


Abstract
The serotonin (5-HT)2A/2c agonist 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI), the 5-HT2C agonist 6-chloro-2-[1-piperazinyl]-pyrazine and the 5-HT2A partial agonist m-chloro-phenylpiperazine (mCPP) were injected bilaterally into the medial prefrontal cortex of male rats. DOI and mCPP, but not 6-chloro-2-[1-piperazinly]-pyrazine, elicited a dose-dependent head-twitch response (HTR). DOI-induced HTR had an ED50 of 12.8 nmoles/0.5 μl/side and was inhibited by the 5-HT2A antagonists ketanserin and MDL 100,907 but was not blocked by pretreatment with the selective 5-HT2C/2B antagonist SDZ SER 082. The HTR to mCPP demonstrated a bell-shaped dose-response curve with an ED50of 1.5 nmoles/0.5 μl/side and a peak effect after 3 nmoles/side. The response to mCPP was greatly diminished by both ketanserin and MDL 100,907 and was partially reversed by SDZ SER 082. These findings suggest that the HTR produced by the direct injection of serotonergic agonists into the medial prefrontal cortex is, in part, mediated by the activation of 5-HT2A receptors. Pretreatment of rats with the 5-HT1A agonist (±)-8-hydroxy-dipropylaminotetralin hydrobromide inhibited the HTR to DOI. This is consistent with other evidence that suggests a functional antagonism between 5-HT1A and 5-HT2A receptor activation. The HTR to DOI was potentiated by the novel 5-HT1A selective antagonist WAY 100,635, which suggests that 5-HT1Areceptors tonically regulate this behavioral response to stimulation of cortical 5-HT2A receptors.
The systemic administration of direct as well as indirect 5-HT agonists to rodents has been shown to produce a characteristic HTR (Peroutkaet al., 1981; Colpaert and Janssen, 1983; Green et al., 1983; Goodwin and Green, 1985; Darmani et al., 1990a, 1990b, 1992). HTR produced by serotonergic agonists can be blocked by selective 5-HT2 receptor antagonists (Luckiet al., 1984; Handley and Singh, 1986). These findings suggest that the HTR is mediated by 5-HT2 receptors. Indeed, a strong correlation exists between the potency of serotonin antagonists to inhibit 5-HT agonist-induced HTR and affinity for the 5-HT2 binding site (Peroutka et al., 1981;Ortmann et al., 1982).
The development of highly selective and potent 5-HT2antagonists, along with advanced molecular biological techniques, has led to the classification of serotonin 5-HT2 receptors into at least three different subtypes. Current nomenclature defines the 5-HT2B site as that which corresponds to receptors that mediate contractile function in the fundus of the stomach. The 5-HT2C receptors were originally classified as “5-HT1C” receptors. Because of similarities to the 5-HT2 receptor family (as determined by molecular biology, pharmacological profiles and links to second messenger systems), these receptors are now classified with the 5-HT2 family. 5-HT2A sites are distributed in high density in the cortex as well as in the hypothalamus, caudate putamen and nucleus accumbens (Pazos et al., 1985; Pazos and Palacios, 1985; Hoyeret al., 1992). The corticolimbic distribution of the 5-HT2A receptors has led to the suggestion that these receptors might be critically involved in the neuropathology and treatment of a variety of psychiatric disorders, including anxiety, depression and schizophrenia. Clinical studies have suggested that compounds with antagonist properties at 5-HT2A may possess anxiolytic, antidepressant and antipsychotic properties (Deakin, 1989;Meltzer et al., 1989). The “atypical” antipsychotics, in particular, have a relatively high affinity for 5-HT2Areceptors (Meltzer et al., 1989). It is possible, therefore, that the antipsychotic effect of the atypical antipsychotic drugs involves the inhibition of 5-HT2A receptors.
More recently, it was reported that the direct administration of the mixed 5-HT2A/2C agonist DOB into the mPFCx in rats produces a dose-dependent increase in the HTR (Granhoff et al., 1992). The effect of (±)-DOB was inhibited by pretreatment with ritanserin, a 5-HT2A/2C antagonist, which suggests that the behavioral response to DOB is mediated by 5-HT2A or 5-HT2C receptors. The co-administration of (±)-DOB with the 5-HT1A agonist 8-OHDPAT was shown to inhibit the HTR, which further suggests that DOB-induced HTR is mediated by 5-HT2A receptors. Numerous studies have demonstrated a specific, reciprocal regulation of 5-HT2A receptors by 5-HT1A receptors (Darmani et al., 1990b; Araneda and Andrade, 1991; Millan et al., 1992; Ashby et al., 1994; Uphouse et al., 1994; Meltzer and Maes, 1995).
In the present study, we evaluated the HTR to a variety of serotonergic agonists administered directly into the mPFCx. One of these, DOI, is structurally similar to the phenylisopropylamine DOB, and two others, mCPP and MK-212, are arylpiperazines. These drugs differ in their selectivity and efficacy at the 5-HT2A and 5-HT2C receptors. A pharmacological analysis of the behavioral effects of direct intracortical administration of 5-HT2 receptor agonists and the selective activation of cortical 5-HT2A and 5-HT2C receptors is currently lacking. In addition to characterizing several different 5-HT2 agonists, we also utilized new, subtype-selective 5-HT2 receptor antagonists to evaluate the 5-HT2 receptors in the mPFCx that mediate the HTR produced by the direct administration of 5-HT agonists into this region. Finally, we extended these studies to demonstrate the existence of a regulatory interaction between 5-HT1A and 5-HT2A receptors that may serve to modulate behavioral responses to endogenous 5-HT.
Materials and Methods
Animals and surgery.
Male, Sprague-Dawley rats (150–250 gm, Zivic-Miller Labs, Alison Park, PA) were anesthetized with 1 ml/kg (i.p.) of a mixture of ketamine and xylazine in a ratio of 70 to 6. Anesthetized rats were mounted in a stereotaxic frame (Stoelting Inst., Wood Dale, IL), and the skull was exposed. Two 1.0-mm holes were drilled through the bone, bilaterally, above the mPFCx (3.2 mm anterior to bregma and about 0.7 mm lateral to the sagittal suture) (Paxinos and Watson, 1992). Two additional holes were made partially through the bone in the posterior region of the skull, and two small set screws were screwed into the skull. A 21-gauge stainless steel guide cannula was then stereotaxically placed into each hole and lowered to a position exactly 1.0 mm beneath the surface of the skull. The guide cannulas were then anchored to the skull using Krazy Glue Gel and cemented into position with cranioplastic cement (Plastics One, Reannex, Virginia). “Dummy” cannulas, made of stainless steel wire, were inserted into each guide to keep it patent. Each cannula was measured and precut to a length of 10 mm. Following cannula implantation, each rat was individually housed and was allowed 2 to 3 days for recovery from surgery. Throughout the experiments, the animals were housed in a facility with a light cycle of 12 h on, 12 h off, and they had free access to food and water. The ambient temperature in the vivarium was maintained at 23 ± 1°C.
Drug administration.
Seventy-two hours after surgical implantation of guide cannulas, rats were placed in behavior-monitoring cages (clear polycarbonate, 38 cm × 33 cm × 34 cm) and allowed a minimum of 30 min to habituate. After habituation, the animals were removed from the cages and gently wrapped in a laboratory towel in order to restrain and calm them during drug administration. Animals were positioned in the towel so that the guide cannulas extended through a small hole to allow access to the injection cannula. The dummy cannulas were removed, and an injection cannula was lowered into each guide. Each drug or vehicle was drawn up into a 10-μl Hamilton microsyringe (Hamilton Inst., Reno, NV) that had a 20-cm length of polyethylene tubing (I.D. 0.8 mm) epoxied to the needle barrel. An injection cannula constructed of a 4-cm piece of 21-gauge stainless steel tubing fitted over a 17-mm length of 26-gauge stainless steel tubing was inserted into the other end of the polyethylene tubing. The injection cannula was cut so that 14 mm of the 26-gauge stainless steel tubing was exposed beyond the end of the 21-gauge tubing. This would allow 4 mm of the injection cannula to protrude from the end of the 10-mm guide cannula that had been surgically implanted 1 mm below the surface of the skull. In this way, drugs could be administered directly into the mPFCx, at a depth of 5 mm beneath the surface of the skull. Drug was administered at a rate of 0.5 μl/min in a total volume of 0.5 μl/side. The injection cannula was then removed, and a dummy cannula was reinserted into each guide. Each animal was then returned to the observation cage, and behavior was monitored for the following 30 min.
For experiments that included a second drug treatment, the drugs were administered either i.p. or s.c. 10 min into the habituation period (20 minutes prior to intracerebral injection). After each experiment, the rats were deeply anesthetized with chloral hydrate and sacrificed by decapitation. Brains were immediately removed and placed in a solution of 4% formaldehyde in water. Then, 24 to 48 h later, fixed brains were cut on a refrigerated microtome, and injection sites were verified histologically from 40-μm coronal sections. In all experiments, each subject was used only once per experiment before dissection. Data from animals in which the needle tracks were found to terminate outside of the mPFCx were discarded (fig. 1). All animal use procedures were in strict accordance with the PHS Guide for Care and Use of Laboratory Animals and were approved by the Case Western Reserve University Institutional Animal Care and Use Committee.

Representative injection sites for the intracranial administration of drugs. •, sites of drug injection (as determined from needle tracks) that were within the mPFCx. ○, sites of drug injection in which the injection cannulas terminated outside of the mPFCx. HTR data from animals in which either the right or the left injection cannula was determined to have terminated outside of the mPFCx were excluded from the study. The sites shown are derived from animals used in generating the 5-HT agonist dose-response data (figs. 2and 3). Illustrations are adapted from the atlas of Paxinos and Watson (1992). Measurements in millimeters refer to distance from bregma. aca, anterior commissure, anterior; Acb, accumbens nu; aci, anterior commissure, intrabulbar; Cg1, cingulate cortex, area 1; Cg3, cingulate cortex, area 3; Cl, claustrum; DP, dorsal peduncular cortex; fmi, forceps minor corpus callosum; Fr2, frontal cortex, area 2; IL, infralimbic cortex; lo, lateral olfactory tracts; MO/VO, medial orbital and ventral orbital cortex; RF, rhinal fissure.
Behavioral observation.
After drug administration (see above), the number of head twitches observed in each 5-min period was recorded for a total of 30 min. In addition, other stereotypical behaviors (including locomotor activity, sniffing, grooming and rearing) were observed and noted.
Drugs.
The following drugs and chemicals were used in this study: DOI and 8-OHDPAT (Research Biochemicals Inc., Natick, MA), MK-212 (Merck, Sharpe and Dome, Wilmington, DE), mCPP (Aldrich Chemical Co., Milwaukee, WI), MDL 100,907 (Merrell-Dow Pharmaceuticals, Cincinnati, Ohio), SDZ SER 082 (Sandoz, Basel, Switzerland) and WAY 100,635 (Wyeth-Ayerst, Princeton, NJ). All drugs were dissolved in double-distilled demineralized water.
Data analysis.
Unless otherwise noted, all data are expressed as the mean ± S.E.M. of the absolute number of head twitches observed in the 30-min observation period immediately after the intracranial administration of drugs or vehicle. The ED50 for DOI and the half-maximal response to mCPP were calculated using the following equation:
Results
The effect of direct, bilateral administration of 5-HT2agonists into the mPFCx on the production of HTR.
DOI and mCPP, administered directly into the mPFCx, elicited a maximal HTR within the first 5-min observation period (fig. 2). The response had a duration of approximately 30 min, after which time the response was not significantly greater than that seen in control animals (fig. 2).

Time course for the production of the HTR by 5-HT2 agonists administered bilaterally into the mPFCx. Each data point represents the mean ± S.E.M. of at least six determinations. A) The time course of DOI-induced HTR. DOI 8.4 nmoles [P < .05 at all time-points except 15 min (not significant)]; DOI 28 nmoles [P < .05 at all time-points except 15 min (not significant)]. B) The time course of mCPP-induced HTR. mCPP 1 nmole [P < .05 for all time-points except 15, 25 and 30 min (not significant)]; mCPP 3 nmoles (P < .05 for all time-points except 25 and 30 min (not significant)]; mCPP 8 nmoles (P < .05 for all time-points except 15 and 30 min (not significant)]; mCPP 30 nmoles (P < .01 for 5 min time-point).
Bilateral administration of DOI into the mPFCx produced a dose-dependent activation of the HTR. At doses of 2.8, 8.4 and 28 nmoles/0.5 μl/side, rats demonstrated an average of 9 ± 2, 32 ± 4, and 42 ± 4 head twitches, respectively (fig.3). The responses to both the 8.4- and the 28-nmole doses of DOI were significantly greater than the response to saline alone (P < .01, fig. 3). The ED50 for the HTR to DOI was calculated to be 12.8 nmoles/side by nonlinear regression analysis.

Dose-response curve for the production of a HTR by 5-HT agonists. The ED50 for DOI was estimated to be 12.8 nmoles, calculated as an internal ED50 (base line to maximal response). The dose of mCPP producing a half-maximal response, in the primary (upward) phase of the dose-response curve, was estimated to be 1.5 nmoles. Vehicle injections elicited an average of 5.7 ± 2.0 twitches in 30 min. Each value represents the mean ± S.E.M. of the total HTR from 6 to 8 animals. * P < .01 with respect to vehicle-injected controls.
The bilateral administration of mCPP into the mPFCx at doses of 1, 3, and 8 nmoles/side produced an average of 18 ± 2, 46 ± 8, and 26 ± 3 head twitches, respectively. Each of these responses was significantly greater than that of vehicle-injected control animals (fig. 3). A dose of 30 nmoles of mCPP, administered bilaterally into the mPFCx, significantly enhanced the HTR in the first 5-min observation period, but by 10 min this response was no greater than that observed in control animals (fig. 2). The total number of head twitches produced by 30 nmoles of mCPP was not greater than that observed in vehicle-treated animals. The half-maximal response to mCPP for the initial phase of mCPP-induced HTR was estimated to be 1.52 nmoles using nonlinear, least-squares regression analysis with the maximal response equal to that produced by a 3-nmole dose of mCPP. Thus mCPP was approximately 8.5 times more potent than DOI in producing the HTR. Rats injected with MK-212 bilaterally into the mPFCx at doses of 3, 8.4, 17 or 30 nmoles/0.5 μl/side demonstrated a nonsignificant increase in the HTR (fig. 3).
Effect of subtype-selective 5-HT2 antagonists on HTR.
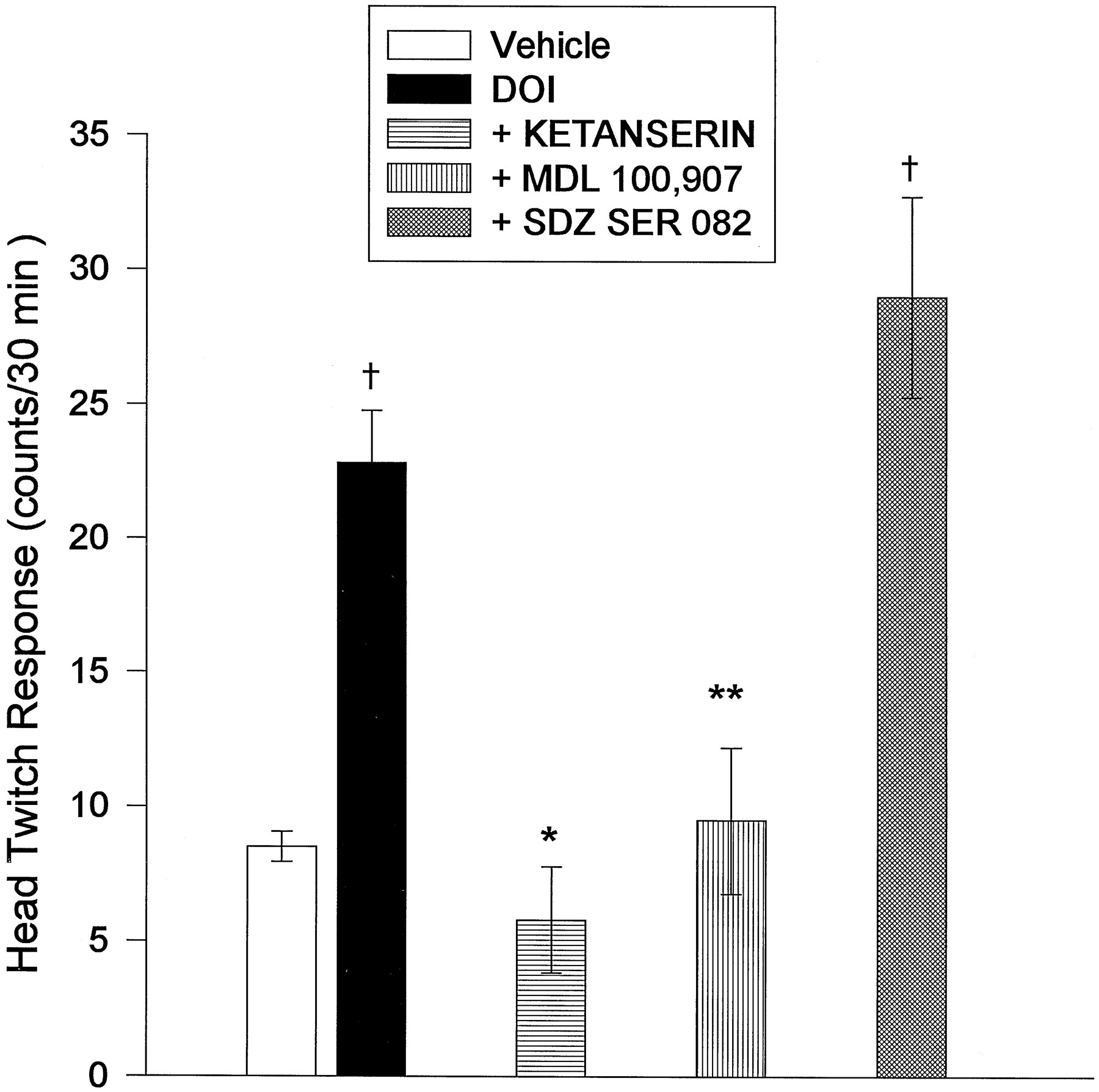
Ketanserin pretreatment (2.5 mg/kg s.c.), completely inhibited the HTR produced by DOI (6 ± 2, n = 6, fig. 4). Ketanserin alone had no effect on the HTR in vehicle-injected controls. In addition, other behaviors, such as locomotor activity and stereotypy, did not appear to be affected by the presence of ketanserin. Pretreatment with the selective 5-HT2A antagonist MDL 100,907 (0.1 mg/kg s.c.) also inhibited the HTR to DOI (fig. 4), with no discernible effect on basal locomotor or stereotypical behavior. Pretreatment with 0.3 mg/kg (s.c.) of the selective 5-HT2C antagonist SDZ SER 082 did not alter the DOI-induced HTR (fig. 4). This dose of SDZ SER 082 did not affect behavior in rats given this drug alone (data not shown).

The effect of pretreatment of animals with 5-HT2 antagonists on HTR produced by bilateral, intra-mPFCx injection of DOI (8.4 nmoles/0.5 μl/side). Each antagonist or saline was administered 20 min before intracortical injection of DOI at the following doses: saline, 0.9%, 1 ml/kg (i.p., n = 6), ketanserin, 2.5 mg/kg (i.p., n = 6); MDL 100,907, 0.1 mg/kg (s.c., n = 4); SDZ SER 082, 0.3 mg/kg (s.c.,n = 6). * P < .001 with respect to DOI group; ** P = .005 with respect to DOI group; † P = .016 with respect to vehicle.
The HTR elicited by 3 nmoles/0.5 μl/side of mCPP, a dose that produced a maximal HTR (fig. 3), was significantly reduced by ketanserin (2.5 mg/kg s.c.), by MDL 100,907 (0.1 mg/kg s.c.) and by SDZ SER 082 (0.3 mg/kg s.c.) (fig. 5).

The effect of pretreatment of animals with 5-HT2 antagonists on HTR produced by bilateral, intra-mPFCx injection of mCPP (3.0 nmoles/0.5 μl/side). Each antagonist or saline was administered 20 min before intracortical injection of DOI at the following doses: saline, 0.9%, 1 ml/kg (i.p., n = 5), ketanserin, 2.5 mg/kg (i.p., n = 5); MDL 100,907, 0.1 mg/kg (s.c., n = 5); SDZ SER 082, 0.3 mg/kg (s.c.,n = 5). * P < .001 with respect to mCPP; ** P = .006 with respect to mCPP group; † P < .001 with respect to vehicle; †† P = .003 with respect to vehicle.
5-HT1A receptors modulate DOI-induced HTR.
Pretreatment with the 5-HT1A selective agonist 8-OHDPAT (100 μg/kg s.c.) completely inhibited the HTR produced by DOI (fig.6). The inhibitory effect of 8-OHDPAT on DOI-induced HTR was reversed by the selective 5-HT1Aantagonist WAY 100,635 (100 μg/kg s.c.). WAY 100,635 also potentiated the DOI-induced HTR (fig. 6). Neither 8-OHDPAT nor WAY 100,635 had any effect on basal HTR in controls.

The effect of pretreatment of animals with the 5-HT1A agonist 8-OHDPAT and the 5-HT1Aantagonist WAY 100,635 on HTR produced by bilateral, intra-mPFCx injection of DOI (3 μg/0.5 μl/side). Saline, 8-OHDPAT or WAY 100,635 was administered 20 min before intracortical injection of DOI at the following doses: saline, 0.9% (i.p., n = 6), 8-OHDPAT, 0.1 mg/kg (s.c., n = 4); WAY 100,635, 0.1 mg/kg (s.c., n = 6). WAY 100,635 (0.1 mg/kg s.c.,n = 6) was also administered to animals that received saline (0.9%, 0.5 μl/side, mPFCx). * P < .001 with respect to vehicle, † P < .001 with respect to DOI.
Discussion
The major finding of the present study was that the direct, bilateral administration of the 5-HT agonists DOI and mCPP into the mPFCx of rats produces HTR. This response was dose-dependent and appears to be mediated by the activation of 5-HT2Areceptors. DOI has similar affinity and efficacy at both the 5-HT2A and the 5-HT2C subtypes of 5-HT receptors, (Glennon et al., 1992; Choudhary et al., 1992). Thus the HTR produced by DOI could be mediated at the 5-HT2A or the 5-HT2C receptor. In order to determine the relative roles of 5-HT2A and 5-HT2C receptors in the production of the HTR to DOI, we tested the ability of MK-212, a 5-HT2 receptor agonist that has a 100-fold greater selectivity for 5-HT2C, compared with the 5-HT2A receptor (Roth et al., 1992), to produce a HTR. Direct, bilateral, intracortical administration of doses of MK-212 ranging from 3 to 30 nmoles/side did not produce a significant HTR. This finding suggests that head-twitch behavior produced by DOI is mediated by the activation of 5-HT2Arather than 5-HT2C receptors.
This conclusion is further supported by our finding that the DOI-induced HTR was reversed by the 5-HT2A receptor antagonists ketanserin and MDL 100,907 but not by the 5-HT2C/2B receptor antagonist SDZ SER 082 (fig. 4). Doses of ketanserin similar to those used in the present study have also been demonstrated to reverse behavioral and neurochemical effects of DOI that have been associated with the activation of 5-HT2receptors (Schechter and Simansky, 1988; Darmani et al., 1992; Pan and Tai, 1992; Schreiber et al., 1995). Ketanserin has been demonstrated to be approximately 1000-fold more selective for the 5-HT2A than for the 5-HT1 receptor, though it is less than 100-fold more selective for the 5-HT2A than for the 5-HT2C receptor (Leysen et al., 1981;1982; Choudhary et al., 1992). Thus the inhibition of the HTR by a moderate dose of ketanserin suggests that the HTR produced by DOI is mediated through the activation of 5-HT2 sites but does not help us determine what receptor subtype is involved in the HTR.
MDL 100,907 is a highly potent and selective antagonist at 5-HT2A receptors that is 300 times more selective for the 5-HT2A receptor than for the 5-HT2C site (Sorensen et al., 1993). In the present study, DOI-induced HTR was inhibited by a dose of MDL 100,907 (0.1 mg/kg) that is one-tenth that shown to have selective neurochemical effects (Schmidt and Fadayel, 1995) and to reverse d-amphetamine-induced slowing of the firing of A9 and A10 DA neurons (Sorensen et al., 1993). Additionally, this dose of MDL 100,907 is 3-fold lower than the ED50 value for inhibition of d-amphetamine-stimulated locomotor activity (Sorensen et al., 1993). The inhibition of DOI-induced head twitch by a low dose of MDL 100,907 suggests that the response is preferentially mediated by the activation of 5-HT2A rather than 5-HT2C receptors. This finding, together with the observation that MK-212 did not produce a HTR, strengthens the conclusion that the DOI-induced HTR is not mediated by 5-HT2C receptors in the mPFCx.
Further evidence that the HTR produced by DOI is not mediated by the activation of 5-HT2C receptors is provided by the finding that the selective 5-HT2C/2B antagonist SDZ SER 082 did not inhibit the response to DOI. SDZ SER 082 has been shown, in autoradiographic studies, to displace 5-HT2C binding from human choroid plexus at a 100-fold lower concentration than from 5-HT2A sites in human claustrum (Waeber and Palacios, 1994). In binding studies, SDZ SER 082 had a 40-fold greater affinity for human recombinant 5-HT2C receptors (pKD = 7.8) than for 5-HT2A receptors (pKD = 6.2) (Nozulak et al., 1995). The dose of SDZ SER 082 used in the present study (0.3 mg/kg s.c.) is equivalent to the ID50 of the compound for inhibition of the hypophagic effect produced by MK-212 in rats and is triple the dose that was demonstrated to reduce significantly MK-212-mediated increases in serum ACTH (Nozulak et al., 1995). Finally, a recent study has demonstrated that a variety of selective 5-HT2A antagonists blocked the HTR to systemically administered DOI and that the relative potency of these antagonists was highly correlated with their affinity at 5-HT2A receptors, but not with their affinity for 5-HT2C sites (Schreiber et al., 1995). Taken together, these findings strongly support the conclusion that the DOI-induced HTR is mediated by a selective activation of 5-HT2A receptors.
Interestingly, the direct, bilateral administration of the phenylpiperazine derivative mCPP into the rat mPFCx also elicited a HTR (figs. 2 and 3). mCPP has been shown to be an antagonist at 5-HT2A receptors in the cerebral cortex (Conn and Sanders-Bush, 1987) and to block the HTR produced by quipazine (Simansky and Schechter, 1988). Recently, however, mCPP has been shown to behave as a partial agonist at cloned 5-HT2A receptors (Grotewiel et al., 1994). It is therefore possible that the HTR to mCPP is a result of the activation of 5-HT2Areceptors. The dose-response curve for the mCPP-induced HTR was a “bell-shaped” dose-response curve. It may be that at different doses, mCPP, which has been demonstrated to have significant affinity for a variety of receptors in addition to 5-HT2A sites, including 5-HT2C (Curzon and Kennett, 1990; Choudharyet al., 1992), 5-HT1B and 5-HT1Areceptors (Sills et al., 1984; Asarch et al., 1985; Hamon et al., 1986), may actually be activating receptors that are functionally antagonistic to the response to 5-HT2A receptor activation (e.g., 5-HT1A sites).
In much the same manner as previously demonstrated for DOI, pretreatment of animals with either ketanserin or MDL 100,907 was shown to decrease the HTR produced by mCPP (fig. 5). This suggests that the activation of 5-HT2A receptors is required for the production of a HTR by mCPP. Although the HTR to DOI was unaffected by pretreatment with the selective 5-HT2C/2B antagonist SDZ SER 082, the HTR to mCPP was significantly reduced by this agent. Because the dose-response curve to mCPP is an inverted-U function, and because the dose of mCPP used in the evaluation of antagonist effects was a dose that produced a maximal HTR, the direction of the curve shift produced by SDZ SER 082 cannot be determined. Further study would be required to determine whether this effect reflects an inhibition of the mCPP response or, alternatively, a potentiation of the response, perhaps through some functional interaction between 5-HT2Aand 5-HT2C receptors. The fact that SDZ SER 082 had any effect at all on the mCPP response without altering behavior on its own further supports the conclusion that mCPP-induced HTR is not solely mediated by the activation of 5-HT2A sites in the mPFCx.
Several studies have suggested that the frontal cortex may not be involved in the HTR produced by systemically administered 5-HT agonists. Lucki and Minugh-Purvis (1987) reported that ablation of the frontal cortex did not inhibit the HTR to either the 5-HT precursor 5-HTP or the direct-acting 5-HT agonist quipazine. Bedard and Pycock (1977) reported that knife cuts at the level of the caudal diencephalon decreased the HTR and that cuts at the level of the posterior commissures eliminated the HTR entirely. Transection at the level of the anterior commissures, however, had no effect on the HTR to 5-HTP. These authors concluded that the ability of 5-HT agonists to elicit a “wet-dog” shake did not depend on the frontal regions of the cortex. Despite these findings, which suggest that the HTR to systemically administered 5-HT agonists is not critically dependent on processes in the frontal cortex, the present study demonstrates that the direct activation of 5-HT2A receptors in the mPFCx can produce a HTR. Although it is possible that after injection directly into the mPFCx, the drugs studied here could have entered the cerebrospinal circulation and thus come into contact with receptors in the spinal cord, the fact that no HTR was observed in animals in which DOI was injected outside of the mPFCx suggests that this is unlikely. In addition, the rapid onset of the drug-induced HTR suggests that the locus of the response to intracortical drug administration is within the mPFCx. Finally, the production of a HTR through the activation of central 5-HT sites is consistent with the report that i.c.v. administration of the 5-HT neurotoxin 5,7-dihydroxytryptamine increased the HTR produced by the 5-HT2A/2C agonist 5-methoxy-N,N-dimethyltryptamine as well as the Bmax for [3H]ketanserin binding in the cortex of adult male mice (Heal et al., 1985). Although this does not preclude a spinal locus for the HTR to systemically administered serotonergic agents, the correlation between receptor up-regulation in the cortex and an increase in the behavioral response to a 5-HT2agonist supports the conclusion that the activation of cortical 5-HT2A receptors can elicit a HTR.
Pretreatment of rats with the 5-HT1A receptor agonist 8-OHDPAT blocked the HTR produced by DOI, which suggests a functional interaction between 5-HT1A and 5-HT2A receptors in the mPFCx. There is considerable evidence for a functional interaction between 5-HT1A and 5-HT2A receptors (Goodwin and Green, 1985; Backus et al., 1990; Berendsen and Broekkamp, 1990; Glennon et al., 1991; Ashby et al., 1994; Meltzer and Maes, 1995). Anatomically, there is a significant overlap between the distributions of these receptors in the mPFCx (Pazos, 1985; Pazos and Palacios, 1985). Electrophysiological studies suggest that cortical 5-HT1A and 5-HT2Areceptors may be co-localized on the same cell and may mediate membrane hyperpolarization and depolarization, respectively (Araneda and Andrade, 1991). More recently, it has been demonstrated that the suppressant effects of 8-OHDPAT on the basal firing rate of spontaneously active cells in the mPFCx is potentiated by concurrent iontophoretic administration of either of the 5-HT2Aantagonists ritanserin and MDL 100,907 (Ashby et al., 1994). In this same study, the potentiating effects of DOI onl-glutamate-induced cell firing were inhibited by the administration of 8-OHDPAT.
There is also behavioral evidence to support a 5-HT1A/5-HT2A interaction. Darmani and co-workers reported that the HTR produced by the systemic administration of DOI in mice was inhibited by pretreatment with 8-OHDPAT (Darmani et al., 1990b). In addition to studies of the interaction of 5-HT1A agonists with 5-HT2Asystems, pretreatment of animals with selective 5-HT1Aantagonists has been shown to enhance the behavioral responses to 5-HT2A agonists (Björk et al., 1991). In the present study, pretreatment of rats with WAY 100,635, a highly selective and potent antagonist at 5-HT1A receptors (Forster et al., 1995; Khawaja, 1995), reversed the effects of 8-OHDPAT on DOI-induced HTR. Thus the inhibitory effect of 8-OHDPAT on DOI-induced head twitch is likely to be mediated through the activation of 5-HT1A receptors. Furthermore, when rats were pretreated with WAY 100,635 alone, a significant potentiation of the HTR produced by DOI was observed. Because WAY-100635 has been identified as a “silent antagonist” (it exhibits no intrinsic agonist activity), this finding suggests that endogenous 5-HT, through the activation of 5-HT1A receptors, may regulate behavioral responses produced by the stimulation of 5-HT2A receptors. This finding further supports the hypothesis that there is a functionally inhibitory interaction between 5-HT1A and 5-HT2A receptors.
In summary, the major finding of the present study is that the HTR produced by the direct administration of serotonergic agonists into the mPFCx of rats is mediated by the stimulation of 5-HT2Areceptors. DOI, a mixed 5-HT2A/2C agonist, produced a dose-dependent HTR. The observation that the selective 5-HT2C receptor agonist MK-212 did not produce head twitch provides additional evidence that the HTR is mediated through 5-HT2A receptors. The mixed serotonin receptor agonist mCPP was also shown to produce a HTR that was dependent on the activation of 5-HT2A receptors. In addition, it was shown that there exists a functional antagonism of the effects of stimulation of 5-HT2A receptors in the mPFCx that is mediated through the activation of 5-HT1A receptors. These findings support a role for 5-HT2A receptors in the mPFCx in 5-HT-mediated behavior and suggest that a critical analysis of 5-HT2Areceptor function in this region may yield pertinent information about the role of this region in the mechanism of action of new, atypical anxiolytic, antidepressant and antipsychotic drugs.
Acknowledgments
We gratefully acknowledge the helpful comments of Dr. Bryan L. Roth during the preparation of this manuscript. We also thank Merrell-Dow Pharmaceuticals for the generous provision of MDL 100,907.
Footnotes
-
Send reprint requests to: David L. Willins, Ph.D., University Hospitals, Cleveland, Hanna Pavilion, Rm. B58, 11100 Euclid Ave., Cleveland, Ohio 44106.
-
↵1 The research reported was supported in part by USPHS MH 41684. D.L.W. is the recipient of a National Alliance for Research on Schizophrenia and Depression Young Investigator Award.
-
↵2 Present address: Psychiatric Hospital at Vanderbilt, Psychopharmacology Division, Vanderbilt University, Nashville, TN 37212.
- Abbreviations:
- HTR
- head-twitch response
- 5-HT
- serotonin
- 5-HTP
- 5-hydroxy-l-tryptophan
- DA
- dopamine
- mPFCx
- medial prefrontal cortex
- DOI
- 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane
- MK-212
- 6-chloro-2-[1-piperazinyl]-pyrazine
- mCPP
- m-chloro-phenylpiperazine
- 8-OHDPAT
- (±)-8-hydroxy-dipropylaminotetralin hydrobromide
- DOB
- dimethoxy-4-bromoamphetamine hydrobromide
- Received January 17, 1996.
- Accepted April 15, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}