


Abstract
The metabotropic glutamate receptor 5 (mGlu5) is a recognized central nervous system therapeutic target for which several negative allosteric modulator (NAM) drug candidates have or are continuing to be investigated for various disease indications in clinical development. Direct measurement of target receptor occupancy (RO) is extremely useful to help design and interpret efficacy and safety in nonclinical and clinical studies. In the mGlu5 field, this has been successfully achieved by monitoring displacement of radiolabeled ligands, specifically binding to the mGlu5 receptor, in the presence of an mGlu5 NAM using in vivo and ex vivo binding in rodents and positron emission tomography imaging in cynomolgus monkeys and humans. The aim of this study was to measure the RO of the mGlu5 NAM HTL0014242 in rodents and cynomolgus monkeys and to compare its plasma and brain exposure-RO relationships with those of clinically tested mGlu5 NAMs dipraglurant, mavoglurant, and basimglurant. Potential sources of variability that may contribute to these relationships were explored. Distinct plasma exposure-response relationships were found for each mGlu5 NAM, with >100-fold difference in plasma exposure for a given level of RO. However, a unified exposure-response relationship was observed when both unbound brain concentration and mGlu5 affinity were considered. This relationship showed <10-fold overall difference, was fitted with a Hill slope that was not significantly different from 1, and appeared consistent with a simple Emax model. This is the first time this type of comparison has been conducted, demonstrating a unified brain exposure-RO relationship across several species and mGlu5 NAMs with diverse properties.
SIGNIFICANCE STATEMENT Despite the long history of mGlu5 as a therapeutic target and progression of multiple compounds to the clinic, no formal comparison of exposure-receptor occupancy relationships has been conducted. The data from this study indicate for the first time that a consistent, unified relationship can be observed between exposure and mGlu5 receptor occupancy when unbound brain concentration and receptor affinity are taken into account across a range of species for a diverse set of mGlu5 negative allosteric modulators, including a new drug candidate, HTL0014242.
Introduction
Glutamate is a major excitatory neurotransmitter playing an important role throughout the nervous system via activation of metabotropic (G protein–coupled) and ionotropic (ion channel) glutamate receptors, including the metabotropic glutamate 5 (mGlu5) receptor. mGlu5 receptors are widely distributed in the brain, including the cortex, striatum, hippocampus, and cerebellum (Patel et al., 2007); mainly concentrated in postsynaptic structures; and, with a few exceptions, almost undetectable in presynaptic structures (Berthele et al., 1999; Ferraguti and Shigemoto, 2006; Hovelsø et al., 2012). In diseases in which glutamatergic signaling is dysregulated [e.g., depression, anxiety, addiction, neuropathic pain, and levodopa-induced dyskinesia; Slassi et al. (2005); Emmitte (2013); Archer and Garcia (2016)], enhanced mGlu5 receptor activation can lead to increased trans-synaptic glutamate release, which could further exacerbate glutamate-mediated excitotoxic processes. Antagonists, or negative allosteric modulators (NAMs), of the mGlu5 receptor have therapeutic potential in a range of psychiatric and neurologic disorders characterized by glutamatergic hyperexcitability (Gregory et al., 2011). NAMs block the effects of glutamate by binding in the seven transmembrane bundle of the receptor, rather than at the orthosteric glutamate binding site in the Venus flytrap domain (Pagano et al., 2000; Christopher et al., 2015). NAMs have avoided the problems associated with targeting the orthosteric site, including lack of selectivity, poor pharmacokinetics, and low central nervous system penetration (Lindsley and Stauffer, 2013).
Several mGlu5 NAMs have progressed to clinical trials. Dipraglurant and mavoglurant showed promising results in reducing levodopa-induced dyskinesia in Parkinson's disease in phase II trials (Tison et al., 2016; Stocchi et al., 2013), although mavoglurant failed to demonstrate efficacy in subsequent phase IIb studies (Trenkwalder et al., 2016). When administered as an adjunctive therapy in major depression (phase IIb), basimglurant failed to demonstrate efficacy on primary endpoints, although there were significant effects on secondary endpoints [e.g., Montgomery-Åsberg Depression Rating Scale; Quiroz et al. (2016)]. Clinical trials are in progress to further evaluate therapeutic opportunities, including levodopa-induced dyskinesia in Parkinson's disease (dipraglurant) and substance-use disorders (mavoglurant). It is worth noting that several mGlu5 NAMs have reported dose-related adverse events in clinical trials (Kågedal et al., 2013; Kalliomäki et al., 2013; Trenkwalder et al., 2016; Jaso et al., 2017; www.clinicaltrials.gov), and it is currently unclear whether it was possible to achieve sufficient mGlu5 NAM exposure to fully explore efficacious potential.
Measurement of receptor occupancy (RO) is useful in designing efficacy and safety studies to ensure that the therapeutic concept is tested and/or to aid interpretation of any adverse events. There have been numerous publications on specific pharmacokinetic exposures and/or measured mGlu5 RO for mGlu5 NAMs across species (Anderson et al., 2002; Hamill et al., 2005; Kågedal et al., 2013 Gregory et al., 2014; Lindemann et al., 2015; Xu and Li, 2019). Different approaches to the determination of RO have been used, including direct binding after exposure in vivo or ex vivo to radioligands (Able et al., 2011) or indirectly by measuring radioligand displacement. Preclinically, mGlu5 radioligands [3H]methoxy-MPEP ([3H]M-MPEP) (Gasparini et al., 2002) and [3H]methoxymethyl-3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine ([3H]M-MTEP) (Anderson et al., 2002) have been used extensively. Positron emission tomography (PET) can be used in both preclinical and clinical settings, and several mGlu5 PET ligands have been developed, allowing quantitative measurement of receptor expression and distribution across brain regions and RO of mGlu5 compounds after systemic drug administration. The most widely used are [11C]ABP688 (Ametamey et al., 2007) and [18F]FPEB (Sullivan et al., 2013; Wong et al., 2013), which have been shown to bind to the same site as MPEP in rodent (Wyss et al., 2007; Hintermann et al., 2007) and monkey brain (Hamill et al., 2005), respectively. A different PET ligand, [11C]RO511232, was developed and used clinically for basimglurant (www.clinicaltrials.gov), although [11C]ABP688 was used preclinically (Lindemann et al., 2015).
Although there is extensive published experience with mGlu5 PET ligands (Wong et al., 2013; Kågedal et al., 2013; Lohith et al., 2017), there is limited understanding of RO in relation to drug exposure, how it aligns across species, and how exposure-response relationships compare between mGlu5 NAMs. As understanding exposure-mGlu5 RO relationships is important in achieving the right level of mGlu5 target engagement for positive efficacy in a therapeutic setting, we studied the plasma and brain exposure-RO relationships of dipraglurant, basimglurant, mavoglurant, and HTL0014242, a new mGlu5-selective NAM (Christopher et al., 2015). The comparative data for this analysis are derived from experimental data and supplemented by published mGlu5 RO and exposure data across species (Bennett et al., 2014; Lindemann et al., 2015; Quiroz et al., 2016; Tison et al., 2016; Cosson et al., 2018). As this study will demonstrate, when accounting for mGlu5 affinity and brain exposure, exposure-RO relationships were similar across species and different mGlu5 NAMs and in agreement with the simple Emax model.
Materials and Methods
Chemicals
Mavoglurant [(3aR,4S,7aR)-4-hydroxy-4(3-methylphenyl)ethynyl-octahydro-indole-1-carboxylic acid methyl ester], dipraglurant [6-fluoro-2-(4-[pyridin-2-yl]but-3-yn-1yl)imidazo[1,2-a]pyridine], and HTL0014242 (3-chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile) were synthesized by Sosei Heptares. Basimglurant ([2-chloro-4-[1-(4-fluoro-phenyl)-2-methyl-1H-imidazol4-ylethynyl]-pyridine) was purchased from MedChemExplorer (catalog number HY-15446).
Radiotracers
For the mouse and rat ex vivo occupancy experiments, [3H]M-MPEP (2-[(3-methoxyphenyl)ethynyl]-6-methylpyridine) was custom-synthesized by Tritec (specific activity 67 Ci/mmol). For the mouse in vivo occupancy experiments, [3H]M-MPEP was purchased from American Radiolabeled Chemicals, Inc. (catalog number art1571; specific activity 80 Ci/mmol; St. Louis, MO).
[18F]FPEB (3-[18F]fluoro-5-[(pyridin-3-yl)ethynyl] benzonitrile) for the rat PET study was synthesized by Invicro, whereas for the cynomolgus monkey PET study, the tracer was prepared at GE Healthcare. For the rat PET study, synthesis of [18F]fluoride at Invicro was done using a Siemens RDS-111 Eclipse cyclotron equipped with a fluoride target loaded with oxygen-18–enriched water by means of 18O(p,n)18F reaction. Optimization of yield was achieved by using a spirocyclic iodonium ylide precursor (Stephenson et al., 2015). For full details, see Varlow et al. (2020). For the cynomolgus monkey PET study, [18F]FPEB was prepared by GE TRACERlab FX-FN using the following methodology. Commercially purchased [18F]fluoride was transferred onto and trapped on an ion exchange cartridge. After elution with K2CO3 (1 mg) and K222 (10 mg), the [18F]fluoride was dried under vacuum and helium flow under azeotropic conditions. After completion, precursor dissolved in DMSO was added (5 mg in 1.0 ml), and the reaction mixture was heated to 150°C for 10 minutes before being cooled and diluted with water. The solution was transferred through a solid-phase extraction cartridge followed by acetonitrile elution (2 ml) into 3 ml of H2O and HPLC injection. Product was collected with HPLC purification [Luna C18(2); Phenomenex] and acetonitrile/water (45:55, 5 ml/min) and subsequently formulated in physiologic solution after solid-phase extraction.
All other drugs, chemicals, cell culture reagents, and consumables were purchased from commercial sources.
Animals
For mouse in vivo RO studies, male C57Bl/6 mice (25–30 g; Charles River, Raleigh, NC) were used. All procedures were approved by the Institutional Animal Care and Use Committee in accordance with The Guide for the Care and Use of Laboratory Animals.
For mouse ex vivo occupancy and ex vivo Ki studies, male CD1 mice (approximately 30 g; Charles River, Margate, Kent) were used. All experiments were performed in accordance with UK Home Office regulations and in line with the Animals Scientific Procedures Act (1986) and the transposed EU Directive 2016/63/EU. Studies were conducted at Royal Veterinary College after institutional review board approval.
For rat ex vivo RO studies, male Sprague-Dawley rats (250–300 g; Charles River) were used. For rat PET imaging, male Sprague-Dawley rats (350–450 g; Charles River) were used. Both experiments were performed in accordance with UK Home Office regulation and in line with the Animals Scientific Procedures Act and transposed EU Directive 2016/63/EU.
Cynomolgus monkey PET imaging was performed on two adult cynomolgus monkeys (Macaca fascicularis), one female (8 years old) and one male (16 years old). This study was conducted in full compliance with Yale University’s Institutional Animal Care and Use Committee policies and procedures, which follow the recommendations of The Guide for the Care and Use of Laboratory Animals.
Ki Determination
[3H]M-MPEP saturation binding and competition binding assays were performed for human, rat, mouse, and cynomolgus monkey mGlu5 following the methods described in Christopher et al. (2015), using either membranes prepared from human embryonic kidney 293 cells transiently transfected with the receptor for human, rat, and cynomolgus monkey mGlu5 or membranes prepared from frontal cortices isolated from adult male CD1 mice prepared according to the method described in Robertson et al. (2011) for mouse mGlu5. The radioligand employed is based on the mGlu5 NAM M-MPEP and has been reported to bind to the same allosteric site as the mGlu5 NAMs tested in the competition binding assay (Doré et al., 2014). Furthermore, complete inhibition of [3H]M-MPEP binding to mGlu5 has been reported for mavoglurant and dipraglurant (Doré et al., 2014), as well as HTL0014242 (Sergeev et al., 2018). Therefore, the Cheng-Prusoff equation could be applied to derive Ki values from the IC50 values that resulted from a four-parameter logistic equation fit of the competition binding data.
Mouse Ex Vivo Binding Using [3H]M-MPEP
Mice (n = 5) were dosed orally with HTL0014242 in vehicle (10% dimethylacetamide, 10% Solutol HS 15, and 80% of 10% aqueous HPβCD). At 2 hours postdose, animals were culled by cervical dislocation, which was followed by collection of blood by cardiac puncture. Blood was collected into EDTA-K2 tubes and centrifuged (2000g; 5 minutes; 4°C) to obtain plasma. Forebrains were halved along the midline, and one half was prepared by homogenization (Homogenizer; 7000 rpm; 20 seconds; Polytron) in 40 volumes of binding buffer (50 mM HEPES, 150 mM sodium chloride, pH 7.5) immediately prior to use in [3H]M-MPEP binding assays [following the methodology described in Christopher et al. (2015) but using 100 µl homogenate per well; 4°C; 10-minute incubation; and 80 nM [3H]M-MPEP (20× Kd)].
The other brain half and plasma were analyzed to quantify levels of HTL0014242. The analytical methods used were identical to those described in the rat ex vivo binding section below.
Mouse In Vivo Binding Using [3H]M-MPEP
C57BL/6 mice were dosed with HTL0014242 (1, 3, or 10 mg/kg, orally; n = 2 per dose group) or intraperitoneally (n = 3 per dose group) with vehicle (10% Solutol HS 15 and 90% of 10% aqueous HPβCD). To define nonspecific binding, a saturating dose of MPEP (50 mg/kg, i.p.) was administered. At 1 hour postdose, [3H]M-MPEP (30 µCi/kg, in water) was administered as an intravenous bolus via the tail vein. At 1 minute later, mice were decapitated, brains were removed, and forebrain was dissected. Tissue was weighed and homogenized in 10 volumes of ice-cold homogenization buffer (10 mM potassium phosphate, 100 mM KCl, pH 7.4) using a Polytron homogenizer. Homogenates were filtered over grade GF/B glass microfibre membrane filters and washed twice with 5 ml ice-cold homogenization buffer. Filters were counted for radioactivity using a liquid scintillation counter, and specific binding was calculated by subtracting the nonspecific binding.
A pharmacokinetic study was conducted to determine brain and plasma exposures in the C57Bl/6 mice over the dose ranges studied for mGlu5 RO. Mice were dosed with HTL0014242 (1, 3, and 10 mg/kg, orally, or 1, 3, 10, and 30 mg/kg, i.p.; n = 3 per time point per group). Blood samples were taken at 0.25, 0.5, 1, 2, 4, and 6 hours postdose, and the plasma layer was separated by centrifugation (2000g; 5 minutes; 4°C). The whole brain was rapidly removed and frozen on dry ice. Brains were homogenized in 10 volumes of ice-cold homogenization buffer (10 mM potassium phosphate, 100 mM KCl, pH 7.4). After protein precipitation with acetonitrile containing an internal standard, the samples were analyzed for test compound via LC-MS/MS using a similar approach to that described in the rat ex vivo binding section. The concentration data at 1 mg/kg, orally, were below quantifiable limits, so dose proportionality was assumed to estimate exposure based on the data for 3 mg/kg.
Rat Ex Vivo Autoradiography Using [3H]M-MPEP
mGlu5 RO was measured in the brain 1 hour after oral administration of HTL0014242 (1, 3, 10 mg/kg) or mavoglurant (3, 10, 30 mg/kg) in Sprague-Dawley rats (n = 5 per dose group). Whole brains were removed, and a coronal block containing the hippocampus was cut, with one half rapidly cooled to −20/−30°C for sectioning and the other stored at −80°C to quantify compound exposure. Sections of the hippocampal CA3 region were prepared using a cryostat and incubated with [3H]M-MPEP for 10 minutes at room temperature followed by rapid washes with ice-cold buffer. The low temperature during sample processing, short [3H]M-MPEP incubation time, and rapid washing were precautions taken to minimize dissociation of dosed compound from mGlu5. Levels of bound radioactivity in the sections were determined using a β imager. Specific binding (counts per minute per square millimeter) was generated by subtraction of mean nonspecific binding (counts per minute per square millimeter) from mean total binding (counts per minute per square millimeter) for each animal. Mean specific binding was used to determine a single RO value for each dose level as outlined below.
Terminal blood and halved brain samples were collected to measure compound concentrations. Blood was centrifuged at 1900g for 5 minutes at 4°C to prepare plasma. Brain was homogenized in water (1:4). Protein was precipitated from 50-μl aliquots of the individual plasma or brain homogenates by adding 150 μl methanol followed by centrifugation for 30 minutes at 4°C. Aliquots of the resulting supernatant were diluted 2:1 with HPLC-grade water in a 96-well plate. A standard curve was prepared by spiking control plasma and brain with varying concentrations of test compound dissolved in DMSO and then treated in an identical manner to the test samples. Samples were then analyzed using LC-MS/MS with electrospray ionization set in positive mode. The system consisted of an Acquity Binary Solvent Manager, Acquity four-position heated column manager, 2777 Ultra High Pressure Autosampler, and a Xevo-TQ MS Triple Quadrupole mass spectrometer (Waters Ltd., Herts, UK). Gradient elution over 1.8 minutes with 10 mM ammonium formate + 0.1% v/v formic acid in water and methanol at a flow rate of 0.6 ml/min was performed. A linear regression was used to generate the calibration curve for HTL0014242. Concentrations of HTL0014242 were calculated using the peak area ratio of analyte to internal standard based on the standard curve.
Data Analysis in the Rodent Ex Vivo and In Vivo Occupancy Assays Using [3H]M-MPEP.
Data were expressed as percent RO (% RO) (data normalized to average specific binding in vehicle samples as 100%, and % HTL0014242 or % mavoglurant mGlu5 RO was calculated as 100% − % [3H]M-MPEP bound in the presence of drug-treated sample). For the mouse in vivo occupancy experiments, the assay was run on three separate occasions, and the receptor occupancy shown is the average receptor occupancy and S.D. from the three combined experiments.
Rat In Vivo PET RO Study Using [18F]FPEB
The aim of this study was to measure mGlu5 RO of HTL0014242 in the rat brain using 3[18F]fluoro5[(pyridine3yl)ethynyl]benzonitrile ([18F]FPEB) PET imaging.
Anesthesia and Dosing.
Rats (3 groups, n = 5 per group) were administered vehicle or HTL0014242 (1 or 10 mg/kg, orally). At 15 minutes after dosing, the rats were anesthetized and maintained under terminal isoflurane anesthesia (2% to 3% isoflurane, 1 l/min O2). Body temperature was kept stable using a heating pad. Indwelling cannulae were surgically implanted in a vein (for [18F]FPEB tracer administrations) and an artery (for blood sampling of tracer kinetics). Approximately 100 IU heparin sodium was given intravenously prior to the scan to aid blood sampling.
PET Scanning.
PET imaging was performed using an Inveon DPET with docked multimodality computed tomography scanner. The brain was placed in the field of view of the scanner, and a computed tomography scan was acquired for attenuation and scatter correction. At 1.5 hours after administration of the vehicle or HTL0014242, a 60-minute dynamic PET scan was acquired after the intravenous administration of 4–18 MBq of [18F]FPEB.
Arterial Sampling.
To generate a [18F]FPEB plasma input function, continuous arterial blood samples were taken at 3-second intervals during the first minute, and discrete samples were taken across a 60-minute period. Blood was collected into tubes coated with heparin. Radioactivity concentration in blood and plasma were determined at all time points. Discrete plasma samples were extracted and analyzed by HPLC to determine the percentage of parent compound.
Compound Exposure Determination and Ex Vivo [18F]FPEB Uptake Distribution.
Blood samples were collected 0, 30, and 60 minutes after the start of the PET scan, equating to 1.5, 2.0, and 2.5 hours post–HTL0014242 dose. The rats were sacrificed 60 minutes after [18F]FPEB injection (after the PET scan) by exsanguination followed by cervical dislocation under terminal anesthetic. Brain hemispheres were separated, with one half used for analysis of HTL0014242 concentration. The other half was used to determine ex vivo [18F]FPEB uptake in regions of interest (ROI) to confirm HTL0014242 competed for the same mGlu5 binding site as [18F]FPEB.
Brain was homogenized in water (4 ml/g). Protein was precipitated from 10-μl aliquots of the individual plasma or brain homogenates by adding 100 μl acetonitrile, followed by mixing (150 rpm, 20 minutes) and centrifugation (3000 rpm, 15 minutes). Aliquots of the resulting supernatant were diluted 2:1 with HPLC-grade water in a 96-well plate. A standard curve was prepared by spiking control plasma and brain with varying concentrations of test compound dissolved in DMSO and then treated in an identical manner to the test samples as described above to provide a final concentration range of 1–5000 ng/ml (for plasma) and 2–10000 ng/g (for brain). Samples were then analyzed using ultra high performance liquid chromatography-tandem mass spectrometry using electrospray ionization. The system consisted of a Shimadzu Nexera ×2 HPLC system coupled with a Shimazdu LCMS 8060 mass spectrometer. Gradient elution over 2 minutes with water containing 0.1% formic acid and acetonitrile containing 0.1% formic acid at an organic flow rate of 0.4 ml/min was performed. A linear regression was used to generate the calibration curve for HTL0014242. Concentrations of HTL0014242 were calculated using the peak area ratio of analyte to internal standard based on the standard curve.
Ex vivo distribution of [18F]FPEB was determined in dissected ROIs (cortex, prefrontal cortex, hypothalamus, thalamus, hippocampus, striatum, superior and inferior colliculus, cerebellar vermis, cerebellum, and rostral and caudal medulla). Tissues were weighed, and radioactivity was measured using a gamma counter to determine standardized uptake values (SUV) after vehicle and 1 and 10 mg/kg of HTL0014242. All radioactivity counts were decay-corrected to the time of tracer injection and expressed as standardized uptake value ratio. SUV was measured as follows: A one-way ANOVA with post hoc analysis (Tukey multiple comparisons) was used to assess dependent differences on [18F]FPEB uptake in ROIs.
A one-way ANOVA with post hoc analysis (Tukey multiple comparisons) was used to assess dependent differences on [18F]FPEB uptake in ROIs.
Image and Data Analysis.
The PET images were acquired in list mode and reconstructed with increasing frame times over the duration of the scan to characterize the radiotracer kinetics. Three-dimensional histograms with span 3 and maximum ring difference of 79 were used. Fourier rebinning was performed, and images were reconstructed using a two-dimensional filtered backprojection algorithm and a ramp filter and zoom of 1 to generate images on a 128 × 128 matrix. Image processing and data analysis were performed using VivoQuant and MIAKAT, an in-house computational pipeline implemented in MATLAB. ROIs (striatum, thalamus, hypothalamus, cerebellum, cortex, prefrontal cortex, hippocampus, and “other”) were defined in VivoQuant and used to generate time-activity curves (TACs) in MIAKAT.
Using the TACs and parent plasma input function, the volume of distribution (VT) was calculated using a two-tissue compartmental model for individual animal in each region and mean at each dose calculated. Mean VT data were expressed as percent RO by first determining percent [18F]FPEB bound and then normalizing this to vehicle as 100% RO, where %[18F]FPEB bound = (x/vehicle VT)*100 and where %HTL0014242 occupancy = (100% − %[18F]FPEB) bound. Average brain mGlu5 RO was determined from the mean of the RO in each region.
Cynomolgus Monkey In Vivo PET RO Study Using [18F]FPEB
Anesthesia and Dosing.
Cynomolgus monkeys were anesthetized (intramuscular injection of Alfaxan 2 mg/kg, dexmedetomidine 0.02 mg/kg, and midazolam 0.3 mg/kg), intubated, and maintained on oxygen and 1.5%–2.5% isoflurane throughout the imaging sessions. PET imaging was performed on the FOCUS-220 PET scanner (Siemens Healthcare Molecular Imaging, Knoxville, TN). Baseline scans were measured over 1 to 2 hours in each monkey after intravenous injection of [18F]FPEB over 3 minutes (155 MBq or 169.6 MBq for each monkey).
HTL0014242 was dosed orally as a suspension in 10% Solutol HS 15 and 9% HPβCD in water 2 hours prior to injection of [18F]FPEB (169.2 or 101.3 MBq for each monkey). Competition scans were conducted over a period of 2 hours after injection of [18F]FPEB. Dynamic PET scanning was preceded by transmission for attenuation and scatter correction.
Arterial Sampling.
The arterial plasma input functions corrected for the presence of radiometabolites were generated for all scans based on blood samples taken from the femoral artery. Manual sequential blood samples (0.5–3.5 ml) were collected at 18 selected time points during the 120-minute scan. Two 3.5-ml samples were collected before tracer administration to evaluate tracer stability in blood. Samples were collected in EDTA anticoagulant tubes and analyzed for radioactivity over time in a gamma counter (Wallac 2480 Wizard 3M Automatic γ-counter, Perkin-Elmer, Waltham, MA). Pretracer standards were used to evaluate the tracer ex vivo stability in blood. Plasma free fraction was determined through ultrafiltration.
Compound Exposure Determination.
Blood samples were collected predose and at 60, 120 (just prior to tracer), and 240 minutes (end of scan) postdose, relative to the HTL0014242 administration. Plasma was prepared and HTL0014242 concentrations were measured by LC-MS/MS using a Shimadzu Nexera ×2 HPLC system coupled with a SCIEX API 5500 Triple Quad mass spectrometer. Chromatograms were integrated using SCIEX Analyst 1.6.2 software. A linear regression was used to generate the calibration curve for HTL0014242. Concentrations of HTL0014242 were calculated using the peak area ratio of analyte to internal standard based on the standard curve.
Image and Data Analysis.
Reconstructed PET images were analyzed using the image processing PMOD software package version 3.802 (PMOD Technologies, Zurich, Switzerland). Volumes of interest were defined on a stereotaxic anatomic cynomolgus brain atlas to which the subject’s own anatomic T1 scans were registered, and masks were created for the caudate, putamen, hippocampus, anterior cingulate cortex and posterior cingulate cortex, frontal cortex, temporal cortex, parietal cortex, occipital cortex, and cerebellum (Ballanger et al., 2013). Masks were applied to dynamic images to extract the average activity concentration (kilobecquerels per cubic centimeter) within each volume of interest and generate time-activity curves representing regional brain activity concentration over time. TACs were expressed in SUV units (gram per milliliter) by normalizing by the weight of the animal and the injected dose.
A two-tissue compartmental model was used to determine the total VT values for each brain region using metabolite-corrected plasma curves (i.e., arterial input function). A Lassen plot (Cunningham et al., 2010) analysis was performed in GraphPad Prism software and used to estimate mGlu5 occupancy as described by the equation 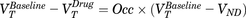 , which, when represented graphically for each (x = VT baseline, y = VT baseline − VT drug), produces a linear relationship, where the x intercept equals VND and the gradient is equal to global target occupancy. A global occupancy was determined graphically as the slope of the line. Occupancy measurements in individual brain regions were determined using the equation above and VND derived from the Lassen plot.
, which, when represented graphically for each (x = VT baseline, y = VT baseline − VT drug), produces a linear relationship, where the x intercept equals VND and the gradient is equal to global target occupancy. A global occupancy was determined graphically as the slope of the line. Occupancy measurements in individual brain regions were determined using the equation above and VND derived from the Lassen plot.
The relationship between percentage mGlu5 occupancy and either plasma concentration or the dose of HTL0014242 was investigated with a single specific binding site (Emax) model with a fixed Hill slope of 1. Occ = X/(X+K), where Occ is the measured mGlu5 occupancy, X is either the HTL0014242 plasma concentration (in nanograms per milliliter) or the dose (in milligrams per kilogram), and K is either EC50 or ED50.
Plasma Protein Binding Measurements
The plasma protein binding assay (in 10% plasma) was performed using a rapid equilibrium dialysis device. HTL0014242, mavoglurant, and dipraglurant prepared in DMSO were added (10 µM; 0.5% DMSO final) to plasma from various species, supplied by B&K Universal, as follows: HTL0014242 for mouse, rat, and cynomolgus monkey; mavoglurant for rat; and dipraglurant for human. Duplicate samples were dialyzed within the device against 4 mM potassium phosphate buffer containing 0.9% NaCl, pH 7.4, for a minimum of 4 hours at 37°C.
After incubation, the contents of each plasma and buffer compartment were removed and mixed with equal volumes of control dialyzed buffer or plasma as appropriate to maintain matrix similarity for bioanalysis. Plasma proteins were then precipitated by the addition of acetonitrile containing an analytical internal standard (50 ng/ml carbamazepine and 200 ng/ml reserpine) and centrifuged, and the supernatant was removed for analysis by mass spectrometry (LC-MS/MS). Test compound was measured in both compartments by LC-MS/MS with concentrations quantified using a calibration curve prepared in assay buffer. The percentage drug bound and unbound were calculated using the following equations:
 Fraction unbound for basimglurant (rat, human) was determined from published plasma protein binding measurements (Lindemann et al., 2015).
Fraction unbound for basimglurant (rat, human) was determined from published plasma protein binding measurements (Lindemann et al., 2015).
Rat Brain Binding Measurements
The brain homogenate binding assay (1:3 dilution) was performed using a rapid equilibrium dialysis device. HTL0014242 prepared in DMSO was added (5 µM; 0.5% DMSO final) to brain homogenate (B&K Universal) and dialyzed within the device (n = 2) against 4 mM potassium phosphate buffer containing 0.9% NaCl, pH 7.4, for 4 hours at 37°C. After incubation, the contents of each homogenate and buffer compartment were removed and mixed with equal volumes of control dialyzed buffer or plasma as appropriate to maintain matrix similarity for bioanalysis. Brain homogenate tissue was then precipitated by the addition of acetonitrile containing an analytical internal standard (50 ng/ml carbamazepine and 200 ng/ml reserpine) and centrifuged, and the supernatant was removed for analysis by mass spectrometry (LC-MS/MS). Test compound was measured in both compartments by LC-MS/MS with concentrations quantified using a calibration curve prepared in assay buffer. The percentages of drug bound and unbound were calculated using the following equations:
 Fraction unbound in rat brain for mavoglurant was estimated from cerebrospinal fluid (CSF) as a fraction of total brain concentration from exposure reported in Bennett et al. (2014). A similar approach was used to determine unbound fraction in rat brain for dipraglurant as determined in a separate pharmacokinetic study (Bennett et al. unpublished). For basimglurant, an average brain fu was computed from the cited rat brain/plasma ratio (Kp 1.7–2.9) and unbound fraction in plasma (0.021) and assuming good passive brain penetration consistent with unbound brain/unbound plasma, Kpuu = 1 (Lindemann et al., 2015), whereby brain fu = Kpuu* plasma fu/Kp. It was assumed the fraction unbound in brain was consistent across species.
Fraction unbound in rat brain for mavoglurant was estimated from cerebrospinal fluid (CSF) as a fraction of total brain concentration from exposure reported in Bennett et al. (2014). A similar approach was used to determine unbound fraction in rat brain for dipraglurant as determined in a separate pharmacokinetic study (Bennett et al. unpublished). For basimglurant, an average brain fu was computed from the cited rat brain/plasma ratio (Kp 1.7–2.9) and unbound fraction in plasma (0.021) and assuming good passive brain penetration consistent with unbound brain/unbound plasma, Kpuu = 1 (Lindemann et al., 2015), whereby brain fu = Kpuu* plasma fu/Kp. It was assumed the fraction unbound in brain was consistent across species.
Additional Characterization of mGlu5 NAMs
The following data and data manipulations were used for calculating the exposure-receptor occupancy for dipraglurant, mavoglurant, and basimglurant.
For dipraglurant, human exposure data were from Tison et al. (2016); on day 1, 50 mg Cmax plasma (793.4 ng/ml) data were normalized to reflect the exposure at 100, 200, and 300 mg at which RO was measured. This day 1 predicted exposure is consistent with the cited exposure after repeat dosing at 100 mg (Cmax plasma 1682.8 ng/ml) since no accumulation would be expected for the short half-life observed. RO data were from https://www.lifescicapital.com/company/addex-therapeutics/ (Wong et al., 2018).
For mavoglurant, rodent exposure and RO were taken from Bennett et al. (2014).
For basimglurant, rat RO were from Lindemann et al., 2015. Human day 1 plasma exposure and median pharmacokinetic half-life in patients with major depressive disorder were taken from Cosson et al., 2018 and mGlu5 receptor occupancy estimated at steady state (Quiroz et al., 2016). Given the recognized long half-life of this compound, it was necessary to estimate the steady-state Cmax by calculating an accumulation ratio using the following equation: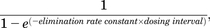 where elimination rate constant = 0.693/quoted half-life of 49 hours.
where elimination rate constant = 0.693/quoted half-life of 49 hours.
This accumulation ratio (3.5) was then applied to the Cmax quoted on day 1 to correlate the steady-state exposure and receptor occupancy. The plasma exposure was used to estimate unbound brain concentration taking into account Kp and unbound fraction in brain.
Predicting RO Using a Simple Emax Model
A simple Emax model was used to predict receptor occupancy as follows: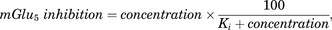 where concentration was either unbound plasma or unbound brain concentration.
where concentration was either unbound plasma or unbound brain concentration.
Unbound concentrations were computed by taking measured concentration * unbound fraction as determined in plasma or brain tissue. For mavoglurant, measured CSF concentrations were considered to represent unbound brain concentrations.
Statistical Analyses
When there are multiple measurements, data are presented as means ± S.D. Exposure-RO data were fitted to a variable-slope four-parameter fit (GraphPad Prism version 8), with basal constrained to “0” and top constrained to “100.” An ANOVA F-test was conducted to determine whether the resulting Hill slope was significantly different from 1.
Results
The physicochemical properties, mGlu5 affinity, human plasma pharmacokinetic half-life, and rat brain penetration properties of HTL0014242, dipraglurant, mavoglurant, and basimglurant are described in Table 1. mGlu5 affinity varies by approximately 260-fold between compounds, with HTL0014242 and basimglurant having the highest mGlu5 NAM affinity. mGlu5 affinity was shown to be very consistent across species. Partitioning into brain was measured in the rat for HTL0014242, dipraglurant, and mavoglurant, indicating approximately 17-fold variation in Kp and 4-fold variation in the estimated Kpuu, with HTL0014242 having the highest relative brain penetration. The precise Kpuu is unknown for basimglurant but assumed to be close to 1 given the cited good brain penetration (Lindemann et al., 2015).
mGlu5 NAM inhibitor properties
cLogP, partition coefficient; Fup, free fraction in plasma; t1/2, half life; N/A, not available; Fubr, free fraction in brain
Total brain and plasma exposure for HTL0014242 in mouse and rat is shown in Table 2. Exposures are taken at the same time point as when mGlu5 RO is measured. For PET studies, assessment of ex vivo [18F]FPEB biodistribution confirmed that HTL0014242 competed for the same binding site as [18F]FPEB. Drug concentrations were measured at the start of the [18F]FPEB scan to reflect the highest exposure. However, it should be noted that previous pharmacokinetic studies showed measurements of exposure to be similar over the same time period as used in this PET study (Bennett et al. unpublished). Plasma exposure and brain exposure for mavoglurant in the rat, basimglurant in rat and human, and dipraglurant in human are given in Table 3. Brain exposure for basimglurant and dipraglurant in human was estimated assuming the same partitioning as for rat. For the dipraglurant dose, proportionality was assumed to estimate exposures at the higher doses of 200 and 300 mg at which mGlu5 RO has been measured.
Measured mGlu5 receptor occupancy and associated exposure dosing for HTL0014242. PK, pharmacokinetics; cyno, cynomolgus monkey; N/A, not available
Mavoglurant, dipraglurant and basimglurant mGlu5 RO and exposure
The mGlu5 RO measured and the dose and exposure levels are illustrated in Tables 2 and 3. mGlu5 RO increased with increasing dose/exposure for all compounds studied when multiple dose levels were profiled. For each compound, there are clear exposure-RO relationships across a range of species, but when the data are plotted together, the relationship for each compound is distinct, with greater than 100-fold difference between the exposures required for a similar level of RO (i.e., not overlaying), as illustrated in Fig. 1A. When both the unbound concentration in plasma and mGlu5 affinity are accounted for, as illustrated in Fig. 1B, the exposure-response relationships for each compound do appear to be closer together, although greater than 10-fold difference was found between the most and least potent relationships.

Relationship between exposure and receptor occupancy across multiple mGlu5 NAM chemotypes. (A) Exposure plotted as plasma Cmax. (B) Exposure plotted as unbound plasma Cmax divided by the affinity (Ki) of each ligand at the mGlu5 receptor. The dotted line represents the theoretical curve fit where 50% occupancy is achieved when the unbound plasma = Ki. (C) Exposure plotted as the unbound brain concentration divided by the affinity (Ki) of each ligand at the mGlu5 receptor. The dotted line represents the theoretical curve fit where 50% occupancy is achieved when the unbound brain = Ki. Data were also fitted to a four-parameter sigmoidal dose-response curve (solid line). Cyno = cynomolgus monkey.
Figure 1C shows, when considering differences in both the unbound concentration in brain and mGlu5 affinity, the relationship between compounds and RO is more unified, with exposure-RO relationships being <10-fold separated. Fitting the data to a variable-slope four-parameter fit (GraphPad Prism version 8) resulted in a curve with EC50 calculated to be 0.67 (confidence interval 0.41–1.10) with a Hill slope of 0.81 (confidence interval 0.38–1.23), which was demonstrated to be not significantly different from 1 based on an F-test. The F-test comparison of data fits (null hypothesis Hill slope = 1 vs. an alternative unconstrained Hill slope) indicated that Hill slope = 1 produced the better fit (P = 0.4, F ratio = 0.74). The theoretical exposure-RO relationship is shown in Fig. 1C in the dotted line, where 50% mGlu5 RO is achieved where ratio of unbound brain concentration/Ki = 1. Both the theoretical curve and the curve fit of the data are remarkably similar given the breadth of compounds and data sets used for the analysis.
HTL0014242 predictions of RO from unbound plasma or brain are given in Table 4, and a graphical representation of the predictions from unbound plasma versus measured RO is given in Fig. 2. The predictions from either matrix are similar and consistent with the good brain penetration observed for this compound in the rat (Kpuu = 1.3; Table 1). Figure 2 does demonstrate a tendency to underpredict mGlu5 RO across all species when predicting from plasma exposure, with predictions being from 8% higher to 41% lower than measured, with an average underprediction of 14%. Table 5 illustrates predictions of RO for mavoglurant, dipraglurant, and basimglurant and indicates similar RO or slight overprediction of observed RO for these compounds.
Measured and predicted mGlu5 RO for HTL0014242.ub, unbound; cyno, cynomolgus monkey; N/A, not available

Predicted vs. measured mGlu5 receptor occupancy of HTL0014242 across species. Comparison between the predicted (pred.) receptor occupancy (open icons) unbound plasma exposure vs. the measured (meas.) receptor occupancy (filled icons), demonstrating that plasma levels underpredict RO.
Measured and predicted mGlu5 RO for mavoglurant, dipraglurant, and basimglurant. ub, unbound
Discussion
The current study is the first to formally investigate exposure-RO relationships of selective mGlu5 NAMs across species in a collective manner. This investigation clearly demonstrated that, irrespective of species, a more unified exposure-response relationship across mGlu5 NAMs was evident when accounting for unbound brain concentration and mGlu5 affinity, rather than total systemic exposure. Consideration is given to whether RO could be predicted for a given level of exposure, with the potential to underpin future clinical trial designs. The strengths and limitations of these approaches are highlighted in this discussion.
Dipraglurant, mavoglurant, basimglurant, and HTL0014242 have all progressed to clinical development despite having distinct structures and physiochemical attributes that contribute to differing pharmacokinetic properties and mGlu5 receptor affinities ranging from Ki values of 0.56 to 117 nM. These NAMs are all selective for mGlu5 (Vranesic et al., 2014; Bezard et al., 2014; Lindemann et al., 2015; Christopher et al., 2015), have no known active metabolites, and all bind to the M-MPEP site. Furthermore, the current study demonstrated that their affinities are conserved across human, mouse, rat, and monkey mGlu5 orthologs, consistent with previously reported data for basimglurant (Lindemann et al., 2015). In combination, this data set is uniquely placed to examine cross-species exposure-RO relationships across mGlu5 NAMs with differential physiochemical properties.
The relationship between total plasma exposure and mGlu5 RO demonstrated distinct exposure-RO profiles for each compound, as illustrated by the oral dose of 3 mg/kg in the rat, for which HTL0014242 demonstrated 80% mGlu5 RO and mavoglurant produced 45% RO despite similar plasma concentrations of 139 and 128 ng/ml, respectively. The free drug hypothesis states that only unbound compound binds the target; therefore, correcting to unbound plasma concentration and compound affinity improved the exposure-occupancy relationship compared with total plasma alone. As mGlu5 RO is measured in the brain, it is not surprising that the most unified exposure-RO relationship emerges when unbound brain concentrations are accounted for as well as mGlu5 affinity. The fit of the data indicated that the Hill slope was not significantly different from 1, indicating that mGlu5 RO likely reflects binding at a single receptor binding site. Consequently, this unified relationship spans the theoretical curve predicted from a simple Emax model with a Hill slope of 1 and for which an unbound exposure/Ki ratio of 1 would be expected to yield 50% RO. This observation is consistent with the published dipraglurant human plasma exposure-RO relationship, which obeyed a first-order Hill equation (Wong et al., 2018). Given the number of data sources for the present analysis covering a range of species and mGlu5 NAMs with differing properties, it is remarkable that the exposure-RO relationships are so close.
However, although a more unified relationship is achieved by using unbound brain corrected for mGlu5 affinity to relate to RO, there is still up to 10-fold variance in exposure relative to Ki for a given level of RO. Therefore, it is important to consider the key factors that may contribute to this variance—namely, measurement of brain penetration, free fraction, and associated experimental design and methodology for determination of exposure and RO.
Firstly, understanding brain penetration is essential but challenging since the concentrations can be influenced by technical factors such as whether brain exposure is derived from CSF or whole-brain measurements (Westerhout et al., 2011; O’Brown et al., 2018). In this study, brain penetration was assessed using total brain concentrations, fraction unbound in rodent brain homogenate, and in one case, CSF concentrations. Observations with mavoglurant using CSF data were consistent, with the overall exposure-RO relationships indicating that any bias was minimal. Furthermore, for the compounds studied here, the Kpuu values observed in rodents were consistent with largely passive distribution, which provided confidence in extrapolation across species.
It was assumed that the degree of brain partitioning of dipraglurant and basimglurant was equivalent to that observed in the rat. The brain unbound/Ki-RO relationship was fitted excluding the human data, and this confirmed a similar EC50 and Hill slope to that observed with the full data set [EC50 = 0.49 (95% confidence interval 0.28–0.84); Hill slope = 0.86 (95% confidence interval 0.41–1.31)], confirming that the overall relationship was not biased by the assumption that human and rat brain penetration was similar for these compounds.
A second factor that may influence variance in exposure-RO relationship is the method used to assess the true unbound concentration available to interact with the target. The established in vitro methods for measuring brain unbound fraction are crude, relying on whole-brain homogenate with no indication of variance in unbound concentrations across brain regions. For passively permeating compounds such as HTL0014242, the unbound plasma concentration would be expected to reflect unbound brain concentration, allowing the use of unbound fraction in plasma as a surrogate. Based on the above observation that these exposure-RO relationships are consistent with a simple Emax model, it was deemed valid to apply this methodology to predicting RO from either matrix, brain, or plasma. HTL0014242 underpredicted the measured mGlu5 RO by an average of 14% based on brain data. Given the similarity in prediction from the two matrices, there is most likely an underestimation of free fraction rather than brain penetration, especially for HTL0014242 because of its high plasma protein and brain tissue binding (>99% bound) whereby it is challenging to accurately measure low unbound concentrations (<1% unbound). Using this approach for dipraglurant, mavoglurant, and basimglurant, RO tends to be similar or overpredicted compared with measured RO. Since these compounds vary in brain penetration, but all have higher unbound fraction than HTL0014242, this observation highlights the challenge in accurately predicting RO from unbound concentrations when binding is high.
Despite the slight underprediction of RO based on animal data, HTL0014242 illustrates that this approach could be used to provide a conservative estimate of mGlu5 RO across a variety of species, including human. Measurement of HTL0014242 mGlu5 RO in human would be a useful next step to determine whether the relationship observed in animals is consistent with human and how plasma concentrations relate to mGlu5 RO in the brain. Despite the caveats around the influence of free fraction, predicted RO for these compounds is in line with observed values, supporting the use of unbound plasma concentrations measured clinically to estimate RO. Such information could aid dose selection and clinical experimental design providing that the relationship between unbound plasma and unbound brain concentrations is understood.
Experimental design factors relating to the dose selection, sampling time for measurement of exposure versus RO, and number of replicates could potentially contribute to the overall variability in the exposure-RO relationships. For new experimental data, doses were deliberately chosen to support determining a full exposure-RO relationship. For in vivo and ex vivo RO measurements, the exposure was measured from the same brain sample as that used for RO, thus removing any potential disconnect between measurements. For PET studies, the scan time for HTL0014242 studies was 1 hour in duration, but plasma and brain concentrations were known to be similar across this period after oral dosing. Scanning periods are not reported for dipraglurant and basimglurant, but the long half-life of basimglurant suggests that brain concentrations would remain consistent. Dipraglurant has a short half-life in humans (<1 hour), so it is possible that the Cmax measured exposure overestimates the average concentration associated with the RO. However, considering the large data set containing compounds with different properties, it is unlikely that the timing of exposure and mGlu5 RO measurements contributed significantly to the variance in exposure-RO relationships. The number of replicates for HTL0014242 RO and exposure studies was relatively small (2–5) but adequate, considering the data were part of a larger data set covering a wide range of mGlu5 RO (12%–95%). Lastly, in the cynomolgus PET study, only one monkey was used for each dose level since RO could be related directly to exposure measured in that animal. Although the exposure-RO relationship was consistent with that observed in other species, it is possible that increasing the number of replicates would improve the accuracy and precision of this data. A further consideration relates to the methodology used to measure mGlu5 RO. mGlu5 NAM PET studies have used a variety of PET ligands. Although there is potential for ligands to bind at different binding sites, it has been established that similar chemical scaffolds can inhibit prototypic MPEP/FPEB sites by interacting at nonidentical but overlapping sites (Gregory et al., 2014; Rook et al., 2015), and [18F]FPEB or [11C]ABP688 PET ligands appear to bind at the M-MPEP binding site (Hamill et al., 2005; Wyss et al., 2007; Hintermann et al., 2007). Therefore, it is unlikely that the choice of PET ligand contributes to the variance observed in the exposure-RO relationships.
PET studies provide distribution and derived ligand binding across several brain regions. HTL0014242 RO ranged from 62.2% to 79.1% at 1 mg/kg and 73.3%–89.1% at 10 mg/kg in four key regions (striatum, cerebellum, frontal cortex, and hippocampus) in the rat. However, this level of variance is insufficient to explain an underprediction of RO. The widespread expression of mGlu5 (Ferraguti and Shigemoto, 2006; Hovelsø et al., 2012) would suggest a consistent RO across the brain, supporting the use of total RO to compare with exposure. It should also be noted that there was some interanimal variability in measured mGlu5 RO, as indicated by the S.D. quoted. As the predicted ROs for HTL0014242 fall within this level of variability (Table 3), the most likely explanation for underprediction is in the estimate of unbound fraction as described above.
A strength of this study is that every effort was taken to minimize the impact of factors acknowledged to contribute to variability in this novel combined analysis of new and published data. In particular, brain penetration was measured in sufficient replicates to provide an average view of the exposure, and when possible, exposure was obtained from the same animals in which RO was measured. mGlu5 NAMs were included that were known to bind to the same MPEP site. RO data were generated using established methods and validated mGlu5 probes to provide consistency with RO incorporated from publications. Finally, for exposure and RO studies, doses were chosen to explore the full exposure-RO curve.
Despite the variability in the exposure-RO relationships, the evidence presented here suggests that the behavior of selective mGlu5 NAMs is consistent with a simple Emax model, thus providing guidance on extrapolation from animal to human. As more data become available, particularly in human, it would be beneficial to further expand these relationships and to encompass mGlu5 NAMs with different binding modes. Notwithstanding the long history of the mGlu5 field, this unified assessment of the exposure-RO relationship across species and mGlu5 NAMs has demonstrated, for the first time, the importance of understanding the concentration in the target organ for the interpretation and design of nonclinical and clinical studies.
Acknowledgments
The authors thank John Christopher, Sarah Bucknell, Miles Congreve, James Hagan, and Eimear Howley, as well as colleagues in the Medicinal Chemistry and DMPK departments at Sosei Heptares, for unlabeled compound synthesis, management of in vivo pharmacokinetic studies, and/or general advice. In addition, the authors also thank RenaSci Ltd for conducting the rat ex vivo binding studies and Ian Brown and colleagues at Teva Pharmaceuticals, 145 Brandywine Pkwy, West Chester, PA 19380, for mouse ex vivo binding studies; Ilan Rabinar, Lisa Wells, and colleagues at Invicro, Hammersmith hospital, Du Cane Road, United Kingdom, for conducting the rat [18F]FBEP PET study; Khanum Ridler and colleagues at Invicro, New Haven, CT, for the cynomolgus monkey [18F]FBEP PET study; and Graham Hagger at Royal Veterinary College, London, for help with the mouse ex vivo binding studies.
Authorship Contributions
Participated in research design: Bennett, Cooper.
Conducted experiments: Bennett, Sergeev, MacSweeney, Bakker.
Performed data analysis: Bennett, Sergeev, Cooper.
Wrote or contributed to the writing of the manuscript: Bennett, Sergeev, MacSweeney, Bakker, Cooper.
Footnotes
- Received October 9, 2020.
- Accepted January 26, 2021.
This work is fully funded by Sosei Heptares, Steinmetz Building, Granta Park, Great Abingdon, Cambridgeshire, CB21 6DG, UK.
Abbreviations
- Emax, maximum effiacy; MPEP, 6-methyl-2-(phenylethynyl)-pyridine; M-MPEP, 2-[(3-methoxyphenyl)ethynyl]-6-methylpyridine; [18F]FPEB, 3-[18F]fluoro-5-[(pyridin-3-yl)ethynyl] benzonitrile; HPLC, high-performance liquid chromatography; HPβCD, (2-hydroxypropyl)-β-cyclodextrin; LC-MS/MS, liquid chromatography with tandem mass spectrometryCSF
- cerebrospinal fluid
- fu
- free fraction
- Kp
- brain/plasma ratio
- Kpuu
- unbound brain/unbound plasma
- mGlu5
- metabotropic glutamate 5 receptor
- NAM
- negative allosteric modulator
- PET
- positron emission tomography
- RO
- receptor occupancy
- ROI
- region of interest
- SUV
- standardized uptake value
- TAC
- time-activity curve
- VT
- volume of distribution
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.




{kind=link}
{kind=link}