing GLP-2 analogs and is an excellent candidate for the treatment of gastrointestinal diseases, such as SBS.
SIGNIFICANCE STATEMENT Apraglutide is a potent and selective GLP-2 agonist with an extremely low clearance and prolonged elimination half-life, which differentiates it from teduglutide (the only approved GLP-2 agonist). The enhanced pharmacokinetics of apraglutide will benefit patients by enabling a reduced dosing frequency and removing the need for daily injections.
Introduction
Short bowel syndrome (SBS) is a severe, chronic condition characterized by malabsorption due to physical or functional loss of significant portions of the small intestine (Jeppesen et al., 2011). Consequently, patients suffer life-threatening malnutrition, dehydration, and imbalances of fluids and salts resulting from severe diarrhea.
SBS results in a process of adaptation that increases mucosal surface area (Weale et al., 2005; Hasosah et al., 2008). Non-nutritional gut stimulators, including growth factors and hormones, contribute to this. One of these, glucagon-like peptide-2 (GLP-2), is released by the intestinal L-cells into the circulation after nutrient ingestion and acts on the intestine to stimulate growth, increase absorption, promote healing, and maintain intestinal epithelial integrity (Orskov et al., 1986; Xiao et al., 1999; Burrin et al., 2000, Brubaker, 2018).
GLP-2 stimulated growth of the small intestine and colon in mice (Drucker et al., 1996; Tsai et al., 1997; Litvak et al., 1998) but had no effect on the growth of other tissues (Drucker et al., 1996). Additional biologic actions of GLP-2 on the gastrointestinal tract include enhancement of nutrient absorption, stimulation of blood flow, anti-inflammatory activity, improvement of barrier function, and modulation of motility (Nagell et al., 2004; Guan et al., 2006; Meier et al., 2006; Stephens et al., 2006; Bremholm et al., 2009; Hsieh et al., 2009; Moore et al., 2010; Rowland and Brubaker, 2011).
In SBS, GLP-2 increases intestinal absorption by promoting intestinal growth and delays accelerated gastric emptying, gastrointestinal transit, and hypersecretions (Brubaker, 2018).
GLP-2 is of limited use as a therapeutic agent because of the very short circulating half-life (only ∼7 minutes in humans) due to cleavage by dipeptidyl peptidase-IV (DPP4), as well as renal clearance (Drucker et al., 1997; Hartmann et al., 2000, Tavares et al., 2000; Hansen et al., 2007). Consequently, pharmacologically active GLP-2 analogs with a longer half-life were developed.
The GLP-2 analog teduglutide (Gattex, Revestive) has an Ala-to-Gly substitution at position 2 to eliminate the DPP4 cleavage site ([Gly2] hGLP-2 (1–33)) and a terminal half-life of 3.0–5.5 hours by the subcutaneous route in humans (Marier et al., 2008). In patients with SBS, once-daily treatment with teduglutide significantly reduced parenteral nutrition requirements and improved intestinal function (Jeppesen et al., 2011; Burness and McCormack, 2013).
Various additional analogs of GLP-2 with reduced clearance relative to hGLP-2 are under development for SBS, including apraglutide (FE 203799, VectivBio; EudraCT numbers 2017-002486-21 and 2017-002487-41) and glepaglutide (ZP1848; Zealand Pharma; EudraCT numbers 2015-002826-38, 2017-004394-14, and 2018-001429-26).
The GLP-2 analog apraglutide ([Gly2, Nle10, D-Phe11, Leu16] hGLP-2 (1–33)-NH2), identified through chemistry structure-activity relationship studies of lipophilic amino acid substitutions in positions 11 and 16 of [Gly2] hGLP-2 (1–33), showed exceptionally low clearance in in vivo rat pharmacokinetic (PK) screening (⇓⇓).
This paper presents the pharmacology and PK of apraglutide compared directly with teduglutide and glepaglutide. In particular, we show that the optimized chemical structure of apraglutide translates to a superior PK profile, allowing intestinal growth to be achieved with less-frequent dosing in rats.
Materials and Methods
Peptide Synthesis
Peptides used in these studies were prepared as trifluoroacetic acid salts by solid-phase peptide synthesis and purified by reverse-phase high-performance liquid chromatography (HPLC), as previously described (⇓⇓). The peptide identities were verified by mass spectrometry. The peptide batches had purity >90% and peptide content >75%, as measured by analytical HPLC and elemental analysis of N, respectively. Amino acid sequences of the peptides used in these studies are shown in Table 1; the amino acid sequence of glepaglutide was identified from a published patent application (WO06117565A2).
Amino acid sequence and molecular weight of hGLP-2 and GLP-2 analogs
Amino acid substitutions relative to native hGLP-2 are shown in bold underlined letters.
Cell Culture
Human embryonic kidney (HEK) 293 cells (CRL-1573; American Type Culture Collection) were maintained in DMEM containing 5% (v/v) heat-inactivated FBS and 4 mM l-glutamine at 37°C under 5% CO2 in a humidified atmosphere. Cells were passaged 1:3 to 1:6 by trypsinization every 3–4 days. G418 (500 μg/ml) was included in the culture medium for the cells stably transfected with the GLP-1 receptor.
Vectors
hGLP-1 receptor activity was studied in a stable cell line generated with the human GLP-1R1b–expressing clone (pcDNA3.1-hGLP-1R encoding NM_002062) and a pCRE-Luc clone expressing a luciferase reporter gene with two tandem repeats of the cAMP response element (CRE) in HEK293 cells.
Transient transfections in HEK293 cells were also carried out with the human GLP-2R1c–expressing clone (pcDNA3.1-hGLP-2R encoding NM_004246) and a pCRE-Luc clone expressing a luciferase reporter gene with two tandem repeats of the CRE.
An HEK293 stable cell line was generated expressing the rat GLP-1 receptor (pCDNA5/FRT-rGLP-1R encoding NM_012728) and transiently transfected with a pCRE-Luc clone expressing a luciferase reporter gene with two tandem repeats of the CRE.
Transient transfections in HEK293 cells were also carried out with the rat GLP-2 receptor–expressing clone (pCEP4-rGLP2R encoding NM_021848) and a pCRE-Luc clone expressing a luciferase reporter gene with two tandem repeats of the CRE.
Cell Transfection
For the GLP-2 receptor assays, HEK-293 cells were transiently cotransfected with human or rat GLP-2 receptor and a reporter plasmid containing a luciferase gene under the control of cAMP responsive elements. Briefly, DNA plasmids containing cDNA encoding human or rat GLP-2 receptor were prepared for transfection with LipofectAMINE 2000 transfection reagent (Invitrogen, Carlsbad, CA). DNA-LipofectAMINE complexes were added to cells at ∼90% confluence and incubated for 24 hours at 37°C under 5% CO2.
For the cell-based hGLP-1 receptor assay, monoclonal HEK-293 cells stably cotransfected with the hGLP-1 receptor and a reporter plasmid containing a luciferase gene under the control of cAMP responsive elements were used.
Luciferase Reporter Gene Assay
Stably or transiently transfected cells grown in T-175 flasks were dispersed with 5 ml of trypsin EDTA solution, harvested in phenol red–free DMEM containing 5% heat-inactivated FBS and 4 mM L-glutamine, and plated onto 96-well white, clear-bottom plates at 4.5 × 104/90 μl per well (GLP-2 receptor assay) or 2.5 × 104/90 μl per well (GLP-1 receptor assay) and incubated overnight at 37°C under 5% CO2.
Each compound was typically tested in serial dilutions in half log units over a final concentration range of 0.3 pM to 30 nM in the GLP-2 receptor assays and at concentrations up to 1000 nM in the GLP-1 receptor assay. The native ligands, hGLP-2 or hGLP-1, were run as positive controls at 30 nM in the respective assays, and maximal efficacy of compounds was expressed relative to the positive controls run within the same assay. The assay was initiated by addition of compounds (10 μl of 10× working solutions) to each well in the 96-well plates. Blanks (compound dilution buffer) were included on each plate. After compound addition, plates were gently mixed and incubated for 5 hours at 37°C under 5% CO2. After incubation, 100 μl of reconstituted luciferase substrate (Luclite; PerkinElmer, Waltham, MA) was added to each well. The plates were sealed, gently mixed, and incubated for 10 minutes at room temperature in the dark. Luminescence was measured on an Analyst (Molecular Devices, Sunnyvale, CA) plate reader using the default luminescence counting protocol. In each assay, compounds were typically tested in duplicate and were tested in three or more independent assays.
Animals and Housing
Rat.
Adult male Sprague-Dawley rats of approximately 225–275 g b.wt. were obtained from Harlan (Indianapolis, IN). Upon delivery, rats were housed in ventilated Plexiglas cages at ambient temperature (22–25°C) and maintained on a 12-hour light/dark cycle. Food (rodent chow, Teklad 2018 global diet; Harlan) and water were provided ad libitum. The rats were acclimatized to the facility for at least 2 days prior to the initiation of experiments. Rats used in the PK studies were surgically catheterized with polyethylene tubing implanted in one carotid artery for blood collection and one jugular vein for intravenous compound administration. All animal procedures were approved by the Ferring Research Institute Institutional Animal Care and Use Committee and were in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Research Council.
Monkey.
Male adult cynomolgus monkeys weighing 4.64–6.39 kg were housed at Valley Biosystems (West Sacramento, CA) according to the US Department of Agriculture Animal Welfare Act (9CFR) and the conditions outlined in the Guide for Care and use of Laboratory Animals. An intravenous catheter was inserted in the saphenous vein for intravenous dosing. Blood samples were taken by venipuncture from a cephalic vein.
Minipig.
Castrated male Yucatan minipigs weighing 62.2–67.6 kg were housed in individual cages without bedding according to the US Department of Agriculture Animal Welfare Act (9CFR) at Sinclair Research Center (Auxvasse, MO). The minipigs had in-dwelling ear vein catheters surgically inserted for intravenous dosing. Blood samples were taken by venipuncture.
Preparation of Dosing Solutions
In Vitro Assays.
For the in vitro cell-based functional assays, peptides were prepared as 1 mM stock solutions in 100% DMSO and stored at −20°C until use. Stock solutions were serially diluted to 10× working solutions in phenol red–free DMEM supplemented with 0.2% fatty acid–free bovine serum albumin and 1% DMSO.
PK Studies.
For the dosing solutions used in the in vivo PK studies, apraglutide and teduglutide were dissolved in 25 or 50 mM phosphate buffer without additional salts at pH 7.4. For the 5 mg/kg s.c. dose of apraglutide, to dissolve apraglutide at 10 mg free base/ml, 0.1 N sodium hydroxide was added stepwise during formulation, resulting in a final pH of 7.8. hGLP-2 was dissolved in 25 mM phosphate buffer (pH 7.4) isotonic with sodium chloride. Glepaglutide was dissolved in normal saline (0.9% w/v sodium chloride). For the intravenous PK studies, dosing solutions were prepared with single compounds or as a cassette containing a total of three to six compounds (the chemical entity being tested plus other GLP-2 analogs from the chemistry structure-activity relationship program in the same dosing solution). For the subcutaneous PK studies, compound dosing solutions were prepared as single compounds, and animals were dosed with two to five compounds in separate dosing solutions at different subcutaneous sites within the same animal.
In Vivo Pharmacodynamic Studies.
For the in vivo rat intestinal growth pharmacodynamic studies, apraglutide, teduglutide and low doses (30 nmol/kg) of glepaglutide were dissolved in 25 mM phosphate buffer without additional salts at pH 7.4. Glepaglutide (high-dose, 300 nmol/kg) was prepared by dissolving the peptide in sterile water. For all peptides, dosing solutions were prepared to deliver doses ≤100 nmol/kg. Dosing vials were prerinsed with peptide solution at the target dosing concentration to minimize peptide adsorption to the vial, and then dosing solution was prepared in the prerinsed vial. Dosing solutions were stored (4°C) for up to 8 days.
Pharmacokinetics.
Dosing solutions were administered by intravenous bolus injection to catheterized animals or subcutaneously. In the intravenous PK studies, blood samples were collected at multiple time points up to 6 hours postinjection. In the subcutaneous PK studies, blood samples were collected at multiple time points up to 169 hours postinjection. Blood was collected into tubes containing EDTA as an anticoagulant, and after centrifugation, plasma was collected, frozen in dry ice, and stored at −80°C until analysis.
Analysis of Plasma Peptide Concentrations in Plasma.
The concentrations of GLP-2 analogs were determined using a liquid chromatography tandem mass spectrometry method. Standard and internal standard solutions were prepared in 50% DMSO in acetonitrile. Samples were injected into a Jupiter 5-µm, 300-Å C18 HPLC column (50 × 2.0 mm) (Phenomenex, Torrance) coupled to a Shimadzu LC-20AD series - (Shimadzu, Kyoto Japan) system. Analytes were eluted using a gradient method with a mobile phase (A: 0.01% trifluoroacetic acid and 1% formic acid in water; B: 0.01% trifluoroacetic acid and 1% formic acid in 70% acetonitrile) at a flow rate of 0.5 ml/min and detected using an API-4000 triple quadrupole mass spectrometer (Applied Biosystems, Ontario, Canada) in the positive electrospray ionization mode. Analyte concentrations were calculated by linear regression analysis using the peak area ratio of analyte to the internal standard on the Applied Biosystems Analyst software version 1.4.2.
Pharmacokinetic parameters were determined by best fitting of compound concentration-time curves using a noncompartmental curve stripping method (PK Solutions 2.0 software; Summit Research Services, Montrose, CO). The PK parameters assessed were area under the curve from time zero to infinity normalized to dose (AUC∞/unit dose), clearance, maximum observed plasma concentration normalized to dose (Cmax/unit dose), subcutaneous bioavailability (Fsc), elimination half-life, time of the maximum observed plasma concentration (Tmax), and initial apparent volume of the central compartment (Vc).
Rat Intestinal Growth.
Intestinal growth, as determined by wet weight of the small intestine, was measured as a primary pharmacodynamic endpoint in normal rats.
In the once-daily dosing studies, Sprague-Dawley rats (N = 6 per group) received apraglutide and teduglutide once daily for 5 days at equivalent subcutaneous doses of 3–1000 nmol/kg. Rats were euthanized within 24 hours after the last dose. Apraglutide, teduglutide, and glepaglutide were also administered subcutaneously to Sprague-Dawley rats (N = 6 per group) at equivalent doses (30 or 300 nmol/kg) with a dosing interval of 24 (compounds injected once daily for 5 days), 48 (two injections at times 0 and 48 hours), or 72 hours (single injection). The rats were euthanized 96 hours after the first dose or 72 hours after the single dose. The small intestine was flushed with saline, patted with gauze to remove excess saline, and weighed. Intestinal wet weight was normalized to body weight [small intestine weight (gram)/body weight (gram)]. Data generated from multiple separate experiments were combined.
Data and Statistical Analysis.
Concentration-response data from the in vitro reporter gene assays were entered into ActivityBase software (IDBS, UK) and fitted using a four-parameter concentration-response nonlinear regression model to generate the concentration-response curves and determine the maximum receptor activation expressed as the percentage of positive control reference compound [maximum response (Emax)], ED50, and Hill slope. For graphical presentation, representative curves were also generated by fitting the data to a nonlinear regression equation for a sigmoidal curve with variable slope (GraphPad Prism software, La Jolla, CA).
Dose-response data from the intestinal growth assay were fitted using a three-parameter Hill equation to determine ED50 and Emax (GraphPad Prism software): where Y is the effect, Emax is the fitted maximal effect, X is the dose (nanomoles per kilogram), nH is the unitless Hill coefficient, and ED50 is the effective dose generating 50% of the Emax.
where Y is the effect, Emax is the fitted maximal effect, X is the dose (nanomoles per kilogram), nH is the unitless Hill coefficient, and ED50 is the effective dose generating 50% of the Emax.
Data are presented as means with S.E.M., S.D., or 95% confidence interval as noted in the tables and figure legends. ANOVA with Newman-Keuls multiple comparison post hoc test was used for comparisons of intestinal growth data treatment effects. P values <0.05 were considered significant.
Results
Apraglutide Is Highly Potent and Selective for Activation of the GLP-2 Receptor.
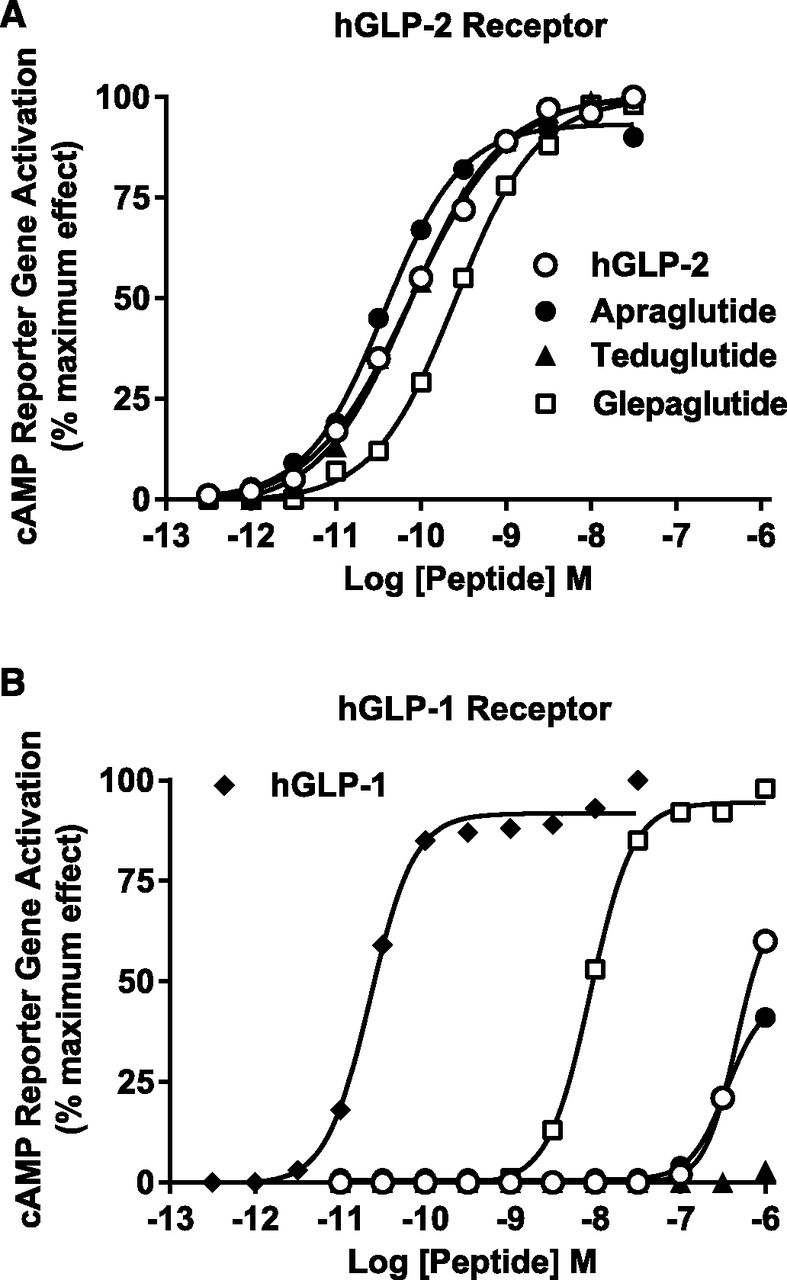
The in vitro activities of apraglutide and the other peptide analogs are summarized in Table 2, and representative concentration-response graphs are shown in Fig. 1. Apraglutide was 2-fold more potent than native hGLP-2 and teduglutide for activation of the hGLP-2 receptor. Apraglutide, hGLP-2, and teduglutide had similar potency at the rGLP-2 receptor. Glepaglutide was less potent at both the hGLP-2 and rGLP-2 receptors relative to apraglutide. All peptides had ≥95% Emax at the hGLP-2 receptor and ≥87% Emax at the rGLP-2 receptor. Apraglutide and teduglutide were highly selective for the hGLP-2 receptor versus the hGLP-1 receptor, with EC50 values >1000 nM (the highest concentration tested) at the hGLP-1 receptor. Glepaglutide fully activated the hGLP-1 receptor and had selectivity of <50-fold for activation of hGLP-2 receptor versus hGLP-1 receptor (GLP-1/GLP-2 ratio = 36 vs. >33,000 for apraglutide and >11,000 for teduglutide) (Table 2). Similar selectivity was observed for the rGLP-2 receptor versus the rGLP-1 receptor. This indicated that GLP-1 receptor would not play a significant role in the pharmacodynamics of apraglutide in the intestine. The peptides tested at concentrations up to 1000 nM had no activity in a recombinant human glucagon receptor assay (data not shown).
Agonist potency and efficacy of peptides for the human and rat GLP-2 receptors and selectivity vs. human GLP-1 receptor in cell-based functional assays of cAMP responsive activation
Values are means with 95% confidence interval in parenthesis or means ± S.E.M. of three or more independent assays.

Apraglutide is potent and selective for the GLP-2 receptor. Apraglutide, hGLP-2, and GLP-2 analogs’ concentration-response curves for functional activation of hGLP-2 receptor (A) or hGLP-1 receptor (B). Data are expressed as a percentage of the maximum response to the native receptor ligand (hGLP-2 or hGLP-1) for the stimulation of a cAMP responsive reporter gene in HEK-293 cells cotransfected with receptor and reporter gene. Each peptide was tested in at least three separate experiments. Data shown are averaged duplicates from a representative experiment. Concentration responses were fitted to the data using nonlinear regression for a sigmoidal curve (GraphPad Software).
Apraglutide (1 µM) was also tested against a panel of 73 receptors, five ion channels, and three transporters (CEREP, Redmond, WA) and had no significant binding affinity (<25% inhibition of control specific binding, results not shown) against receptors other than the GLP-2 receptor. The panels of receptors, ion channels, and transporters against which apraglutide was evaluated are included as Supplemental Data.
Apraglutide Has Unique Pharmacokinetic Properties of Very Low Clearance and Long Elimination and Terminal Half-Lives.
Rat intravenous PK data for apraglutide compared with hGLP-2, teduglutide, and glepaglutide are summarized in Fig. 2A and Table 3. As expected, native hGLP-2 had very rapid clearance (25 ml/kg per minute) and a short elimination half-life (6.4 minutes). Stabilization of the DPP4 cleavage site, as exemplified by teduglutide, decreased clearance by 2.5-fold to 9.9 ml/kg per minute. A further 4-fold reduction in clearance was observed with the C-terminal polylysine analog glepaglutide (clearance 2.8 ml/kg per minute). The clearance of apraglutide was 10-fold lower than that of glepaglutide. Initial intravenous PK studies, in which apraglutide was tested at 0.2 mg/kg and blood samples were taken through 2-hour postdose, demonstrated a clearance of only 0.27 ml/kg per minute. The elimination half-life of apraglutide was 159 minutes (Table 3). This very low clearance and long elimination half-life of apraglutide was confirmed in a follow-up intravenous PK study conducted with apraglutide dosed at 0.1 mg/kg and blood samples collected through 5-hour postdose (data not shown). The clearance of apraglutide in the follow-up intravenous PK study was almost 50-fold lower than the clearance of teduglutide (Table 3).

Apraglutide has low clearance in rat, monkey, and minipig PK studies. Graphs show the time course of plasma peptide concentrations after intravenous bolus injection (mean ± S.D.). Peptides were administered to (A) Sprague-Dawley rats, dose normalized to 0.2 mg/kg (n ≥ 3); (B) adult male cynomolgus monkeys (n = 4), at a dose of 0.1 mg/kg; and (C) adult male Yucatan minipigs (n = 3), apraglutide 0.025 mg/kg and teduglutide 0.1 mg/kg.
PK parameters after intravenous bolus administration
Rat: PK studies were conducted in male Sprague-Dawley rats, and PK parameters were determined by analyzing compound concentration-time curves using a noncompartmental curve stripping method. Doses are milligrams free base per kilogram body weight. Values are means ± S.D.
Very low clearance and long elimination half-life were also shown for apraglutide in direct PK comparisons of intravenous apraglutide and teduglutide in the monkey and minipig. Apraglutide clearance after intravenous administration was 0.046 ml/kg per minute in the monkey PK study and 0.032 ml/kg per minute in the minipig PK study. The clearance of apraglutide was more than 50-fold lower than that observed for teduglutide in monkey and 30-fold lower in minipig (Table 4). Similarly, the elimination half-life of apraglutide after intravenous administration was 9- to 11-fold longer than that of teduglutide in both monkey (474 vs. 43 minutes) and minipig (782 vs. 88 minutes). Average plasma concentrations of apraglutide and teduglutide in monkey and minipig over time are shown in Fig. 2, B and C, respectively.
PK parameters after intravenous bolus administration
Monkey and minipig: PK studies were conducted in male cynomolgus monkeys and castrated male Yucatan minipigs, respectively. PK parameters were determined by best fitting of compound concentration-time curves using a noncompartmental curve stripping method and presented. Doses are milligrams free base per kilogram body weight. Values are means ± S.D.
The PK profiles of apraglutide and teduglutide were also characterized after administration by single-dose bolus subcutaneous injection (Fig. 3; Table 5). In rats, the subcutaneous terminal half-life of apraglutide was appreciably longer than its elimination half-life by the intravenous route (197 minutes at 0.1 mg/kg i.v. vs. 701 minutes at 1 mg/kg s.c. or 1349 minutes at 5 mg/kg), suggesting slow release from the subcutaneous space into the circulation. The observed long value of Tmax ≥400 minutes postdose may reflect contributions of the long half-life of the peptide in the circulation and its slow absorption from the subcutaneous injection site (Fig. 3A; Table 5). The subcutaneous bioavailability of apraglutide was good (74% and 54% at 1 and 5 mg/kg, respectively). In contrast, teduglutide had a terminal half-life of about 31 minutes, suggesting that it was absorbed fairly rapidly and also with good bioavailability.

Apraglutide has sustained exposure levels after subcutaenous dosing in rat, monkey, and minipig. Graphs show the time course of apraglutide and teduglutide concentrations in plasma (mean ± S.D.) by the subcutaneous route. S.E. bars that are not visible are encompassed within the symbol. (A) Figure shows PK profile of apraglutide or teduglutide after subcutaneous bolus injection at 5 mg/kg in Sprague-Dawley rats (n ≥ 3). (B) Adult male cynomolgus monkeys received apraglutide (n = 4) or teduglutide (n = 4) at 0.25 mg/kg by subcutaneous bolus injection into the lower back. (C) Adult male Yucatan minipigs received apraglutide (n = 5) and teduglutide (n = 4) at a dose of 0.15 mg/kg, administered by subcutaneous bolus injection into the neck.
PK parameters after subcutaneous bolus administration
Rat: PK studies were conducted in male Sprague-Dawley rats. PK parameters were determined by best fitting of compound concentration-time curves using a noncompartmental curve stripping. Doses are milligrams free base per kilogram body weight. Values are means ± S.D.
Terminal half-life and Tmax were also notably longer for apraglutide than for teduglutide in the monkey and minipig subcutaneous PK studies (Table 6). The subcutaneous terminal half-life of apraglutide [1941 minutes (32 hours) in monkey and 1808 minutes (30 hours) in minipig] was additionally longer than the intravenous elimination half-life (474 minutes in monkey and 782 minutes in minipig), suggesting slow release from the subcutaneous injection site also in these species. Accordingly, the observed apraglutide Tmax was over 1300 minutes (22 hours) in both monkey and minipig (6- to 10-fold longer than those observed for teduglutide). The bioavailability of apraglutide was 32% in monkey and 43% in minipig. Average plasma concentrations of apraglutide and teduglutide over time after subcutaneous administration to monkey and minipig are shown in Fig. 3, B and C, respectively.
PK parameters after subcutaenous bolus administration
Monkey and minipig: PK studies were conducted in male cynomolgus monkeys and castrated male Yucatan minipigs, respectively. PK parameters were determined by best fitting of compound concentration-time curves using a noncompartmental curve stripping. Doses are milligrams free base per kilogram body weight. Values are means ± S.D.
Apraglutide Is Potent and Long-Acting in a Pharmacodynamic Assay of Rat Intestinal Growth.
To determine pharmacodynamic activity, apraglutide and the other GLP-2 analogs were tested in a rat model of intestinal growth (intact animals, gain in small intestine wet weight). In once-daily dosing studies, complete dose-response curves were generated for apraglutide and teduglutide (dose range tested was 3–1000 nmol/kg injected s.c. once daily for 5 days). At each dose, apraglutide had significantly greater effect on intestinal growth than teduglutide (P < 0.05; Fig. 4). The dose-response data were fitted to a three-parameter Hill equation to estimate Emax and potency (ED50). Apraglutide was more potent and had a greater Emax than teduglutide (ED50 25 vs. 68 nmol/kg and Emax 63% vs. 41%, respectively, Fig. 4).

Effect of apraglutide and teduglutide on stimulation of rat small intestine growth. Peptides were administered by subcutaneous injection once daily for 5 days at the doses of 3–1000 nmol/kg. On day 5 (96 hours after the first dose), small intestines were collected and weighed. Intestine weight for each rat was normalized to body weight and expressed as the percent increase over the mean of a vehicle control group run in the same experiment. A typical experiment involved six animals per group, and the results of multiple experiments were combined. Values are plotted as means with S.E.M. (n = 17–36). S.E. bars that are not visible are encompassed within the symbol. To compare the response of apraglutide- to teduglutide-treated groups, data for each dose were analyzed by two-way ANOVA and Newman-Keuls test for post hoc pairwise multiple comparisons. *P < 0.05 for apraglutide treatment group vs. the corresponding teduglutide treatment group at the same dose. Dose-response curves were fitted to sigmoidal curve with variable slope. Fitted parameters of ED50 and Emax with 95% confidence intervals (CI) are summarized in the accompanying table.
We also tested the intestinotrophic activity of apraglutide, teduglutide, and glepaglutide at less-frequent dosing intervals to gain insight into their duration of action. The peptides were administered once daily (five doses given at a dosing interval of 24 hours) or every 48 hours (two doses at times 0 and 48 hours) at doses of 30 or 300 nmol/kg, with the small intestine collected 96 hours after the first dose. Additionally, peptides were tested by administration of a single dose, and intestines were collected 72 hours postdose. Relative to vehicle control, all peptides significantly increased small intestine weight at the 24- and 48-hour dosing intervals at both doses (Fig. 5, A and B). In the single-injection study, apraglutide was active at both doses, glepaglutide was active only at the high dose, and teduglutide was not active at either dose (Fig. 5C). For all three dosing intervals, at both doses, apraglutide had a significantly greater effect on small intestine weight compared with the other GLP-2 analogs (Fig. 5).

Effect of apraglutide and teduglutide on stimulation of rat small intestine growth with less-frequent dosing. Apraglutide, glepaglutide, or teduglutide were administered to rats by s.c. injection at doses of 30 or 300 nmol/kg once daily (A) or every 48 hours at 0 and 48 hours (B) for 5 days, and small intestines were collected and weighed on day 5 (96 hours after the first dose). The peptides were also tested after a single injection, and small intestines were collected and weighed 72 hours postinjection (C). Intestine weight for each rat was normalized to body weight and expressed as percent increase over the mean of a vehicle control group run in the same experiment. A typical experiment involved six animals per group, and the results of multiple experiments were combined. The data are presented as means with S.E.M. (n = 6–42). Data for vehicle control groups are not plotted. The values for the mean of the vehicle groups were 0%, with an S.E.M. value of 0.6 (n = 108) for the 24-hour interval, 1.1% (n = 42) for the the 48-hour dosing interval, and 1.0% (n = 24) for the single injection with 72-hour postinjection collection. Data for each dosing interval were analyzed by one-way ANOVA and Newman-Keuls test for post hoc pairwise comparisons. *P < 0.05 vs. corresponding vehicle control; ^P < 0.05 vs. corresponding apraglutide treatment group.
Similarly, administration of apraglutide (3 mg/kg; three times per week) to male and female mice significantly increased small intestinal weight (P < 0.001) and length (P < 0.001) at termination at 3 weeks (Wheeler et al., 2018). Further, gains were observed when animals were terminated after 10 weeks (P < 0.01). In addition, both crypt depth and villus height and colon weight and length were markedly increased at 3 weeks of apraglutide administration (P < 0.001), but although these effects were maintained, they did not improve further with continued apraglutide administration.
Increases in intestine weight and length were also observed in a 26-week study in rats administered apraglutide (0.5–12 mg/kg per dose). Intestine length was increased by up to 23.5%, and intestine weight was increased by up to 113.8% (data not shown).
Discussion
Apraglutide is a novel GLP-2 analog with very low clearance. Its few amino acid substitutions (Table 1; Wiśniewski et al., 2011, 2016) confer unique PK properties, namely very low clearance and high protein binding, that enable a long in vivo half-life without conjugation or other major modifications to the peptide. The direct comparisons reported here for apraglutide versus native hGLP-2 and other GLP-2 analogs currently in the clinic (teduglutide) or in clinical trials (glepaglutide) for the treatment of SBS show that apraglutide has superior PK and pharmacodynamic profiles. In addition, they demonstrate that these translate to greater biological activity in vivo.
The attractive physiological and pharmacological activities of GLP-2 have resulted in many efforts to slow down the very rapid clearance of the native peptide so that it can be developed into a therapeutic agent. GLP-2 is cleaved by the enzyme DPP4, which is widely distributed in the circulation and various tissues, including capillaries of the small intestine (Hansen et al., 1999). A variety of strategies can increase elimination and terminal half-lives of peptides to create therapeutic agents, including amino acid substitutions to prevent enzyme binding or promote protein binding, and conjugation with larger molecular entities, such as fusion proteins or polyethylene glycol, to increase peptide size and reduce renal clearance (Werle and Bernkop-Schnürch, 2006; Nestor, 2009). These have been applied to the GLP-2 peptide. The GLP-2 analog teduglutide, which is approved for the treatment of SBS, has amino acid substitutions that eliminate the DPP4 enzymatic cleavage site. Another GLP-2 analog currently being investigated in clinical trials for the treatment of SBS, glepaglutide, has a polylysine moiety incorporated at the C terminus to reduce renal clearance. In addition, the introduction of specific lipophilic amino acid substitutions at positions 11 and 16 unexpectedly conferred very low systemic clearance to a third peptide analog in development for the treatment of SBS, apraglutide.
The structural modifications of apraglutide were targeted at improving the PK profile while preserving potency and selectivity. In cell-based assays of receptor activation, apraglutide retained potency, efficacy, and selectivity at the GLP-2 receptor comparable to native hGLP-2 and teduglutide (Fig. 1; Table 2). The cell-based reporter gene assays also showed marked selectivity of apraglutide and teduglutide for rat GLP-2 receptors over GLP-1 receptors. The GLP-1 receptor thus does not play a significant role in the pharmacodynamics of apraglutide in the intestine. Glepaglutide was less potent than apraglutide at the GLP-2 receptor and less selective versus the GLP-1 receptor (Fig. 1; Table 2).
An extensive preclinical toxicology program has not revealed any safety concerns with apraglutide. After exposure of apraglutide (0.5–12 mg/kg) for up to 26 weeks in rat (n = 160), all rats that received ≥3 mg/kg per dose showed minimal to moderate hyperplasia of the crypt/villus in the duodenum and jejunum compared with controls. The effects were dose-dependent, indicating that this was a treatment effect. Treatment-related minimal hyperplasia of the crypt was also observed in the colon, cecum, and rectum, but fewer animals were affected. Some bile duct hyperplasia was observed in female rats, but this was not dose-related, so the relationship of this finding to apraglutide treatment is unclear (data not shown). No colonic polyps were observed. However, in light of the potential risk of accelerated growth of colon polyps and neoplasms with teduglutide treatment, a comprehensive preclinical carcinogenicity program has been developed for apraglutide.
PK studies of apraglutide administered intravenously to rat, monkey, and minipig clearly demonstrated its low clearance (Fig. 2). Native hGLP-2 administered to rat was rapidly cleared from the circulation as expected due to known mechanisms of DPP4 cleavage and renal clearance (Drucker et al., 1997; Tavares et al., 2000; Hansen et al., 2007). Stabilization of the DPP4 cleavage site of native hGLP-2, as exemplified by teduglutide, reduced clearance, and further reductions in clearance were observed for glepaglutide (∼10-fold reduction relative to native hGLP-2). Clearance of apraglutide was ∼100-fold lower than for native hGLP-2 in rat and 30- to 50-fold lower than for teduglutide in rat, monkey, and minipig (Fig. 2; Table 3 and 4). These results suggest that elimination of DPP4 cleavage only partly reduces clearance of native GLP-2; it must be augmented with additional modifications to obtain GLP-2 analogs with a low clearance and long half-life. The mechanisms responsible for the unique PK profile of apraglutide, including the contribution of plasma protein binding to lowering clearance, are under investigation.
Rat PK studies demonstrated that apraglutide has good subcutaneous bioavailability (54%–74%). Apraglutide subcutaneous terminal half-life in rat, monkey, and minipig was longer than the equivalent intravenous elimination half-life, probably due to delayed absorption from the subcutaneous space. Tmax and terminal half-life of apraglutide were considerably longer than those of teduglutide after subcutaneous administration in rat, monkey, and minipig (Fig. 3; Table 5 and 6).
The present studies, in addition to confirming that the very low clearance of apraglutide is observed in several animal models, demonstrated that the prolonged terminal half-life of apraglutide translated to greater pharmacodynamic activity relative to GLP-2 analogs with shorter terminal half-lives. Intestinal growth was the primary pharmacodynamic endpoint in vivo. The stimulation of intestinal growth by apraglutide supports that previously reported in a mouse model of gluten sensitivity (Wheeler et al., 2018). In once-daily dosing studies in normal rats, apraglutide induced dose-related increases in small intestine growth (Fig. 4), with an ED50 of 25 nmol/kg and a 63% increased Emax over vehicle control. Consistent with its prolonged terminal half-life, apraglutide showed superior stimulation of small intestine growth in vivo relative to comparator GLP-2 analogs. In the complete dose-response study, a direct comparison of once-daily dosing of apraglutide and teduglutide showed that apraglutide had significantly greater effect at each dose, and the fitted dose-response curves showed greater Emax and potency for apraglutide (Fig. 4). Intestine weight was normalized by body weight to decrease the rat-to-rat variation in the assessment of intestine weight. Although there was a moderate increase (≈10%) in body weight at some of the higher doses of apraglutide, this does not impact the interpretation for intestinal trophic effects.
In studies examining the efficacy of GLP-2 analogs administered either once daily or less frequently, apraglutide showed greater ability to stimulate small intestine growth in rat at each dosing interval relative to teduglutide and glepaglutide when directly compared at two doses (30 and 300 nmol/kg; Fig. 5). These results show that the longer terminal half-life of apraglutide translated to greater pharmacological activity in vivo and suggest the potential for less-frequent dosing to achieve efficacy.
Indeed, the efficacy of apraglutide administered less frequently has been demonstrated in a neonatal piglet SBS model (Slim et al., 2019). Apraglutide was administered subcutaneously on days 0 and 4 to preterm piglets with 75% intestinal resection with jejunocolic anastomosis. After 7 days, significant intestinal lengthening from the postresection measurement was observed in piglets receiving apraglutide compared with piglets fed saline (11.5 cm vs. −4.5 cm; P = 0.001). Similarly, piglets treated with apraglutide had significantly greater small intestinal weight on day 7 (0.15 g/cm vs. 0.12 g/cm; P = 0.004), despite equivalent weight gain.
Apraglutide is expected to provide a major contribution to patient care by enabling once-weekly (instead of daily) administration, which in turn improves quality of life and treatment compliance. The reduced injection frequency provides significant clinical effects while increasing patient comfort and acceptability and decreasing the risk of injection site reactions. Clinical trials of once-weekly subcutaneous apraglutide in adult patients are currently underway (NCT 03415594/NCT 03408132). Data from these clinical trials will help determine whether the benefits of the increased half-life and elevated potency of apraglutide are offset by any unwanted effects. Apraglutide had a positive safety profile in a phase 1A trial that completed in 2017, being well tolerated in healthy human volunteers (Bolognani et al., 2019). Clinical evidence demonstrates that GLP-2 agonists have the potential to address unmet medical needs for patients with serious gastrointestinal disorders. Patients receiving teduglutide for SBS in clinical trials had significant and meaningful reductions in the requirement for parenteral nutritional support (Jeppesen et al., 2011; Burness and McCormack, 2013). Teduglutide was also shown to improve the symptoms of Crohn’s disease, but the results were not statistically significant (Buchman et al., 2010; Blonski et al., 2013).
An additional potential therapeutic indication for GLP-2 agonists is the prevention or treatment of chemotherapy-induced intestinal damage (mucositis) that causes gastrointestinal side effects, including diarrhea and infections from intestinal bacteria. Data from animal models suggest that GLP-2 agonists can ameliorate the deleterious effects of chemotherapy and radiation on the gastrointestinal tract (Tavakkolizadeh et al., 2000; Boushey et al., 2001; Booth et al., 2004; Torres et al., 2007; Rasmussen et al., 2010; Kissow et al., 2012).
GLP-2 agonists may thus have several valuable therapeutic applications. An attractive feature of GLP-2 treatment is that its biological effects are largely specific for the gastrointestinal tract. One of the first identified biological actions of GLP-2 was stimulation of intestinal growth. Although GLP-2 increased proliferation of crypt cells and decreased enterocyte apoptosis, resulting in increased mucosal surface area and villus height with normal morphology, GLP-2 did not impact the growth of the intestinal muscle layers or stimulate proliferation in other organs (Drucker et al., 1996; Tsai et al., 1997; Litvak et al., 1998). In subsequent studies, GLP-2 agonists demonstrated anti-inflammatory effects in rodent models of intestinal inflammation (Boushey et al., 1999; Alavi et al., 2000; Sigalet et al., 2007; Arda-Pirincci and Bolkent, 2011; Alters et al., 2012).
The mechanisms involved in the intestinotrophic and anti-inflammatory effects of GLP-2 are not completely understood. GLP-2 receptor expression is most abundant in the small intestine, colon, and stomach (Munroe et al., 1999), with an expression pattern that is highly localized to specific cell types, including intestinal subepithelial myofibroblasts, enteroendocrine cells, and enteric and nodose ganglia of vagal neurons (Yusta et al., 2000; Bjerknes and Cheng, 2001; Ørskov et al., 2005; Guan et al., 2006; Nelson et al., 2007; Koopmann et al., 2008). Since the GLP-2 receptor is not expressed on intestinal epithelial cells, the major site of GLP-2 proliferative and protective actions, the effects of GLP-2 are thought to be mediated indirectly through release of growth factors, including insulin-like growth factor 1, keratinocyte growth factor,receptor tyrosine-protein kinases (ErbB) ligands, and other mediators, such as vasoactive intestinal peptide, that act in a local paracrine fashion to elicit biological actions (Rowland and Brubaker, 2011).
In summary, apraglutide is a potent and selective agonist at the GLP-2 receptor with remarkably low clearance and prolonged elimination half-life, endowing it with superior in vivo pharmacodynamic activity as demonstrated by its ability to induce intestinal growth. Such reduction in peptide clearance by substitution of a limited number of amino acid residues is unprecedented in the field of peptide therapeutics and may lessen challenges presented by high clearance of the native peptide. Apraglutide is in development for SBS. From a clinical perspective, the improved pharmacokinetic profile of this peptide enables less-frequent drug administration and would thus provide considerable benefits in the care of patients with SBS.
Acknowledgments
We thank Denise Riedl and Michael Dunn for data auditing; Leslie Callejas-Dominguez for managing the vivarium; and Scott Rey, Jeannine Toy, and Erin La for technical assistance with the experiments.
Authorship Contributions
Participated in research design: Hargrove, Alagarsamy, Croston, Laporte, Qi, Srinivasan, Schteingart, Rivière.
Conducted experiments: Hartwig, Lu, Posch, Wiśniewska.
Contributed new reagents or analytical tools: Sueiras-Diaz, Wiśniewski.
Performed data analysis: Hargrove, Laporte, Qi, Srinivasan, Lu, Posch.
Wrote or contributed to writing of the manuscript: Hargrove, Croston, Qi, Srinivasan, Wiśniewski, Schteingart, Rivière, Dimitriadou.
Footnotes
- Received August 22, 2019.
- Accepted February 18, 2020.
The work herein described was funded by Ferring Pharmaceuticals.
Portions of this work were previously presented at Digestive Diseases Week (2011), American Peptide Symposium (2011), AAPS National Biotech Conference (2011).
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- apraglutide
- [Gly2, Nle10, D-Phe11, Leu16] hGLP-2 (1–33)-NH2
- AUC∞
- area under the concentration curve from time zero to infinity
- CRE
- cAMP response element
- DMEM
- Dulbecco’s modified Eagle’s medium
- DPP4
- dipeptidyl peptidase-IV
- Emax
- maximum response
- Fsc
- subcutaneous bioavailability
- GLP-1
- glucagon-like peptide-1
- GLP-2
- glucagon-like peptide-2
- GLP-1R
- glucagon-like peptide-1 receptor
- GLP-2R
- glucagon-like peptide-2 receptor
- HEK
- human embryonic kidney
- hGLP-1
- human glucagon-like peptide-1
- hGLP-2
- human glucagon-like peptide-2
- HPLC
- high-performance liquid chromatography
- PK
- pharmacokinetics
- rGLP-1
- rat glucagon-like peptide-1
- rGLP-2
- rat glucagon-like peptide-2
- SBS
- short bowel syndrome
- Tmax
- time of the maximum observed plasma concentration
- Vc
- apparent volume of the central compartment
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}