


Abstract
Dopamine D3 receptors have eluded definitive linkage to neurologic and psychiatric disorders since their cloning over 20 years ago. We report a new method that does not employ a radiolabel for simultaneously defining in vivo receptor occupancy of D3 and D2 receptors in rat brain after systemic dosing using the tracer epidepride (N-[[(2S)-1-ethylpyrrolidin-2-yl]methyl]-5-iodo-2,3-dimethoxybenzamide). Decreases in epidepride binding in lobule 9 of cerebellum (rich in D3 receptors) were compared with nonspecific binding in the lateral cerebellum. The in vivo occupancy of the dopamine D3 receptors was dose dependently increased by SB-277011A (trans-N-[4-[2-(6-cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide) and U99194 (2,3-dihydro-5,6-dimethoxy- N,N-dipropyl-1H-inden-2-amine). Both antagonists increased extracellular levels of acetylcholine (ACh) in the medial prefrontal cortex of rats and modified brain-tissue levels of ACh and choline. Consistent with these findings, the D3 receptor antagonists enhanced the acquisition of learning of rats either alone or in the presence of the norepinephrine uptake blocker reboxetine as with the attention-deficit–hyperactivity disorder (ADHD) drug methylphenidate. Like reboxetine, the D3 receptor antagonists also prevented deficits induced by scopolamine in object recognition memory of rats. Mice in which the dopamine transporter (DAT) has been deleted exhibit hyperactivity that is normalized by compounds that are effective in the treatment of ADHD. Both D3 receptor antagonists decreased the hyperactivity of DAT−/− mice without affecting the activity of wild type controls. The present findings indicate that dopamine D3 receptor antagonists engender cognition-enhancing and hyperactivity-dampening effects. Thus, D3 receptor blockade could be considered as a novel treatment approach for cognitive deficits and hyperactivity syndromes, including those observed in ADHD.
Introduction
Dopamine D3 receptors are a member of the D2 family of proteins. D3 receptors are localized with high density in ventral striatum (Levant, 1997) and in lower concentration in other central nervous system sites. The receptor was cloned in 1990 (Sokoloff et al., 1990) and yet, after more than twenty years and much investigation, the functional significance of this receptor has not been fully elucidated. D3 receptors have been implicated in the control of drug dependence disorders, schizophrenia, and Parkinson’s disease (Joyce et al., 2004; Micheli and Heidbreder, 2006; Newman et al., 2012). A number of selective ligands have been discovered but only the D3 receptor–preferring agonist pramipexole is in clinical use for the treatment of Parkinson’s disease and restless leg syndrome (Rascol et al., 2011; Scholz et al., 2011). Pramipexole has been reported to have a range of selectivities at D3 to D2 receptors that range from none (Sautel et al., 1995) to about 100-fold (Millan et al., 2002) with both studies using iodosulpiride as a radioligand.
There have been a host of obstacles blocking the definitive identification of the functional significance of D3 receptors. The relatively low density of these receptors and the lack of a range of selective pharmacological agents with high affinity and selectivity for D3 receptors have been one set of limitations impeding success in this area. The lack of a method for identification of D3 receptor activity in vivo has been another serious impediment. Although several methods have been proposed, many of these have been shown to be inconsistent when pharmacological data and data in D3 receptor-deficient mice are compared, or they are not consistent across the range of D3 receptor antagonists studied (c.f., Perachon et al., 2000). Recently two new in vivo assays have been proposed to define D3 receptor blockade (Witkin et al., 2004; Collins et al., 2005).
Recent work has also enabled the measurement of the occupancy of dopamine D3 receptors by ligands in vivo. For example, imaging and ex vivo binding with [11C]- (+)-4-propyl-9-hydroxynaphthoxazine ([11C]-PHNO) has been successfully employed for dopamine D3 receptors in vivo (McCormick et al., 2010; Gallezot et al., 2012). Additionally, techniques have been established leveraging the discreet expression of dopamine D3 receptor in lobules 9 and 10 of the cerebellum in vivo (Kiss et al., 2011). We now report a new liquid chromatography–tandem mass spectrometry (LC–MS/MS) based method for defining the occupancy of compounds at D3 receptors after systemic dosing that enables simultaneous assessment of D2 receptor occupancy by using this regional specificity. This method uses a nonlabeled tracer measured by LC–MS/MS, as previously reported for dopamine D2, NK1, 5HT2A, and opioid receptors mu, kappa, and delta (Chernet et al., 2005; Barth et al., 2006; Need et al., 2007). In the present study, LC–MS/MS receptor occupancy data were used to guide investigation of the in vivo activities of two D3 receptor antagonists, SB-277011A and U99194. Specifically, we inquired as to whether blockade of D3 receptors might impact acetylcholine (ACh) efflux and turnover, and whether these neurochemical effects were associated with the ability of the D3 receptor antagonists to affect the acquisition of behavior (learning) and to prevent memory deficits induced by the muscarinic receptor antagonist scopolamine, similar to effects produced by attention-deficit–hyperactivity syndrome (ADHD)-treatment-related drugs. Since these neurochemical and behavioral data were aligned, we then showed that the D3 receptor antagonists also significantly attenuated the hyperactivity of mice without dopamine transporters (DAT−/− mice), a model that detects the activity of drugs used in the therapeutic resolution of ADHD symptoms (Wickens et al., 2011; Fan et al., 2012).
SB-277011A and U99194 are structurally distinct antagonists of D3 receptors (Fig. 1) with nanomolar affinities for the receptor. SB-277011A is 100-fold more selective for D3 receptors in its binding profile over D2 receptors (Reavill et al., 2000), and U99194 displays only a 20-fold selectivity (Waters et al., 1993). Binding at these receptors however does not necessarily define in vivo selectivity and does not always even predict functional selectivity in vitro (Sautel et al., 1995). Previous data have also suggested that some molecules might not function as selective D3 receptor antagonists in vivo (Perachon et al., 2000). However, both molecules have recently been reported to prevent yawning induced by the D3 receptor agonist (+)-PD128,907, which has been pharmacologically validated as a D3-mediated event in rats (Collins et al., 2005). Consistent with these observations, we now report that both SB-277011A and U99194 occupy D3 receptors in rat brain in vivo after systemic dosing with relatively low levels of D2 receptor occupancy. Using doses of these antagonists that selectively impact D3 receptors in vivo, we were able to produce neurochemical and behavioral effects of these D3 receptor antagonists that are consistent with the potential for beneficial effects in disorders of cognitive impairment and of ADHD.
Materials and Methods
Compounds
Epidepride, SB-277011A, and reboxetine SO4 were synthesized at Eli Lilly and Company. U99194 was purchased from Tocris (Bristol, UK); scopolamine HBr and methylphenidate HCl were purchased from Sigma-Aldrich (St. Louis, MO). Structures of the dopaminergic ligands are shown in Fig. 1. SB-277011A was dissolved in 25% β-cyclodextrin; U99194, methylphenidate, and reboxetine were dissolved in water; and scopolamine was dissolved in 0.9% NaCl. For behavioral studies, compounds were injected i.p., or s.c., 30 minutes prior to testing (except for the object recognition in rats experiments, see below). Compounds were dosed in a volume of 1 ml/kg for rats except in the behavioral acquisition studies (5 ml/kg) and 10 ml/kg (mice).

Structures of epidepride, SB-277011A, and U99194.
Receptor Occupancy
“Cold” epidepride was used as an occupancy tracer in the LC–MS/MS-based experiments and was administered at an intravenous dose of 3 μg/kg dissolved in sterile water (Chernet et al., 2005; Barth et al., 2006). The D3 tracer used, epidepride (Fig. 1), had an affinity for D3 receptors of 0.12 nM ([3H]-7-OH-DPAT binding) and for D2 receptors of 0.80 nM ([3H]-7-OH-DPAT binding).
Adult male Sprague Dawley rats (Harlan Laboratories, Indianapolis, IN) weighing 240–260 g were housed six to a cage in a room using a 12-hour on/off lighting schedule (lights on at 6 AM). Room temperature was maintained at 21 ± 3°C. Animals had ad libitum access to food and water and were permitted at least 2 days after arrival at our site to adapt to housing conditions before testing.
Dopamine D3 Receptor Occupancy in Vivo
Tracer dose (3μg/kg) and survival interval (40 minutes) were optimized for epidepride to generate detectable and quantifiable in vivo levels in lateral cerebellum, lobules 9 and 10 of the cerebellum, and dorsal striatum by LC–MS/MS (Barth et al., 2006). Tissue levels of epidepride in the lateral cerebellum represent nonspecific binding, those in lobules 9 and 10 of the cerebellum represent total binding for D3 receptor–occupancy measurements, and epidepride levels in the dorsal striatum represent the total binding for D2 receptor–occupancy measurements. To examine D3 dose–occupancy relationships, groups of 4–6 rats were pretreated with SB-277011A (s.c.) or U99194 (i.p.) or compound vehicle. One hour later, animals were briefly restrained and administered a low intravenous dose of nonlabeled epidepride tracer via the lateral tail vein. Rats were sacrificed by cervical dislocation, and brain lateral cerebellum, lobules 9 and 10 of the cerebellum, and the dorsal striatum were dissected and weighed.
Analysis of Nonlabeled Epidepride Levels
Previously weighed brain tissue samples were placed in conical 1.5 ml polypropylene centrifuge tubes to which 4 volumes (w/v) of acetonitrile containing 0.1% formic acid was added. Samples were then homogenized using an ultrasonic dismembrator probe (Fisher Scientific model 100, Pittsburgh, PA), vortexed and centrifuged for 16 minutes at 16,000g (Eppendorf model 5417R, Westbury, NY). 100µL of supernatant was then added to 300 μl of water in 1.5 ml autosampler vials and vortexed.
Epidepride concentration measurements were made using a high-performance liquid chromatograph (HPLC, Agilent Technologies model 1100, Wilmington, DE) with triple-quadrupole mass spectral detection. The HPLC system employed a C18 column (ZORBAX Eclipse, 2.1 × 50 mm, 3.5-micron particle size; Agilent Technologies, Santa Clara, CA) with an aqueous mobile phase consisting of 23% acetonitrile with 0.1% formic acid. Epidepride was quantified after elution from the HPLC column using an API 3000 triple-quadrupole mass spectrometer (Applied Biosystems, Foster City, CA) in positive electrospray mode using multiple reaction monitoring methods to monitor the transition from precursor to fragment ions with mass-to-charge ratios of 419.2 and 112.1, respectively. Chromatographic assays were calibrated using a standard curve generated by extracting a series of brain tissue samples from nontreated animals to which known quantities of epidepride had been added.
Receptor Occupancy Calculation
Receptor occupancy calculations were made for each animal employing the widely used ratio method (Farde et al., 1988; Kapur et al., 1999; Wadenberg et al., 2000) and the following equation: 100 *{1– [(Ratiot –1)/(Ratioc – 1)]} = % Occupancy. The “Ratiot” represents the ratio of epidepride concentrations measured in the striatum or lobules 9 and 10 to those measured in the lateral cerebellum in individual animals pretreated with compounds or vehicle. The “Ratioc” represents the average ratio of epidepride levels measured in the striatum or lobules 9 and 10 to that measured in the lateral cerebellum for the vehicle-pretreated group.
In Vivo Microdialysis in Rats
All studies were performed according to the guidelines set forth by the National Institutes of Health and implemented by the Animal Care and Use Committee of Eli Lilly and Company. Male Wistar or Sprague Dawley rats (250–300 g, purchased from Harlan) were used for the experiments.
Surgical Procedures
Two weeks prior to the microdialysis experiments, the rats were anesthetized with a mixture of chloral hydrate and pentobarbital, placed in a stereotaxic apparatus, and implanted with a guide cannula [Bioanalytical Systems (BASi), West Lafayette, IN] in the medial prefrontal cortex (AP, 3.2; ML, 0.6; DV, –2.2), according to Paxinos and Watson (1996). Twenty-four hours before testing, a 4-mm concentric microdialysis probe (BASi, model BR-4) was inserted through the guide cannula. The actual location of the probes was verified histologically at the end of the experiment.
Acetylcholine Measurements
ACh determination in dialysates from the different brain regions was performed as described previously (Shirazi-Southall et al., 2002; Tzavara et al., 2006a). On the day of the experiment, a modified Ringer’s solution (147.0 mM NaCl, 3.0 mM KCl, 1.3 mM CaCl2, 1.0 mM MgCl2, 1.0 mM Na2HPO4 ⋅ 7H2O, 0.2 mM NaH2PO4 ⋅ H2O, pH 7.25) supplemented with 0.1 μM neostigmine was perfused at a rate of 2.4 μl/min in the medial prefrontal cortex. Samples were collected every 15 minutes and analyzed immediately, online, with HPLC coupled to electrochemical detection, with a 150 × 3-mm ACH-3 column [Environmental Sciences Associates, Inc. (ESA), Thermo Scientific, Sunnyvale, CA] maintained at 35°C. The mobile phase (100 mM disodium hydrogen phosphate, 2 mM 1-octanesulfonic acid, and 50 μl/l of a microbicide reagent (ESA); pH 8.0, adjusted with phosphoric acid) was delivered by an HPLC pump (ESA) at 0.4 ml/min. The potentiostat used for electrochemical detection (ESA; model Coulochem II) was connected with a solid-phase reactor for ACh (ESA; model ACH-SPR) and with an analytical cell with platinum target (ESA; model 5041).
Data Analysis
Data were expressed as multifold change from baseline, which is the average of the five basal values before any manipulation, and were analyzed either with one-way, i.e., treatment (between subjects variable), two-way, i.e., treatment (between subjects variable) × time (within subjects variable), or three-way (treatment 1 × treatment 2 × time) ANOVA (analysis of variance) followed by Duncan’s test. The effects of each of the drugs are presented both over a course of time every 15 minutes after the injection of the drug, as well as overall average effects during the 3-hour observation period after the injection of the drug (index of area under curve); data analyzed with one-way ANOVA followed by Bonferroni’s test).
Acetylcholine and Choline Tissue Levels in Rats
Male Sprague Dawley rats (175–225 g, purchased from Harlan, Indianapolis, IN) were used for the experiments. Rats were injected i.p. with the compounds under study and 30 minutes later they were decapitated with a guillotine and their brains quickly removed and placed on an ice-cold plate. The cortex, hippocampus, and striatum were dissected freehand on ice, weighed, and stored at –80°C until they were assayed. Tissue preparation, extraction, and measurement of ACh and choline were made essentially as reported previously (Bymaster et al., 1985). Results for ACh and choline were expressed as nmol/g tissue from which the ratio of choline:ACh (turnover index) was calculated; all data from each brain region were analyzed with one-way ANOVA followed by the Tukey’s least significant difference test for multiple comparisons.
Acquisition of Behavior
Male Sprague Dawley rats (Harlan) weighing 40–50 g upon arrival were randomly placed five per cage with free access to food and water. Each rat was studied in only one experiment.
The shuttle boxes, 21 cm on each side separated by a guillotine door, were equipped with metal grid floors through which electrical stimuli could be delivered and scrambled across the grids (Med Associates).
The experimental protocol was a modification of that used previously (Fox et al., 2003). After dosing, the rats were put into individual cages for the duration of pretreatment interval after which they were placed into the right side of shuttle box. Ten seconds later the light was turned on and the door opened. If the animal did not cross over into the left-hand side of the shuttle box within 180 seconds, the trial ended the door closed; 10 seconds later the door was reopened and the next trial began. When the animal crossed to the other side of the box, the timer was stopped, the door closed, and 7 seconds later the grid floor was electrified (0.2 mA, 1 second). After current delivery, the rat was manually removed and placed back into the right-hand side of the shuttle box and the procedure was repeated until five trials were completed. All events and timers were controlled by a computer operating Med Associates software.
The latency to cross over to the left-hand side was recorded for each of the 5 trials. Trial latencies were analyzed using ANOVA followed by Dunnett’s test. Comparison of vehicle to drug treatments across trials was evaluated by two-tailed, two-way ANOVA. For this analysis, three to five trials were used. Probabilities <0.05 were considered significant.
Object Recognition in Rats
Male Sprague Dawley rats (250–300 g, Harlan) were used as per Tzavara et al. (2006a). A 3-minute interval between the training trial and the test trial was used to maximize the memory-retention baseline. Each rat was dosed i.p. with either vehicle, reboxetine (3, 10, 30 mg/kg), SB-277011A (10, 30 mg/kg), or U99194 (10, 30 mg/kg) 40 minutes before the session. Scopolamine HBr (1 mg/kg, sc) was then administered 30 minutes before the session. Each rat was placed in a clear 25 × 25-cm Plexiglas observation box with two identical objects, each designated object A. The rat was allowed to explore for 2 minutes and the time interacting with the objects (sniffing, gnawing, and behavior oriented to an object) was recorded. After 3 minutes the rat was returned to the observation box for the test trial. The test box contained a familiar object (object A) and a novel object (object B).
A separate study was conducted in which the ability of SB-297011A to augment the protective effects of reboxetine against scopolamine degradation was evaluated. In this study, a noneffective dose of reboxetine (3 mg/kg) was given alone or in combination with with low, noneffective doses of SB-297011A (1 or 3 mg/kg). In an additional study, rats were studied for effects of SB-2977011A in the absence of scopolamine. The study was conducted as above with the exception that the interval between exploration periods 1 and 2 was 3 hours instead of 3 minutes.
The amount of time spent interacting with each object during the 2-minute test was recorded. Results are expressed as the percentage of time spent interacting with the new object B and were analyzed with one-way ANOVA. The ability of compounds to produce effects statistically differentiated from that of either vehicle, scopolamine, or reboxetine plus scopolamine were evaluated by post-hoc Dunnett’s tests.
Dopamine Transporter Knockout Mice
Breeding, Genotyping, and Maintenance of the Mice.
Dopamine transporter (DAT) knockout (KO) mice were obtained by homologous recombination (Giros et al., 1996) and maintained on two inbred genetic backgrounds: C57BL/6JOrl (B6) and DBA/2JOrl (D2). Homozygous null mutants and their wild type (WT) littermates of a hybrid B6xD2F1 genetic background were obtained from the mating of B6-DAT-heterozygous females with D2-DAT-heterozygous males. All mice used in this study were on a B6xD2F1 background. The genotype of the mice was determined by polymerase chain reaction (PCR) analysis as follows. Genomic DNA (50 ng) from tail biopsies was amplified with primers DAT-1 (CCCGTCTACCCATGAGTAAAA), DAT-2 (CTCCACCTTCCTAGCACTAAC), and NEO2 (TGACCGCTTCCTCGTGC), generating a 870-bp product (DAT-1/NEO2) for the recombined DAT gene and a 580-bp product (DAT-1/DAT-2) for the wild type DAT gene. After weaning, mice were housed two to four per cage and maintained under standard housing conditions with food and water available ad libitum. All experiments were carried out in accordance with the European Communities Council Directive (86/809/EEC) regarding the care and use of animals for experimental procedures and approved by the local ethical committee.
Locomotor Activity.
Locomotor activity in DAT KO mice and WT littermates was measured after an acute administration of the following drugs: SB-277011A (1, 3, and 10 mg/kg), U99194 (10, 30, and 60 mg/kg), d-amphetamine (3 mg/kg), and reboxetine (5 and 10 mg/kg), all injected i.p. in a volume of 10 ml/kg. Thirty minutes after the injection, horizontal locomotor activity of DAT KO and WT mice, injected with one of the above compounds or respective vehicles, was measured in an actimeter over 60 minutes. For this, mice were placed in individual transparent cages (20 cm × 10 cm × 10 cm) and their horizontal movements (measure of ambulatory behavior) were recorded by four infrared sensors (Imetronic, Bordeaux, France) as in a previous study (Tzavara et al., 2006b). Results are expressed as ambulations over the 60-minute period. Statistical analysis was performed by two-way ANOVA (genotype × treatment) and Duncan’s post-hoc test.
Results
Receptor Occupancy.
The in vivo occupancy of D3 receptors was increased in a dose-dependent manner by both SB-277011A and U99194 (n = 6 and n = 5, respectively) (Fig. 2). Greater than 90% occupancy was measured 1 hour after s.c. administration of 10 mg/kg SB-277011A. At the highest dose tested, SB-277011A had a D3 receptor occupancy of 111 ± 5.0% (mean ± S.E.M.) The occupancy measured 1 hour after i.p. administration of U99194 at 60 mg/kg was 81 ± 9.9%. Selectivity for the D3 receptor relative to the D2 receptor was demonstrated, as these doses generated D2 occupancies of only 3 ± 5.6% and 20 ± 14% for SB-277011A and U99194, respectively. The high variability at low receptor-occupancy values is not uncommon for these methods (e.g., Chernet et al., 2005). Regardless, it is clear that the separation of occupancies of D2 and of D3 dopamine receptors at the higher doses of both SB-277011A and U99194 is substantial and statistically significant.

Occupancy of dopamine D3 and D2 receptors by SB-277011A (s.c.) and U99194 (i.p.). Rats were injected with the D3 antagonists and 60 minutes later epidepride was administered at 3 μg/kg (i.v.) followed by a 40-minute survival interval. Occupancy of D3 and D2 receptors were then determined as described in Materials and Methods. The percent occupancy of D2 receptors is represented by the horizontal line within each bar. Data represent the mean ±S.E.M. of five to six rats.
Neurochemistry.
Both SB-277011A and U99194 increased cortical dialysate levels of ACh in a dose-dependent manner (Fig. 3). Significant increases in ACh-microdialysate levels were achieved at multiple 15-minute time points postdosing of SB-277011A (15–90 minutes) and U99194 (0–45 minutes) at 30 mg/kg (Fig. 3). At this dose level, the effect of SB-277011A on cortical ACh levels was more pronounced than the effect of U99194, when the data were expressed and analyzed as overall average effects (inserts in Fig. 3), which was due to the fact that SB-277011A exerted a longer lasting effect on ACh compared with the effect of U99194 (Fig. 3).

Temporal effects of SB–277011A and U99194 on microdialysate levels of acetylcholine (ACh) from the medial prefrontal cortex of rats. Data in inserts represent the overall average effects on ACh levels. Each point represents the mean ±S.E.M. of five to six rats. *P < 0.05 compared with vehicle.
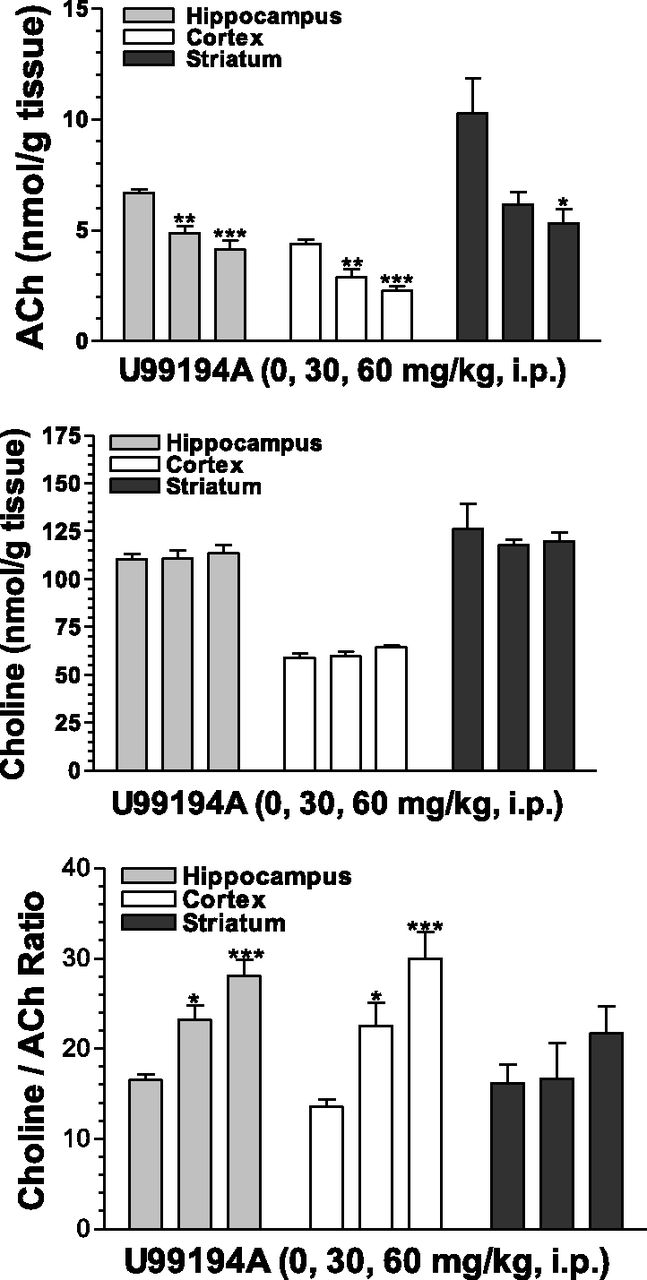
To further explore the effects of these D3 receptor antagonists on cholinergic neurochemistry, tissue levels of ACh and choline were examined. Fig. 3 shows, in accord with the increases in extracellular levels of Ach in the medial prefrontal cortex, corresponding significant and dose-dependent decreases in tissue levels of ACh in the hippocampus, the cortex, and the striatum after SB-277011A administration. SB-277011A also increased choline levels in the hippocampus and the striatum at 60 mg/kg (Fig. 4). The SB-277011A-induced increases in the ratio choline:ACh were found significant in the hippocampus and cortex at 30 and 60 mg/kg, and in the striatum at 60 mg/kg. Generally, comparable effects were observed after dosing with U99194 at doses of 30 and 60 mg/kg, although it did not significantly affect choline in any of the brain regions studied or the ratio of choline:ACh in the striatum (Fig. 5).

Effects of SB-277011A on tissue levels of ACh, choline, and the ratio choline:ACh in the hippocampus, cortex, and striatum of rats. Each point represents the mean ±S.E.M. of six rats. *P < 0.05, **P < 0.01 compared with vehicle (0 mg/kg).
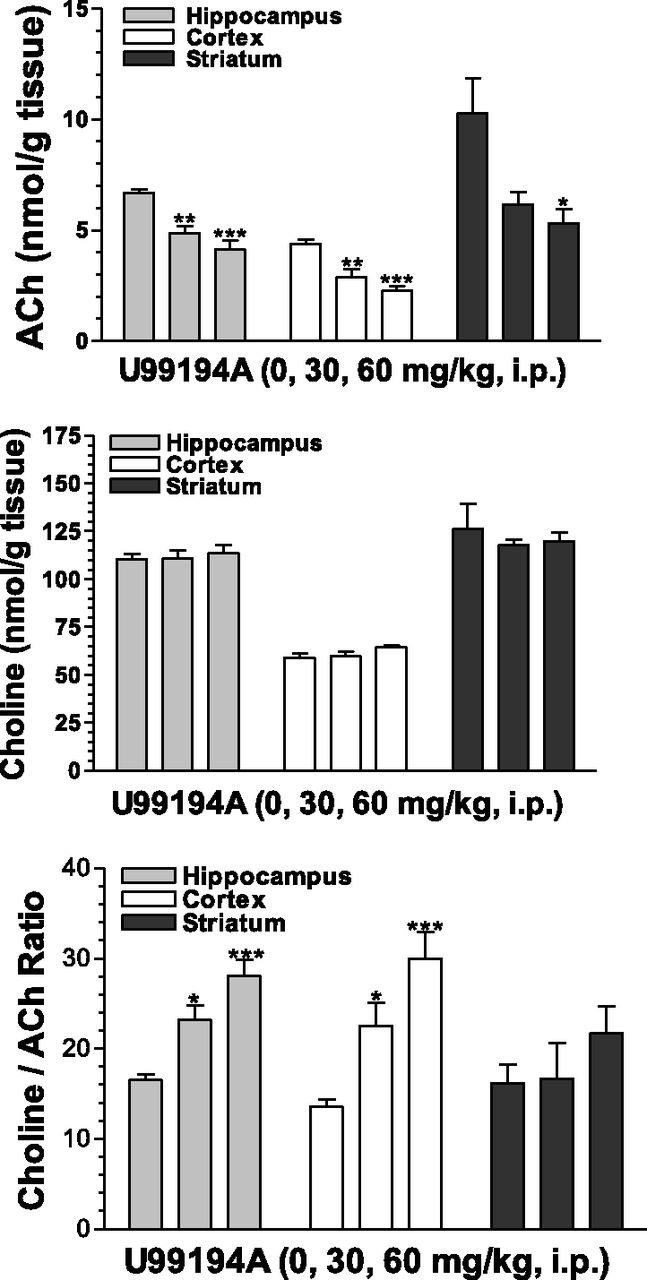
Effects of U99194 on tissue levels of ACh, choline, and the ratio choline:ACh in the hippocampus, cortex, and striatum of rats . Each point represents the mean ±S.E.M. of six rats. *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle (0 mg/kg).
Acquisition of Behavior.
Drugs were evaluated for their ability to enhance the acquisition of behavior of rats. Under the conditions studied, the performance of the rats increased over successive trials (F4,49 = 6.7, P < 0.001) (Fig. 6). Post-hoc Dunnett’s test revealed that performance on trials 3, 4, and 5 were significantly better than on trial 1 (P < 0.05).

Effects of methylphenidate, reboxetine, SB-277011A, and U99194 on the acquisition of behavior of male Sprague Dawley rats. All compounds were dosed i.p., 30 minutes prior to testing. Each point represents the mean ± S.E.M. of 7–10 rats. Solid circles represent vehicle values. Methylphenidate: open circles, 1 mg/kg; inverted triangle, 3 mg/kg. Reboxetine: inverted triangle, 3 mg/kg; triangle, 10 mg/kg. SB-277011A: inverted triangle, 10 mg/kg; triangle, 30 mg/kg. U99194: inverted triangle, 10 mg/kg; triangle, 30 mg/kg.
Methylphenidate significantly enhanced the acquisition of behavior (F2,69 = 7.5, P < 0.05). For reboxetine, a trend toward significance was achieved (F2,81 = 5.9, P = 0.07). Analysis of the 10-mg/kg dose of reboxetine relative to vehicle revealed a significant enhancement of behavioral acquisition (F1,54 = 7.6, P < 0.05). A similar trend for enhancement was seen with U99194 (F2,63 = 5.9, P = 0.08); when the 30-mg/kg dose of U99194 was compared with vehicle, a significant augmentation in acquisition of behavior was observed (F1,45 = 7.6, P < 0.05). SB-277011A did not significantly enhance behavioral acquisition (F2,66 = 1.8, P = 0.5).
Object Recognition.
Scopolamine produced a significant degradation in performance in the object recognition task (Fig. 7). Both SB-277011A and U99194 prevented these effects of scopolamine at both 10 and 30 mg/kg. Reboxetine was also effective in preventing scopolamine-induced performance effects at 10 and 30 but not at 3 mg/kg (Fig. 7).
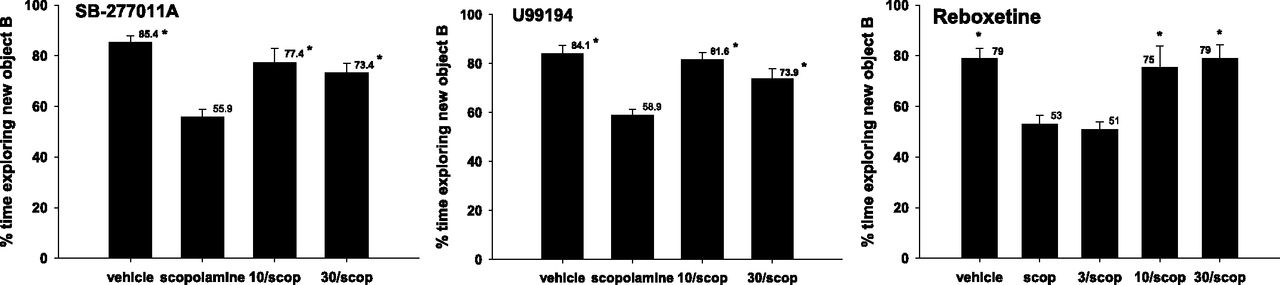
Effects of SB-277011A, U99194, and reboxetine on scopolamine-disrupted novel object–recognition performances of rats. Each bar represents the mean ± S.E.M. of six to eight rats. Statistical comparisons were made by Dunnett’s test after significant ANOVA with P < 0.05 (*) compared with scopolamine (scop).
The ability of SB-277011A to enhance novel object recognition on its own was also evaluated. In this experiment, a 3-hour instead of 3-minute delay was imposed before presentation of the novel object. Under these conditions, performance compared with the 3-minute condition was degraded to a level like that imposed by scopolamine. The percent time exploring the novel object was: vehicle = 48.9%, 10 mg/kg SB-277011A = 56.7%, and 30 mg/kg SB-277011A = 46.4%. Although there was a trend for an effect with 10 mg/kg, these effects were not significantly different than vehicle control values.
To evaluate the potential for D3 receptor blockade to enhance the efficacy of reboxetine, lower doses of SB-277011A were tested. Neither 1 nor 3 mg/kg SB-277011A significantly prevented the effects of scopolamine (Fig. 8). When these doses of SB-277011A were tested against a noneffective dose of reboxetine (3 mg/kg), SB-277011A dose-dependently augmented the protective effects of reboxetine (Fig. 8).

Augmentation of the behavioral effects of reboxetine by SB-277011A on novel object recognition of rats after scopolamine disruption. SB-277011A (i.p.) was given alone 40 minutes prior to testing or with reboxetine (3 mg/kg s.c. + 40 minutes) and tested against a scopolamine challenge (1 mg/kg s.c. + 30 minutes). Each bar represents the mean ± S.E.M. of four rats. Statistical comparisons were made by Dunnett’s test after significant ANOVA with P < 0.05 (*) compared with vehicle (veh); #, compared with scopolamine (s); $, compared with reboxetine (r) plus scopolamine.
Dopamine Transporter Null Mice.
DAT−/− mice had markedly enhanced basal locomotor activity levels than age-matched WT controls (Fig. 9). d-Amphetamine (3 mg/kg, i.p.) significantly decreased activity levels in the KO mice and significantly increased activity of WT mice (two-way ANOVA shows significant interaction between genotype and treatment, F1,31 = 52.9; P < 0.0001). In contrast to the effects of d-amphetamine, SB-277011A (3–30 mg/kg, i.p.) (two-way ANOVA showed significant interaction between genotype and treatment with F3,39 = 13; P < 0.0001) and U99194 (30 and 60 mg/kg, i.p.) (two-way ANOVA showed significant interaction between genotype and treatment, with F3,34 = 3.02; P < 0.05) significantly attenuated the hyperactivity of DAT KO mice without significantly altering the activity of WT mice. Reboxetine (an analog of atomoxetine, a nonstimulant drug with proven efficacy in patients with ADHD) was also studied. Under these conditions, reboxetine significantly decreased locomotor hyperactivity of the DAT KO mice at doses of 5 and 10 mg/kg i.p. without affecting levels of activity of WT mice (two-way ANOVA shows significant interaction between genotype and treatment of F2,61 = 5.1; P < 0.001).

Effects of d-amphetamine, reboxetine, and dopamine D3 receptor antagonists U99194 and SB-277011A on locomotor activity of DAT−/− mice (unfilled bars) or DAT+/+ mice (filled bars). Results represent total horizontal locomotor activity (ambulations) over a 60-minute period. Each bar represents the mean ±S.E.M. of mice (n = 5–7 for SB-277011A and U99194; 7–11 for d-amphetamine, and 6–18 mice/group for reboxetine). *P < 0.05, **P < 0.01, ***P < 0.001 as compared with vehicle of the same genotype.
Discussion
The present report provides a new method for determining in vivo receptor occupancy of dopamine D3 receptors, the first study to use an antagonist ligand and a technique that does not require radioactivity. The method capitalizes on the acceptable tracer characteristics of epidepride for D3 receptors in combination with the selective expression of D3 receptor in lobules 9 and 10 of the cerebellum. In vivo receptor occupancy was readily measured by comparison of the binding of epidepride to lobules 9 and 10 of cerebellum with nonspecific binding in the lateral cerebellum. A powerful addition to the present method was the ability to simultaneously measure the occupancy of D2 receptors in the same rats by comparisons of epidepride binding in dorsal striatum versus cerebellum. Although with more painstaking efforts, simultaneous D2 and D3 receptor occupancy can be measured by the agonist radioligand [11C]-PHNO, a second radioligand (e.g., 3H-raclopride) is often employed for establishment of dopamine D2 occupancy (Kiss et al., 2011). Availability of a method for detection of the occupancy of D3 receptor ligands to D3 receptors and their selectivity for the closely related D2 receptors, as described here, provides another important tool for making inroads into the discovery and development of novel dopamine D3 receptor compounds. By leveraging the LC–MS/MS version of this technique, simultaneous measurements were possible, linking occupancy at multiple central receptor sites. Additionally, this technique alleviates the costs associated with using radioactive ligands and increases the speed of compound evaluation relative to similar recently published techniques (Kiss et al., 2011).
Using these methods, we determined that both SB-277011A and U99194 can achieve significant occupancy of D3 receptors in rat brain upon systemic administration. Further, both SB-277011A and U99194 can selectively occupy D3 over D2 receptors. SB-277011A was more potent than U99194 in vivo in accord with its higher affinity for D3 receptors. Significant D3 receptor occupancy (∼60%) was achieved at 3 mg/kg SB-277011A and 90% occupancy was observed at 10 mg/kg. In contrast, U99194 was about 10-fold less potent in occupying D3 receptors and at the highest dose tested of (30 mg/kg) only occupied ∼80% of the D3 sites. In addition, SB-277011A was more selective for D3 than D2 receptors. At the lowest dose of each molecule that occupied at least 80% D3 receptors, the occupancy of D2 receptors by SB-277011A was <5%, whereas the occupancy of D2 receptors by U99194 was about 20%. The ability to measure occupancy of receptors in vivo is key to defining the relationship of target engagement to biologic effect. The receptor occupancy data on these molecules extends the limited findings in the published literature. The occupancy of D3 receptors in vivo by SB-277011A is thus consistent with one dose–response finding reported with an agonist radioligand (Rabiner et al., 2009). We cannot find any data in the literature on the occupancy of dopamine receptors in vivo by U99194. Our current data on U99194 is a key testament to the need for such in vivo target-engagement bioassays in assessing the value of research tools. It is generally appreciated that high in vitro potency or selectivity do not necessarily translate in vivo (e.g., Perachon et al., 2000). Although SB-277011A is 100-fold more selective for D3 receptors in its binding profile than for D2 receptors (Reavill et al., 2000), U99194 is only 20-fold selective (Waters et al., 1993), suggesting that the compound at higher doses will cross over significantly to D2 receptors in vivo. Indeed, it is often assumed that low potency or selectivity in vitro translates to poor potency or selectivity in vivo. However, the high occupancy of D3 receptors by U99194 at 60 mg/kg (81%) and the ability of U99194 to block a D3-mediated behavioral effect (Collins et al., 2005) indicate that the low selectivity of this molecule in vitro is less of a liability than previously supposed.
Both of the D3 receptor antagonists studied produced large increases in ACh efflux in medial prefrontal cortex of rats as did the ADHD medication atomoxetine (Tzavara et al., 2006a). Lacroix et al. (2003) had also reported increases in ACh efflux measured from rat cingulate cortex. To understand these effects further we examined ACh and choline tissue levels and determined the ratio of choline:ACh that is considered a measure of ACh turnover rate in three brain areas (hippocampus, striatum, and cortex). Both D3 receptor antagonists dose-dependently decreased ACh tissue levels, probably due to its enhanced release, and increased ACh turnover rate, most likely due to an increase in its synthesis and release, in all three brain regions studied. Combined, these neurochemical results, thus, demonstrate that the two D3 receptor antagonists given at doses that engage the receptor stimulate ACh release and synthesis in the brain, as captured by both in vivo and ex vivo methods. To the best of our knowledge the effects of D3 receptor blockade on brain tissue levels of ACh and choline have not previously been reported. Importantly, the fact that the effects on tissue ACh and choline levels mirror the findings in ACh efflux in response to D3 receptor antagonists offers an additional robust and reliable tool in the study of neurochemical coupling to dopamine D3 receptor blockade in the brain.
Corresponding to the increases in cortical ACh and increases in brain ACh turnover, the dopamine D3 receptor antagonists prevented the effects of scopolamine in a novel object recognition task while occupying D3 receptors. The positive effects of both compounds, in this behavioral model of cognitive impairment, that are associated with enhancements in ACh function in the brain were also observed with reboxetine, an analog of the ADHD-treatment drug atomoxetine, which also prevents scopolamine-induced deficits (Tzavara et al., 2006a). SB-277011A also enhanced the effects of reboxetine in this task. In addition, U99194 enhanced the learning of rats in a five-trial shuttle box procedure that is also sensitive to the ADHD drug methylphenidate and to reboxetine (present study) as well as to other procognitive mechanisms (e.g., Fox et al., 2003). These findings implicate D3 receptors as negative regulators of attentional processing and are consistent with a literature of cognitive improvements with D3 receptor antagonism (Laszy et al., 2005; Braszko, 2010; Micale et al., 2010) opposing the cognitive deficits impacted by D3 receptor agonists (Smith et al., 1999). A polymorphism of the D3 receptor has been associated with the cognitive impairments in schizophrenia (Szekeres et al., 2004; Bombin et al., 2008) and D3 receptor blockade might be associated with some of the beneficial cognitive-impacting effects of atypical antipsychotic agents and of selective D3 receptor antagonists (Lacroix et al., 2003; Lumme et al., 2007; Millan and Brocco, 2008). However, clinical data also have suggested cognitive benefit from the anti-Parkinson D3/2 receptor agonist pramipexole (Levin et al., 2009).
In vivo models for predicted drug efficacy in ADHD are imperfect. However, although no disease models exist, multiple in vivo readouts do detect effects of drugs used in ADHD therapeutics such as methylphenidate, d-amphetamine, and atomoxetine (Wickens et al., 2011). In the present study, effects on cognitive aspects of the disorder, including attentional deficits, were studied in rats, where methylphenidate and the atomoxetine analog reboxetine were used as positive controls. Modeling of the hyperactivity symptoms of ADHD has used the dopamine transporter (DAT)−/− mouse. Indeed, several genetic studies show an association between a polymorphism in the noncoding regions of the DAT gene and ADHD, suggesting that DAT-mediated processes could significantly contribute to the pathogenesis of this disorder. In the DAT KO mice, a five-fold increase in extracellular dopamine in the brain is associated with dramatic hyperactivity. This hyperactivity, paradoxically, is inhibited by psychostimulants like amphetamine and methylphenidate, thereby providing a simple model in which the effects of ADHD pharmacological agents can be assessed (Gainetdinov and Caron, 2003).
In the present study, we showed that the ADHD drugs d-amphetamine and reboxetine decreased the profound hyperactivity of DAT−/− mice. This dampening effect was also observed with two structurally distinct D3 receptor antagonists SB-277011A and U99194. In contrast to d-amphetamine but comparable to that of reboxetine, the D3 antagonists produced this effect in DAT−/− mice without stimulating locomotion in WT control mice (DAT+/+). Thus, in conjunction with the binding to D3 receptors, the subsequent rise in ACh efflux in the medial prefrontal cortex, augmentation of ACh turnover rate, reversal of the cognitive impairments induced by the known amnestic drug scopolamine, and the augmentation of the procognitive effects of reboxetine, these antihyperactivity data support the potential application of dopamine D3 receptor antagonists in the treatment of ADHD.
Despite the potential therapeutic applications of dopamine D3 receptor antagonists, there are no selective drugs approved for clinical use. The present findings encourage further discovery and development on this interesting biologic mechanism. The promise of bringing such compounds into clinical use (Micheli and Heidbreder, 2006) has also been improved recently by the identification of a crystal structural complex of D3 receptors with eticlopride (Chien et al., 2010).
Acknowledgments
The authors thank Dei Zhang for help in chemical synthesis and Dr. H. C. Fibiger for encouragement in this area.
Authorship Contributions
Participated in research design: Barth, Need, Tzavara, Giros, Overshiner, Gleason, Wade, Johansson, Perry, Nomikos, Witkin.
Conducted experiments: Need, Tzavara, Overshiner, Gleason, Wade, Perry.
Contributed new reagents or analytic tools: Johannson.
Performed data analysis: Barth, Need, Tzavara, Giros, Overshiner, Gleason, Wade, Johansson, Perry, Nomikos, Witkin.
Wrote or contributed to the writing of the manuscript: Barth, Need, Tzavara, Giros, Overshiner, Gleason, Wade, Johansson, Perry, Nomikos, Witkin.
Footnotes
A portion of the data presented was previously presented in abstract form at the following conference: J. M. Witkin, S. D. Gleason, C. Overshiner, M. R. Wade, K. A. Svensson, K. Perry, B. Giros, E. T. Tzavara, and G. G. Nomikos (2005) In vivo occupancy of dopamine D3 receptors by antagonists produces neurochemical and behavioral effects of potential relevance to attention-deficit-hyperactivity-disorder. American College of Neuropsychopharmacology; 2005 Dec 11–15; Waikaloa, HI.
Abbreviations
- ACh
- acetylcholine
- ADHD
- attention-deficit–hyperactivity disorder
- ANOVA
- analysis of variance
- DAT
- dopamine transporter
- HPLC
- high performance liquid chromatography
- KO
- knockout
- LC–MS/MS
- liquid chromatography–tandem mass spectrometry
- 7-OH-DPAT
- 2-dipropylamino-7-hydroxy-1,2,3,4-tetrahydronaphthalene
- PHNO
- (+)-4-propyl-9-hydroxynaphthoxazine
- SB-277011A
- trans-N-[4-[2-(6-cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide
- U99194
- 2,3-dihydro-5,6-dimethoxy-N,N-dipropyl-1H-inden-2-amine
- WT
- wild type
- Received July 31, 2012.
- Accepted November 28, 2012.
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}