


Abstract
Abicipar pegol (abicipar) is a novel DARPin therapeutic and highly potent vascular endothelial growth factor (VEGF) inhibitor intended for the treatment of neovascular age-related macular degeneration (nAMD). Here we develop a translational pharmacokinetic/pharmacodynamic (PK/PD) model for abicipar to guide dosing regimens in the clinic. The model incorporated abicipar-VEGF binding kinetics, VEGF expression levels, and VEGF turnover rates to describe the ocular and systemic PK data collected from the vitreous, aqueous humor (AH), choroid, retina, and serum of rabbits after a 1-mg abicipar intravitreal (IVT) dose. The model was translated to humans using human-specific mechanistic parameters and refitted to human serum and AH concentrations from patients with diabetic macular edema and nAMD. The model was then used to simulate 8-, 12- (quarterly), and 16-week dosing intervals in the clinic. Simulations of 2 mg abicipar IVT at 8-week or quarterly dosing in humans indicates minimum steady-state vitreal concentrations are maintained above both in vitro IC50 and in vivo human IC50 values. The model predicted virtually complete VEGF inhibition for the 8-week and quarterly dosing schedule during the 52-week treatment period. In the 16-week schedule, clinically significant VEGF inhibition was maintained during the 52-week period. The model quantitatively described abicipar-VEGF target engagement leading to rapid reduction of VEGF and a long duration of VEGF inhibition demonstrating the clinical feasibility of up to a 16-week dosing interval. Abicipar is predicted to reduce IVT dosing compared with other anti-VEGF therapies with the potential to lessen patient treatment burden.
SIGNIFICANCE STATEMENT Current anti‐VEGF treatments for neovascular age-related macular degeneration require frequent (monthly) intravitreal injections and monitoring, which increases patient burden. We developed a mechanistic pharmakinetic/pharmadynamic model to describe the interaction between abicipar (a novel VEGF inhibitor) and VEGF to evaluate the duration of action. The model demonstrates extended abicipar-VEGF target engagement leading to clinical feasibility of up to a 16-week dosing interval. Our model predicted that abicipar 8-week and quarterly dosing schedules maintain virtually complete VEGF inhibition during the 52-week period.
Introduction
Age-related macular degeneration (AMD) represents the major cause of irreversible moderate or severe visual impairment in people 50 years of age or older worldwide (Flaxman et al., 2017). An advanced form of the disease, neovascular AMD (nAMD), accounts for 90% of the legal blindness caused by AMD (Ferris et al., 1984). Antivascular endothelial growth factor (VEGF) intravitreal (IVT) injections remain the preferred treatment of nAMD (Schmidt-Erfurth et al., 2014; American Academy of Ophthalmology Retina/Vitreous Panel, Preferred Practice Pattern®Guidelines, 2015). VEGF-A mediates retinal and choroidal angiogenesis, and anti-VEGF treatments are the cornerstone therapies for AMD. In patients with an early form of AMD, VEGF expression has been shown to increase in the retinal pigmented epithelium (Famiglietti et al., 2003), leading to increased VEGF concentrations in the vitreous (Kliffen et al., 1997). Secretion and diffusion of retinal VEGF into the vitreous (Adamis et al., 1994; Aiello et al., 1994; Miller et al., 1994) and then to the aqueous humors (Becerra et al., 2004) contributes to intraocular neovascularization (Bressler, 2009). The current IVT anti-VEGF therapies for nAMD include ranibizumab and bevacizumab (antibody-mediated therapeutics), aflibercept (a recombinant fusion protein), and brolucizumab (a single-chain antibody fragment that inhibits VEGF-A). These treatments demonstrate efficacy in maintaining visual acuity but require dosing every 4–8 weeks (Martin et al., 2011; Heier et al., 2012; Papadopoulos et al., 2012). The frequency of IVT injections poses a time and economic burden for patients, physicians, and health care systems (Prenner et al., 2015; Brown et al., 2016). A 12-week (quarterly) dosing interval has long been thought to be desirable to physicians and patients. However, ranibizumab clinically tested in a quarterly dosing interval showed inferior visual outcomes to the ones reported with monthly regimens (Abraham et al., 2010; Schmidt-Erfurth et al., 2011). Recognizing the high bar for demonstrating maintenance of vision on a quarterly dosing interval, both aflibercept and brolucizumab have explored less frequent dosing in a proportion of patients. Aflibercept recently gained US Food and Drug Administration approval for a quarterly treatment interval in patients with nAMD that respond to therapy for 1 year after 8-week dosing. Results from the HAWK and HARRIER phase III trials showed that brolucizumab dosed at a quarterly treatment interval in a proportion of patients with nAMD achieved the primary endpoint of noninferiority to aflibercept given at an 8-week treatment interval (Dugel et al., 2017). Approximately 57% of patients in HAWK and 52% of patients in HARRIER receiving 6 mg of brolucizumab were extended to quarterly dosing if they showed no disease activity as assessed starting in week 16. At week 96, approximately 46% of patients in HAWK and 38% of patients in HARRIER receiving 6 mg of brolucizumab were maintained on quarterly dosing (Dugel et al., 2020). Thus, it is critical for emerging therapies to balance treatment outcomes and patient burden.
DARPin molecules are a new class of uniquely engineered proteins exhibiting high stability, high potency, and extended duration of action (Pluckthun, 2015; Smithwick and Stewart, 2017). When targeted against VEGF, the DARPin therapeutic has the potential to reduce the treatment burden for retinal neovascular diseases. Abicipar pegol (abicipar), is an antiangiogenic therapy that maintains vision gain with less frequent dosing through an optimized combination of molar dose, a long vitreous half-life, and high binding affinity for VEGF-A isoforms (Souied et al., 2014; Smithwick and Stewart, 2017). Compared with ranibizumab, the current gold standard treatment of patients with nAMD, abicipar provides higher binding affinity and longer vitreous half-life (Rodrigues et al., 2018). In a rabbit model of VEGF-induced vasculopathy, abicipar provided a longer duration of effect than ranibizumab at an equimolar dose (Rodrigues et al., 2018). In phase II and III trials, abicipar 1 and 2 mg demonstrated sustained effectiveness in patients with nAMD. In the REACH phase 2 study, abicipar was effective in patients with nAMD at 8–12 weeks after receiving the last injection of abicipar (Callanan et al., 2018). Both 1 or 2 mg of abicipar produced greater improvements in best-corrected visual acuity with fewer IVT injections administered than 0.5 mg ranibizumab (Callanan et al., 2018). In the CEDAR and SEQUOIA identical phase III studies, abicipar (2 mg 8-week or quarterly dosing) demonstrated noninferiority compared with ranibizumab (0.5 mg dosed monthly) on the primary endpoint of stable vision at week 52 (Rodrigues et al., 2018). The purpose of this analysis is to develop a translational pharmacokinetic/pharmacodynamic (PK/PD) model of abicipar to provide a quantitative understanding of the drug disposition and target engagement in the rabbit and use the model to translate to humans to explore the feasibility of longer dosing intervals in the clinic.
Materials and Methods
Animals and Dosing.
This study complied with all requirements of the US Department of Agriculture, and all regulations issued by the US Department of Agriculture implementing the Animal Welfare Act, Title 9 of the Code of Federal Regulations, Parts 1, 2, and 3. The animal procedures used have been approved by Allergan’s Animal Care and Use Committee and are in accordance with the United States National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
For modeling, data were obtained from two in vivo rabbit PK studies in which drug-naïve Dutch Belted rabbits (Covance, San Diego, CA; n = 18) were single-housed with water and food available ad libitum. Animals were dosed IVT with a single dose of 1 mg of abicipar into each eye. Animals were first anesthetized with ketamine (15 mg/kg) and acepromazine (1 mg/kg) administered intravenously. One to two drops of Ophthetic (0.5% proparacaine HCL; Allergan, Irvine, CA) was applied to both eyes. Test materials formulated in PBS at a final concentration of 20 mg/ml were administered in a single IVT injection of 50 µl into the midvitreous region. The eye was flushed with sterile saline and treated with ophthalmic antibiotic, e.g., Zymaxid (0.5% gatifloxacin ophthalmic solution; Allergan) once after injection, and for at least three consecutive days after injection.
Blood and Ocular Tissue Collection and Handling.
Blood was collected via the central ear artery at 6 and 72 hours and on days 7, 14, 21, and 28 postdose for serum analysis (0.5–0.8 ml/sample). Ocular tissues including whole vitreous, aqueous humor (AH), retina tissue, and choroid tissue were collected at 6 and 72 hours and on days 7, 14, 21, and 28 postdose. Three rabbits per time point were sacrificed by intravenous injection of sodium pentobarbital. All ocular tissues were placed on dry ice upon processing and stored at or below −20°C or colder until sample analysis.
Determination of Free Abicipar Concentration.
The quantitative determination of abicipar in rabbit serum, AH, vitreous, retina, and choroid tissue samples was performed using an ELISA with the range of quantitation between 0.313 and 7.50 nM in serum with 1:2 minimal required dilution, 1.25–30.0 nM in AH and vitreous with 1:10 minimal required dilution, and 0.625–10.0 nM in 10% retina and choroid homogenates. The immunoassay for abicipar is based on a sandwich format in which recombinant canine VEGF (R&D Systems, Minneapolis, MN) is used to capture abicipar, after which a biotinylated antiabicipar antibody (Allergan) is added. A secondary detection reagent, Poly-HRP Steptavidin (Thermo Scientific, Rockford, IL) is added to the complex and absorbance is detected using the SpectraMax M5 Plate Reader (Molecular Devices, Sunnyvale, CA).
Modeling Method (Rabbit Model).
A mechanistic PK/PD model incorporating abicipar-VEGF binding kinetics was implemented to describe the in vivo ocular and systemic PK of abicipar and VEGF inhibition after IVT administration to rabbits. The components of the structural model relating to the binding between abicipar and VEGF are based on the setup of a target-mediated drug disposition (TMDD) model (Mager et al., 2003) wherein abicipar binds to the target, VEGF, in the vitreous and AH with a second-order binding affinity rate (kon) and first-order dissociation rate (koff). The VEGF turnover kinetics (zero-order ksyn and first-order kdeg; ksyn = kdeg*baseline), the receptor abundance of VEGF and the degradation of the drug-receptor complex (kINT) in the vitreous and AH were accounted for in the model.
The equations for the model are described below: (1)
(1) (2)
(2) (3)
(3) (4)
(4) (5)
(5) (6)
(6) (7)
(7) (8)
(8) (9)where VEGFAH and VEGFV, are the VEGF concentrations in the AH and vitreous, respectively, DRAH and DRV are the concentrations of the drug-receptor (abicipar-VEGF) complex in the AH and vitreous, respectively, and AAH, AV, AR, AC and AS are the drug amounts in the AH, vitreous, retina, choroid, and serum, respectively. The rate constants for the set of differential equations (eqs. 1–9) of the model are further defined in Tables 1 and 2. For eqs. 5–9, the drug amounts in the ocular and serum compartments (AAH, AV, AR, AC and AS) were divided into their respective volumes to convert to concentration units which were used for model fitting. Tissue volumes (VV, VR, VC and VAH) were fixed based on actual physiologic values (Table 1) obtained from the literature. Serum volume (VS) was estimated from the model fitting. Table 1 lists the parameters that were fixed during model fitting and Table 2 lists the parameters that were estimated from the model optimization.
(9)where VEGFAH and VEGFV, are the VEGF concentrations in the AH and vitreous, respectively, DRAH and DRV are the concentrations of the drug-receptor (abicipar-VEGF) complex in the AH and vitreous, respectively, and AAH, AV, AR, AC and AS are the drug amounts in the AH, vitreous, retina, choroid, and serum, respectively. The rate constants for the set of differential equations (eqs. 1–9) of the model are further defined in Tables 1 and 2. For eqs. 5–9, the drug amounts in the ocular and serum compartments (AAH, AV, AR, AC and AS) were divided into their respective volumes to convert to concentration units which were used for model fitting. Tissue volumes (VV, VR, VC and VAH) were fixed based on actual physiologic values (Table 1) obtained from the literature. Serum volume (VS) was estimated from the model fitting. Table 1 lists the parameters that were fixed during model fitting and Table 2 lists the parameters that were estimated from the model optimization.
Fixed physiologic and mechanistic model parameters
Estimated model parameters
Parameter precision values are not available.
The model scheme is presented in Fig. 1. Where applicable, mechanistic and physiologic parameter values for the rabbits were obtained from the literature or previously conducted experiments (for binding kinetics). Pigment binding in small molecule therapeutics can affect distribution and target binding. Abicipar’s size of 34 kDa restricts its access to melanin binding, minimizing concerns about pigment binding. Thus, pigment binding was not considered for model development. The PK portion of the rabbit model was fitted to rabbit concentration data collected from vitreous, AH, choroid, retina, and serum, and the intercompartmental rate constants were estimated from the model fitting. The descriptions of the model parameters are listed in Table 1.

Modeling Method (Human Model).
The rabbit model was translated to humans using human-specific VEGF parameter values (Table 1) and refitting to human serum and AH concentrations obtained from patients with DME and nAMD administered single IVT doses of 0.4 and 2 mg, respectively. The structural components (Fig. 1) of the rabbit model are the same as the human model (i.e., the same number of compartments and the same designation for all the rate constants). For the human model, human-specific VEGF target values were used as reported in the literature for patients with DME and/or AMD (Table 1). After the recalibration of estimated model parameters, a 0.4 mg single-dose administration was simulated to determine how well the model predicted the external data of VEGF-A measured from a previously conducted clinical study in patients with DME (Campochiaro et al., 2013). From this validation step, it was necessary to manually adjust the KINT to a value of 9.25 day−1 (see Results) before proceeding with performing the simulations for different clinical dose schedules.
The simulations from the human model were made for 2-mg abicipar (the dose studied in the CEDAR and SEQUOIA phase 3 studies) for three dose schedules: 8-week (Q8) quarterly (Q12), and 16-week (Q16). For the Q8 schedule, doses were administered on day 1 and weeks 4, 8, 16, 24, 32, 40, and 48. For the Q12 schedule, doses were administered on day 1 and weeks 4, 12, 24, 36, and 48. For the Q16 schedule, doses were administered on day 1 and weeks 4, 16, 32, and 48. Simulated human vitreous concentrations were predicted for up to 52 weeks. The vitreous VEGF levels were also predicted based on these dose regimens.
NONMEM 7.3 (ICON Development Solutions, Dublin, Ireland) was used to run the modeling (first-order conditional estimation method) and simulation steps. Graphical outputs were generated using R 3.4.2.
A naïve pool approach was used to fit a mean curve through the data set of each matrix assayed and the model fit was performed for all sampled matrices simultaneously. Because ocular PK sampling was a terminal procedure, serial PK sampling within a single animal was not feasible. Thus, the intersubject variability in the PK parameters was fixed to zero. However, for the simulation of the human vitreous PK time courses, both the intersubject variability for each parameter and the residual error were arbitrarily set at the assumed level of 30% (0.09 for each omega term, and 0.3 for the epsilon, which was proportional) to account for the uncertainty and variability associated with the human PK prediction.
Results
PK/PD Modeling: Rabbit Model.
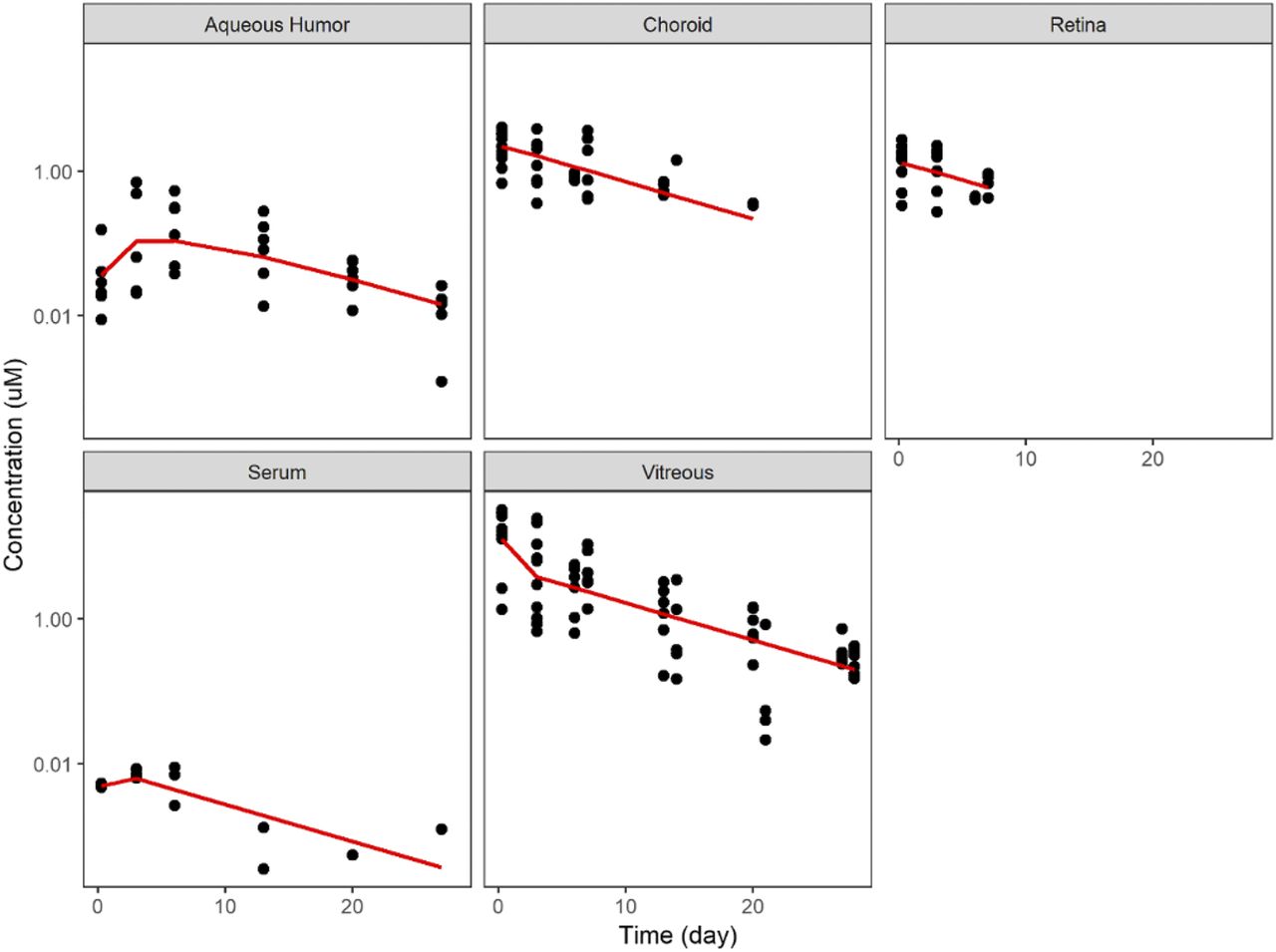
The model predictions showed good correspondence to the experimental results obtained after IVT administration of 1 mg abicipar to rabbits (Fig. 2). The observed concentration-time profiles of free abicipar in the vitreous, AH, choroid, retina, and serum were well described by the model. Table 2 lists the resulting parameters and estimated values for rabbits and human obtained from the model fitting. Several studies have demonstrated that the rabbit is a good model for successful translation to intravitreal human ocular pharmacokinetics (Montezuma et al., 2009; Del Amo and Urtti, 2015; Zernii et al., 2016).

Comparison between model predictions (red lines) and experimentally measured concentration (dots) after intravitreal administration of 1 mg abicipar to rabbits.
PK/PD Modeling: Human Model.
The final rabbit model was scaled to human by changing the mechanistic parameters based on human-specific physiologic values (Table 1) and recalibrated against human AH (0.4 mg dose level; Fig. 3A) and serum (2-mg dose level; Fig. 3B) concentration-time data obtained from abicipar-treated patients with DME or nAMD, respectively. As shown, the model had reasonably good correspondence to the observed PK human data.

Refitting of the model against human AH (A) and human serum (B) abicipar concentration data after 0.4 mg (A) or 2 mg (B) single intravitreal administration. Black dots are the observed data and red solid lines are the fitted (mean) model prediction.
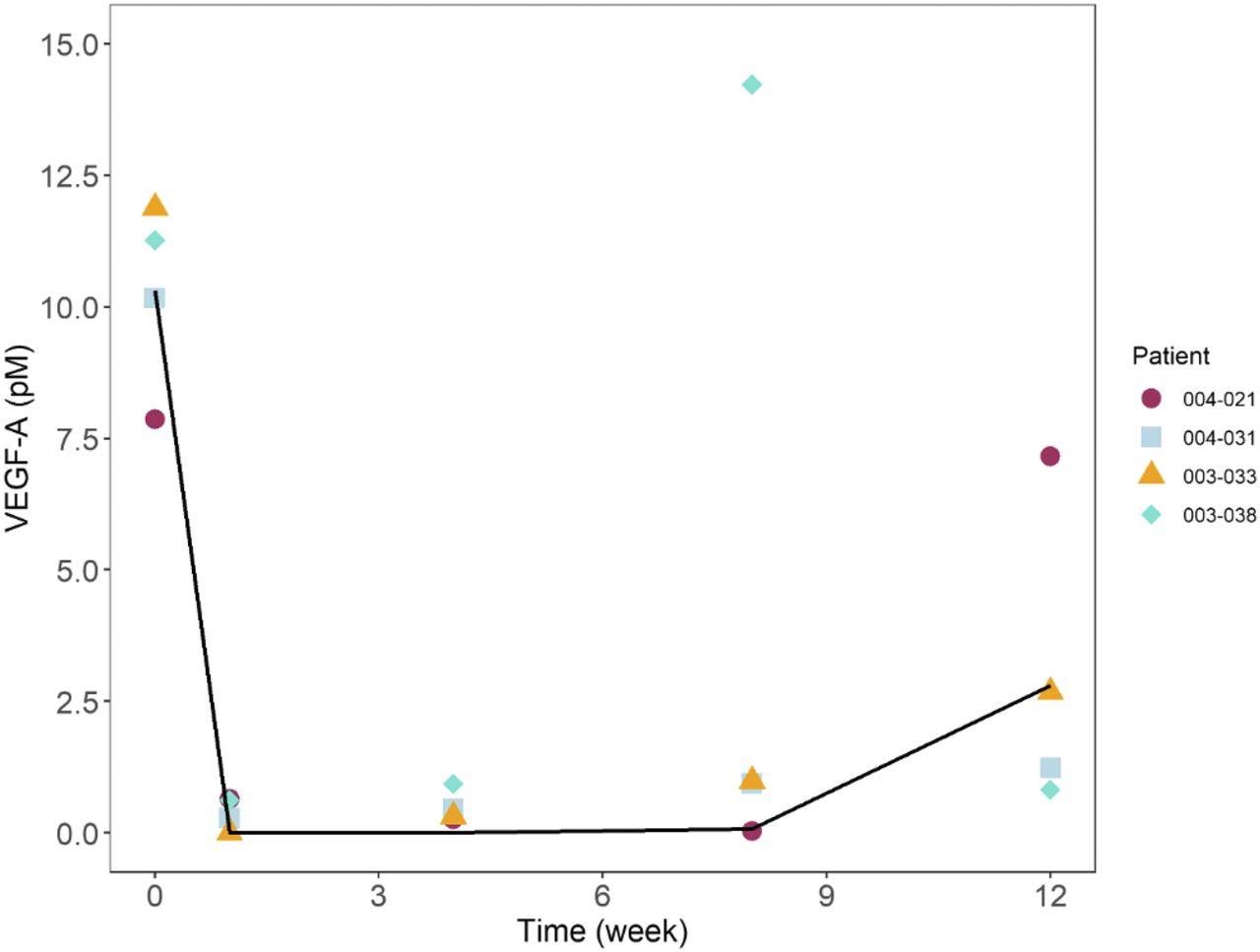
To perform an external validation, the model was used to simulate the VEGF levels in the AH after a single, 0.4-mg IVT administration and determine how well it predicted actual VEGF-A levels measured in a separate clinical study (in patients with DME). The results (not shown) indicated that the model well predicted the extent of the VEGF inhibition but slightly underpredicted the rate of recovery of VEGF. Sensitivity analysis was conducted and showed that KINT was one of the most sensitive parameters for VEGF levels. To correct for the underprediction in the rate of VEGF recovery, KINT was manually adjusted from a value of 55.4 day−1 (estimated from only from PK data) to a value of 9.25 day−1 before proceeding with performing the simulations for different clinical dose schedules. No other parameters were adjusted. As shown in Fig. 4, after adjusting for just a single parameter (KINT), the model predicted the observed VEGF-A levels in the DME study reasonably well. The final human model parameters based on this fitting are shown in Table 2.

Human VEGF-A time course after a single IVT administration of abicipar. Black line is the model prediction and symbols are observed data from the phase I/II DME study (Campochiaro et al., 2013).
Simulations of Abicipar After Human IVT Dosing.
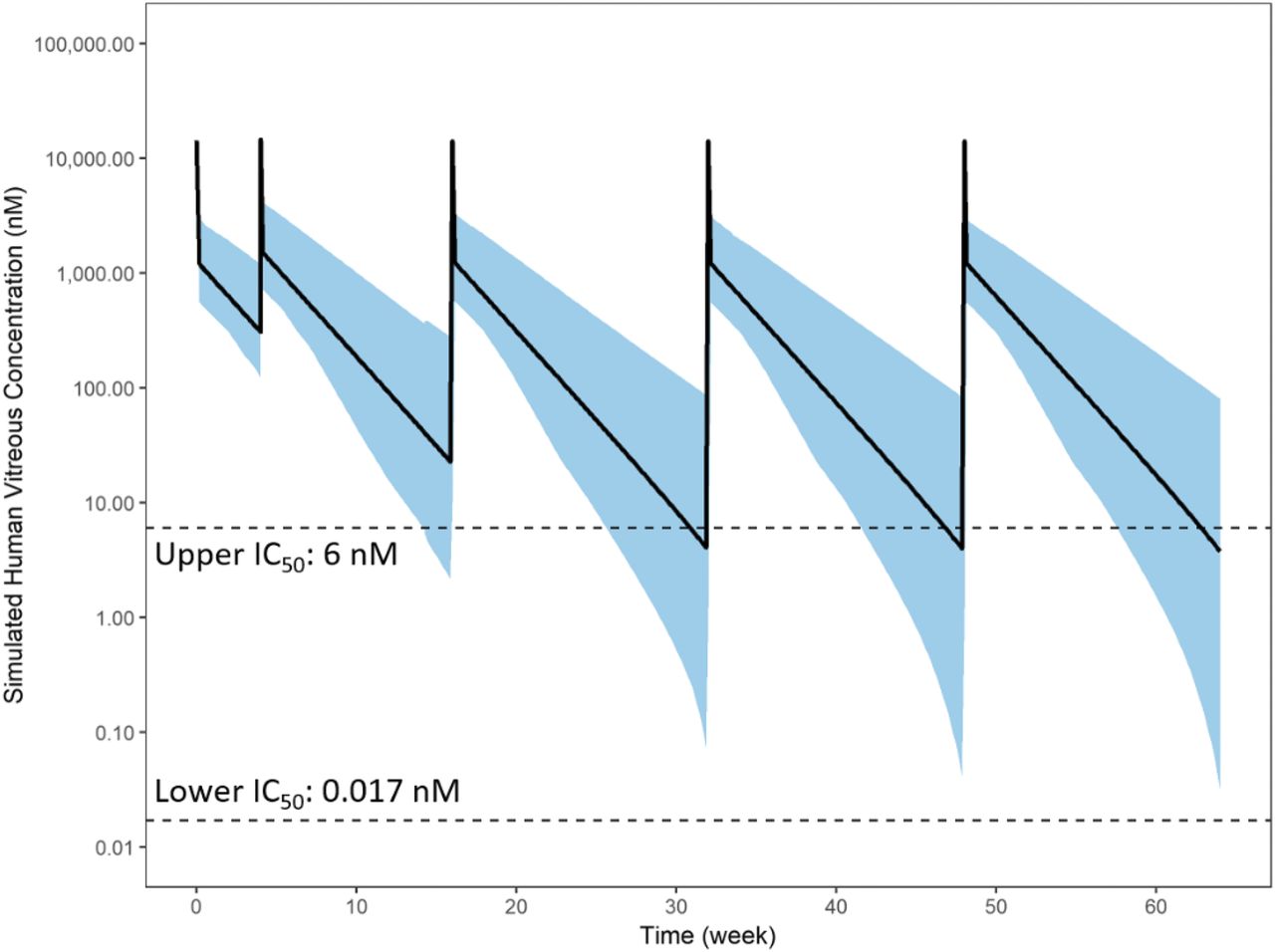
The human model was used to simulate human vitreal concentrations using the same dose (2 mg) and dosing regimens (Q8 and Q12) as studied in the abicipar phase 3 studies CEDAR and SEQUOIA. In addition, the 2 mg, 16-week (Q16) were simulated to test the feasibility of the 4-month interval. The simulated vitreous exposure was compared with in vitro and in vivo IC50 values estimated from a HUVEC-sprouting assay (0.017 nM) (Rodrigues et al., 2018) and from AH VEGF data (6 nM) obtained from patients with DME treated with abicipar, respectively. After both a Q8 and Q12 dosing regimen, mean steady-state minimal concentrations (Cmin,ss) were maintained above the upper end of the IC50 range (Figs. 5 and 6). When accounted for the assumed level of variability (30% between-subject variability in all model parameters) the fifth percentile of the Cmin,ss dips below 6 nM for the Q12 regimen (Fig. 6). For the Q16 regimen, the mean Cmin,ss dips below 6 nM, and the fifth percentile of the Cmin dips below the 6 nM level but stays above the in vitro IC50 (0.017 nM; Fig. 7). In each of the three regimens, both the mean and the entire band of the prediction interval are well above the in vitro IC50. VEGF sequestration and mean AH VEGF levels are simulated for the Q8, Q12, or Q16 regimen (Fig. 8). For the Q8 and Q12 regimens, free AH VEGF is almost completely sequestered after 2 mg abicipar dose levels. For Q16, VEGF-A levels showed some fluctuations, recovering up to 40% of baseline but only briefly at the predose times. Overall, VEGF was inhibited the majority of the time and was maintained significantly below baseline at all times.

Human-predicted vitreous exposure of abicipar after 2 mg intravitreal injections on day 1 and weeks 4, 8, 16, 24, 32, 40, and 48 (Q8 regimen). Black line is the mean and the blue shade is the 5th and 95th percentiles of the prediction.

Human-predicted vitreous exposure of abicipar after 2 mg intravitreal injections on day 1 and weeks 4, 12, 24, 36, and 48 (Q12 regimen). Black line is the mean and the blue shade is the 5th and 95th percentiles of the prediction.

Human-predicted vitreous exposure of abicipar after 2 mg intravitreal injections on day 1 and weeks 4, 12, 24, 36, and 48 (Q16 regimen). Black line is the mean and the blue shade is the 5th and 95th percentiles of the prediction.

Human-predicted AH VEGF levels after 2 mg intravitreal injections of abicipar based on a Q8 (administered on day 1 and weeks 4, 8, 16, 24, 32, 40, and 48), Q12 (administered on day 1 and weeks 4, 12, 24, 36, and 48) or Q16 (administered on day 1 and weeks 4, 16, 32, and 48) regimens. Arrows indicate dosing events.
Discussion
Intravitreal administration of anti-VEGF therapies represents the preferred treatment option for nAMD (Yonekawa and Kim, 2014). Current therapies such as Lucentis and Avastin use frequent monthly IVT injections to improve visual outcomes (Martin et al., 2011; Singer et al., 2012; Brown et al., 2016). However, frequent visits for IVT treatment and monitoring pose a significant emotional and financial treatment burden for patients and create a major economic burden for physicians and health care systems (Holekamp et al., 2014; Prenner et al., 2015; Brown et al., 2016). Currently, there is an unmet need for patients with AMD and health care providers to maintain or improve vision but with a reduced frequency of injections.
The extent and duration of action of anti-VEGF therapies is a function of the PK properties (e.g., half-life or clearance) of the drug, binding affinity (association and dissociation rates) of the drug to VEGF, VEGF abundance in the target tissue, VEGF turnover kinetics, and the dose of the administered drug. Understanding that these factors contribute in a complex way to the overall efficacy of abicipar, we developed a mechanistic PK/PD model to quantitatively characterize the drug-target interaction and PK of abicipar in rabbit and human ocular tissues and serum. The model was built using species-specific in vivo PK data, drug-target binding rate constants, target abundance and target turnover rates, and physiologic values for ocular tissue volumes. The human model was used to explore various clinical dosage regimens and its effect on the duration of action, thereby testing the feasibility of longer-than monthly administrations of abicipar to patients with nAMD.
The model simulations followed the dosing schedules specified in the CEDAR and SEQUOIA study protocols, which investigate the clinical efficacy of abicipar administered to patients with nAMD at 2 mg 8-week or quarterly dosing in comparison with ranibizumab. Additionally, we performed simulations for the Q16 schedule (not studied in CEDAR or SEQUOIA) to explore the theoretical possibility of the 4-month dosing interval. The model demonstrated that abicipar vitreous PK exposure (Cmin,ss) was maintained above the in vitro and in vivo IC50 values for VEGF inhibition at both the Q8 and Q12 dose intervals. In addition, complete inhibition of vitreous VEGF concentrations was maintained during the treatment period for Q8 and Q12 schedules. For the Q16 schedule, although the mean Cmin,ss dipped below the in vivo IC50 value, it is far above the in vitro IC50 value. Although the vitreous VEGF for the Q16 schedules showed some fluctuations, VEGF was inhibited the majority of the time and was maintained significantly below baseline at all times. Taken together, these simulations predict that abicipar may have a duration of VEGF suppression of up to 4 months in some patients, which could reduce frequent IVT dosing and lessen patient treatment burden.
Our model predictions align with the visual acuity outcomes in REACH phase 2 study and CEDAR and SEQUOIA phase 3 studies. In the REACH phase 2 study, abicipar groups had visual acuity improvements that were numerically greater than ranibizumab at week 16, corresponding to 8 weeks after the final abicipar injection (Callanan et al., 2018). This difference for treatment groups was sustained at week 20, corresponding to 12 weeks after the final abicipar injection (Callanan et al., 2018). Our model simulation also suggests that the Q16 treatment may be a feasible dose in some patients in terms of maintaining the VEGF inhibition during a 52-week treatment period. The efficacy endpoint analysis at week 52 of the CEDAR and SEQUOIA phase 3 studies investigated the effects of extended treatment with abicipar 2 mg at 8-week or quarterly intervals after the three initial loading doses. Results from the two pivotal trials demonstrated that abicipar maintained stable vision in greater than 91% of patients on an 8-week and quarterly regimen and achieved the primary endpoint of noninferiority to monthly ranibizumab at week 52.
Integrating mechanistic parameters that were experimentally measured into a TMDD model construct to describe the PK and PD of a biologic drug has previously been successfully applied (Luu et al., 2012). Similarly, we took a quantitative-systems pharmacology approach by using literature and experimentally derived parameter values and directly incorporated them into the model. Because literature information on the transfer kinetics between ocular tissues is not readily available, nor has there been a previously developed ocular PK model for abicipar, we allowed for the transfer kinetics between ocular compartments to be estimated from ocular PK data. Initially, bidirectional kinetics were designated to occur between all ocular compartments. However, this led to model instability and the number of transfer kinetics included in the final rabbit model were reduced to a unidirectional flow between AH and serum, vitreous and AH, retina and serum, and no direct transfer kinetics between vitreous and serum (Fig. 1).
An obvious limitation of the model is the sheer number of model parameters relative to the amount of data available to accurately estimate each model parameter individually. Our original intent, however, was to take a systems-approach by fixing known parameters, thus reducing the number of parameters to be estimated. However, NONMEM was unable to provide parameter precision for the estimated parameters. Attempts to bootstrap the data to obtain parameter precision was also unsuccessful and time intensive. This was somewhat expected considering the model assumes the eye to contain only the tissues that were sampled and excluded many other potentially important tissues in the eye that could uptake the drug. Due to the potential uncertainty in the estimated model parameters, individual parameter values reported herein should be interpreted or implemented with caution. Nevertheless, there was good correspondence between the model prediction and the observed PK profiles in all five ocular tissues sampled, enabling us to proceed with a certain level of confidence with the simulation exercises to explore the feasibility of longer-interval injections for abicipar. Notwithstanding, as standard practice, we varied the initial estimates of the model to ensure the global minimum was obtained. Future work would include the refinement of the PK portion of the model and reoptimizing the transfer kinetic parameters or to take a full physiologically based PK modeling approach. In addition, because abicipar binds to systemic VEGF, future work should explore the inclusion of the TMDD components in the serum compartment. Abicipar is cleared rapidly from the systemic circulation, and most of the data are below the limit of quantitation. Our current model did not include systemic VEGF binding due to the limited range of concentrations to verify nonlinearity associated with TMDD.
In summary, we performed mechanism-based modeling and simulation to better understand the pharmacology, PK, and PD of abicipar after IVT injections in rabbits and in humans. The model was used to explore the feasibility of longer dose intervals than monthly injections in patients with nAMD. The results confirm that abicipar 8-week and quarterly dosing schedules provide sufficient exposure for maintaining virtually complete VEGF inhibition during the 52-week period—results from the CEDAR and SEQUOIA phase 3 studies confirmed our prediction. In addition, simulations showed that abicipar Q16 schedule also maintains significant VEGF inhibition during the 52-week period, lending feasibility for investigation of this schedule in the clinic. The extended duration of action for abicipar compared with existing therapies may reduce the treatment burden for patients and would be an important advancement in the treatment of retinal ischemic diseases.
Acknowledgments
The authors wish to acknowledge Swati Gupta for the bioanalytical development of abicipar. Writing and editorial assistance was provided to the authors by Nayna Sanathara of Allergan plc.
Authorship Contributions
Participated in research design: Luu, Seal, Attar.
Conducted experiments: Seal.
Performed data analysis: Luu.
Wrote and contributed to the writing of the manuscript: Luu, Seal, Attar.
Footnotes
- Received October 9, 2019.
- Accepted February 12, 2020.
This study was sponsored by Allergan plc, Dublin, Ireland. K.T.L., PhD, J.R.S., PhD, and M.A., PhD are full-time employees and stockholders of Allergan plc.
Previous presentation of work at meetings: Pegylation of Abicipar Increases Vitreal Half-Life, Supporting a Potential for up to 3 Month Duration of Action in the Clinic. (2018) Seal JR, Ekawardhani S, Schlegel A, Stumpp MT, Binz HK, Attar M.
DARPin is a registered trademark owned by Molecular Partners AG.
Primary laboratory of origin: Luu (Allergan plc, Irvine, CA).
Abbreviations
- AH
- aqueous humor
- AMD
- age-related macular degeneration
- DME
- diabetic macular edema
- IVT
- intravitreal
- nAMD
- neovascular age-related macular degeneration
- PD
- pharmacodynamic
- PK
- pharmacokinetic
- Q12
- quarterly
- Q16
- 16 week
- Q8
- 8 week
- TMDD
- target-mediated drug disposition
- VEGF
- vascular endothelial growth factor
- Copyright © 2020 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}