


Abstract
2-[(Diphenylmethyl) sulfinyl]acetamide (modafinil), prescribed principally to treat narcolepsy, is undergoing assessment for other neuropsychiatric disorders and medical conditions. The neurochemical substrates of modafinil are unresolved. We postulated that modafinil enhances wakefulness by modulating dopamine (DAT), norepinephrine (NET), or serotonin (SERT) transporter activities. In vivo, we determined DAT and NET occupancy by modafinil by positron emission tomography imaging; in vitro, we determined modafinil activity at the DAT, NET, SERT, and rhesus monkey trace amine receptor 1 (TA1). In rhesus monkey, modafinil occupancy of striatal DAT was detected by [11C]2β-carbomethoxy-3β-4-(fluorophenyl)tropane and of thalamic NET by [11C](S,S)-2-(α-(2-methoxyphenoxy)-benzyl)morpholine. In vitro, modafinil effects in DAT-human embryonic kidney (HEK), NET-HEK, and SERT-HEK cells were investigated alone or combined with the TA1 receptor. Modafinil (i.v.) occupied striatal DAT sites (5 mg/kg: 35 ± 12%, n = 4; 8 mg/kg: 54 ± 3%, n = 3). In thalamus, modafinil occupied NET sites (5 mg/kg: 16 ± 7.8%, n = 6; 8 mg/kg: 44 ± 12%; n = 2). In vitro, modafinil inhibited [3H]dopamine (IC50 = 6.4 μM), [3H]norepinephrine (IC50 = 35.6 μM), and [3H]serotonin (IC50 > 500 μM) transport via the human DAT, NET, and SERT. Modafinil did not activate the TA1 receptor in TA1-HEK cells, but it augmented a monoamine transporter-dependent enhancement of phenethylamine activation of TA1 in TA1-DAT and TA1-NET cells, but not in TA1-SERT cells. The present data provide compelling evidence that modafinil occupies the DAT and NET in living brain of rhesus monkeys and raise the possibility that modafinil affects wakefulness by interacting with catecholamine transporters in brain.
2-[(Diphenylmethyl) sulfinyl]acetamide (modafinil) produces a unique spectrum of pharmacological effects, the most prevalent being enhanced vigilance, arousal, and wakefulness in human subjects (Bastuji and Jouvet, 1988). The drug is widely used to treat narcolepsy (Banerjee et al., 2004), but it is also undergoing clinical trials in an expanding range of psychiatric, neurological, and medical illnesses, including cocaine addiction (Dackis and O'Brien, 2003; Vocci and Ling, 2005; Sofuoglu and Kosten, 2006) and attention deficit hyperactivity disorder (Biederman et al., 2005). Notwithstanding the expanding clinical indications for modafinil, the neurochemical mechanisms that produce therapeutic improvement remain unresolved. This void is particularly relevant for cocaine addiction, because clarity on the underlying mechanisms may yield important leads for candidate therapeutics.
In the original patent description, modafinil (CRL 40476) promoted hyperactivity and hypermotility, potentiated apomorphine-induced stereotypy, and reduced sleep length (Lafon, 1979), all of which are characteristics of hyperdopaminergic activity. Thereafter, several investigators questioned the contribution of dopamine neurotransmission to modafinil pharmacology, on the basis of overlapping, but not identical behavioral and neurochemical responses of modafinil compared with amphetamine, a psychomotor stimulant with robust dopaminergic effects (Duteil et al., 1990; Akaoka et al., 1991; Simon et al., 1995; Lin et al., 1996; Ferraro et al., 1997a,b; Engber et al., 1998; Engele, 1998). Modafinil was a weak inhibitor of the DAT (>1 μM affinity) and displayed no affinity for dopamine receptor subtypes (Mignot et al., 1994; Bryan Roth, personal communication). Enhancement of glutamate release and inhibition of GABA release in various brain regions were proposed as alternative modes of action (Ferraro et al., 1997a,b, 1999).
Notwithstanding these important observations, accumulating behavioral and biochemical evidence converge on dopaminergic and noradrenergic mechanisms as plausible contributors to modafinil pharmacology (Wisor et al., 2001; Boutrel and Koob, 2004; Wisor and Eriksson, 2005). Modafinil engenders behavioral effects consistent with enhanced dopamine transmission, because it increases motor activity in normal or parkinsonian monkeys (Jenner et al., 2000; van Vliet et al., 2006), partially generalizes to a cocaine-like discriminative stimulus in rodents, and serves as a reinforcer at high doses in monkeys or human subjects (Gold and Balster, 1996; but see Deroche-Gamonet et al., 2002; Stoops et al., 2005). Modafinil also increases striatal dopamine efflux as effectively as amphetamine in dog brain, with dose ratios comparable with clinical dose ratios. Significantly, the wake-promoting effects of modafinil are abolished in DAT null mutant mice (Wisor et al., 2001).
The rapid expansion of new therapeutic indications for modafinil, in the face of unexplained mechanisms, creates an exigency to clarify modafinil pharmacology. On the basis of modafinil-induced dopamine efflux and the DAT dependence for the wake-promoting effects of the drug (Wisor et al., 2001), we postulate that modafinil modulation of the DAT (or NET) contributes to enhanced wakefulness and therapeutic benefit. This hypothesis is predicated on whether modafinil occupies the DAT or NET in living brain, an objective that, to our knowledge, has not been addressed. We used PET imaging of the DAT ([11C]CFT) and of the NET with [11C]MeNER (Madras et al., 1989, 2001; Morris et al., 1996; Schou et al., 2003; Spencer et al., 2006) to investigate whether modafinil binds to these transporters in living brain. PET imaging revealed that therapeutic doses of modafinil occupied DAT and NET sites in vivo, leading us to investigate the in vitro pharmacology of modafinil at monoamine transporters. In vitro, we measured modafinil potencies for inhibiting transport of dopamine, norepinephrine, and serotonin by the human DAT, NET, and SERT. Modafinil inhibited dopamine and norepinephrine transport by the DAT and NET, respectively, in agreement with the DAT-dependent increase in extracellular dopamine levels elicited by modafinil administration.
The trace amine PEA promotes dopamine release via the DAT (Sotnikova et al., 2004), an effect potentially linked to its activation of the trace amine receptor 1 (TA1; Borowsky et al., 2001; Bunzow et al., 2001; Miller et al., 2005). PEA is also a potent agonist at TA1, and our previous in vitro work has demonstrated that the response of TA1 to PEA is greatly enhanced when the DAT is also present (Miller et al., 2005). In this regard, TA1 activity may be affected by modafinil, because DAT enhances TA1 activation by PEA (Miller et al., 2005). Modafinil did not act as a direct agonist at TA1, nor did it significantly affect PEA stimulation of TA1 in transfected HEK cells lacking monoamine transporters. Modafinil did, however, augment the enhancement of PEA activation of TA1 in the presence of DAT or NET, indicating a potential indirect contribution of TA1 receptors with regard to modafinil action that warrants further investigation. Accordingly, we provide evidence that modafinil-induced wakefulness is partially attributable to DAT and NET occupancy in the brain, resultant from a cascade of events associated with functional catecholamine transporters.
Materials and Methods
PET Imaging of the Dopamine and Norepinephrine Transporter. Adult female (n = 4) and male (n = 1) rhesus monkeys (Macaca mulatta) were used for PET imaging procedures. Animal care and treatment were supervised by veterinarians under the guidelines and in accordance with Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, National Academy Press, Washington, DC, 1996). The animal care protocol was approved by the Harvard Animal Care Committee and was in compliance with the Harvard Medical School animal management program, an institution accredited by the American Association for the Accreditation of Laboratory Animal Care.
PET imaging of the DAT in caudate putamen was used to determine DAT occupancy by modafinil. DAT was quantified with the selective DAT probe [11C]CFT (Kaufman and Madras, 1991; Hantraye et al., 1992; Morris et al., 1996; Madras et al., 1998; Saka et al., 2004). PET imaging was conducted with monkeys initially anesthetized with ketamine/xylazine (15.0/1.5 mg/kg) and then maintained under general anesthesia with halothane. Monkeys were positioned on the imaging bed of a PC 4096 PET camera (GE/Skanditron AB, Uppsala, Sweden). A stereotactic head-holder was used for head immobilization. CFT was demethylated in the C-2 position, and [11C]methyl was inserted by the methyl iodide reaction. After stabilization in the PET camera, ∼10 mCi of [11C]CFT (specific activity >1500 mCi/μmol) was injected through the venous catheter and sequential images were acquired in 15-s time frames for the first 2 min and in 1-min frames for 58 min. DAT or NET occupancy was measured by administering modafinil i.v. 1 h after baseline image acquisition was completed and then reintroducing radioligand via an i.v. indwelling catheter 1 h later. Parallel studies were conducted with the NET probe [11C]MeNER. At the conclusion of each imaging study, the emission and transmission images were reconstructed using a conventional filtered back-projection algorithm to an inplane resolution of 6-mm full-width half-maximum. All projection data were corrected for nonuniformity of detector response, dead time, random coincidences, and scattered radiation. A sum image was generated by adding all of the frames from frame 10 to the end of the study. Regions of interest were drawn on the summed image in the coronal projection as follows: one 4-pixel region was drawn on each caudate putamen on the slice of maximal intensity. For cerebellum, three 4-pixel regions were drawn at various levels on cerebellar slices. Time-activity data were produced using the regions of interest on all time frames of the PET data. The same set of regions of interest was used to analyze each scan for an individual subject on the same day. When necessary, new regions were drawn to compensate for repositioning. Binding potential was calculated by published methods. Parallel experiments were conducted with the PET ligand [11C]MeNER (Schou et al., 2004; Ghose et al., 2005) to image the NET in the thalamus and monitor modafinil occupancy.
Dopamine, Norepinephrine, and Serotonin Transporter Assays. The generation of stable human DAT-, NET-, and SERT-transfected HEK293 cells has been described previously (Goulet et al., 2001; Yatin et al., 2001; Verrico et al., 2005). Assays to measure drug inhibition of [3H]monoamine transport were performed to determine IC50 values, as described previously (Miller et al., 2001; Verrico et al., 2005). Cells were grown to 70 to 85% confluence in Biocoat 24-well tissue culture plates coated with polylysine. The medium was removed by aspiration, and cells were washed with ice-cold assay buffer: Tris–HEPES, pH 7.4, at 25°C containing 4 mM Tris base, 8.5 mM HEPES, 120 mM NaCl, 5.4 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 10 mM glucose, 100 μM ascorbic acid, and 100 μM tropolone. Cells were harvested by pipetting the medium for cell detachment and resuspended in the assay buffer at approximately 3 × 105/ml. Each assay tube contained ∼1.5 × 105 cells, [3H]DA, [3H]norepinephrine, or [3H]5-hydroxytryptamine and various concentrations of unlabeled inhibitors in a total of 0.4 ml of solution. Uptake activities were initiated by transferring the tubes from ice to 37°C water bath and terminated 3 or 10 min later by transferring the tubes to ice. Each experiment was performed in triplicate and repeated three to six times. All experiments were conducted in darkness. Nonspecific monoamine transport was measured with 10 μM mazindol for DAT, 10 μM nisoxetine for NET, or 10 μM fluoxetine for SERT. In competition experiments, 0.2 ml of drug was added for a 10-min preincubation period. Monoamine transport was initiated by the addition of [3H]monoamine combined with various concentrations of unlabeled monoamine to achieve an appropriate specific activity (Sigma-Aldrich, St. Louis, MO). The assay was terminated by aspiration. The plate was placed on ice and rinsed three times with cold (4°C) uptake assay buffer. After the final wash was aspirated, 0.5 ml of 1.0% SDS was added to each well. After 20 min of gentle shaking, 0.25 ml was removed from each well, and radioactivity was measured for 5 min in vials containing 4 ml of ReadySafe scintillation fluid. IC50 values were obtained by nonlinear curve fitting using Prism version 3.0 (GraphPad Software Inc., San Diego, CA).
Rhesus TA1 Activity, as Determined by Dual-Luciferase Reporter Assay. Stable DAT, NET, and SERT cells were placed in 48-well plates 2 days before transfection. Transfections were performed on 70 to 80% confluent cells as described previously (Miller et al., 2005; Xie et al., 2005). In total, three vectors were cotransfected: 1) a cAMP-response element (CRE)-driven firefly luciferase reporter construct, pTLNC121-3 (from Dr. Walter Borne, University of Zurich, Zurich, Switzerland), which contains a 21×CRE cassette positioned upstream from a TATA box minimal promoter element, as a highly sensitive and quantitative reporter of total cAMP accumulation; 2) an optimized Renilla luciferase construct, pGL4.73 (Promega, Madison, WI), which is unresponsive to increases in cellular cAMP (G. M. Miller, unpublished data), to control for transfection efficiency and experimental variability; and 3) either a TA1 expression vector (Miller et al., 2005) or pcDNA3.1(+) (Invitrogen, Carlsbad, CA) to control for total DNA amounts used in transfections. Transfections into different cell lines were performed simultaneously for each experiment. Dual-luciferase assay (Promega) was performed according to the manufacturer's protocol. Cells were incubated with transfection solution for 12 h and then exposed to drugs at different concentrations for 18 h at incubation conditions. Monoamine transporter inhibitors, when used, were applied to the cells at 15 min before the drug treatment. Cell lysates were prepared by adding 100 μl of lysis buffer into each well and shaking on a rotator platform for 30 min at 25°C. Then, 20 μl of each cell lysate was transferred into a 96-well microplate for determination of luciferase concentration. Luciferase assay substrate reagents (25 μl) were injected into each sample well, and after a 2-s delay, luciferase concentration was measured as relative light units (RLUs) for 12 s at Wallac 1420 multilabel counter-Victor 3V (PerkinElmer Life and Analytical Sciences, Shelton, CT). The experiments were performed three times in triplicate for each concentration of the drug treatment. Percentage of RLU increase was calculated from the ratio of firefly to Renilla luciferase in cell lysates. To assess the effect of monoamine transporter blockers on TA1 in cells that coexpress a monoamine transporter, we defined the enhancement of TA1 activation as the percentage of increase of RLU in TA1-tranfected DAT, NET, or SERT cells subtracted by the percentage of increase of RLU in TA1-transfected HEK293 cells.
Drugs. [3H]Dopamine, [3H]norepinephrine, and [3H]serotonin were purchased from PerkinElmer Life and Analytical Sciences (Wellesley, MA). Modafinil and PEA were purchased from Sigma-Aldrich.
Results
PET Imaging: DAT and NET Occupancy by Modafinil. To investigate whether clinically relevant doses of modafinil accumulate at the DAT, we labeled the DAT with the PET imaging probe [11C]CFT or WIN 35,428 (Madras et al., 1989; Morris et al., 1996). [11C]MeNER was used to label the NET (Schou et al., 2004). In a double-injection study, rhesus monkeys were scanned to obtain baseline measures of DAT or NET binding potential. After decay of [11C]CFT or [11C]MeNER, monkeys were injected i.v. with 2, 5, or 8 mg/kg modafinil, and PET imaging was repeated 1 h later (Table 1). DAT occupancy was insignificant with 2 mg/kg modafinil (n = 1), but 5 mg/kg modafinil occupied 35 ± 11.9% of DAT sites (n = 4), and 8 mg/kg modafinil occupied 53 ± 3% (n = 3) of DAT sites in striatum (Figs. 1, left, and 2; Table 1). Modafinil (8 mg/kg) occupancy of the DAT (Fig. 2) or the NET (Fig. 3) was observable within 60 min after administration. To verify that modafinil occupancy occurred regardless of the DAT probe, a single parallel experiment was performed with the DAT probe [11C]altropane (Fischman et al., 2001). Again, 8 mg/kg modafinil occupied 67% of DAT sites, indicating that DAT occupancy by modafinil was not an artifact of the PET probe. For comparative purposes, a parallel study was conducted with methylphenidate, again at a clinically relevant dose (0.3 mg/kg). Methylphenidate occupied comparable levels of DAT at a dose of 0.3 mg/kg (Table 1).
PET occupancy of modafinil administered i.v. at various doses
Data shown are mean ± S.E.M. (n).
The NET also accumulated modafinil (Figs. 1, right, and 3), with a 5-mg/kg dose occupying 16 ± 7.6% of NET sites (n = 6) and an 8-mg/kg dose occupying 56% of the NET (n = 1). At 8 mg/kg, the percentages of occupancy of DAT and NET were similar, but with a lower modafinil dose (5 mg/kg), occupancy of the DAT was twice as high as the NET (Fig. 1, right; Table 1). These data provided a strong impetus to investigate modafinil effects on DAT and NET in vitro.

Modafinil occupancy of the DAT in monkey striatum (left) or NET in monkey thalamus (right). PET imaging of the DAT was conducted with [11C]CFT (five rhesus monkeys) and of the NET with [11C]MeNER (4 rhesus monkeys). Occupancy was calculated as described under Materials and Methods. For each dose of modafinil, PET imaging was conducted in two to five different monkeys, with each point representing the mean ± S.E.M. of two to six independent experiments, conducted on different days. For representative images, please see Figs. 2 and 3. For 8 mg/kg modafinil, one data set is shown for %NET occupancy.
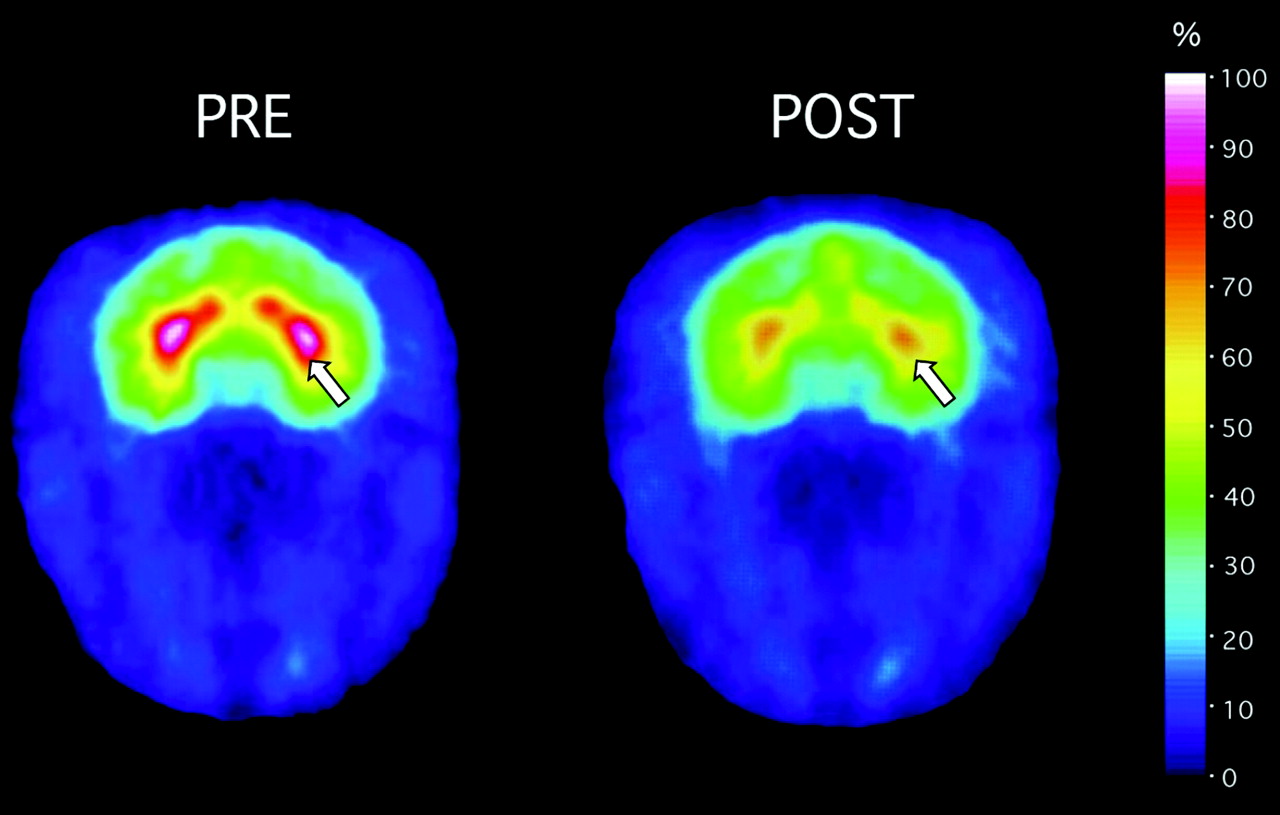
Modafinil (8 mg/kg) occupancy by the DAT in caudate putamen, as detected by PET imaging of the DAT with [11C]CFT. Left, an adult rhesus monkey was injected with [11C]CFT and scanned over 60 min to develop baseline measures of DAT binding potential in the caudate putamen. Images were color-transformed to display occupancy of the DAT with [11C]CFT, with highest levels detected in caudate putamen (white-red), as designated by the arrow, and lowest levels in blue-purple. Regions of interest are drawn over the caudate putamen. Right, after decay of [11C]CFT radioactivity, modafinil was injected i.v., and [11C]CFT was injected again 1 h later. [11C]CFT accumulation was significantly lower compared with baseline levels of accumulation (left). Data are summarized in Table 1 and Fig. 1.

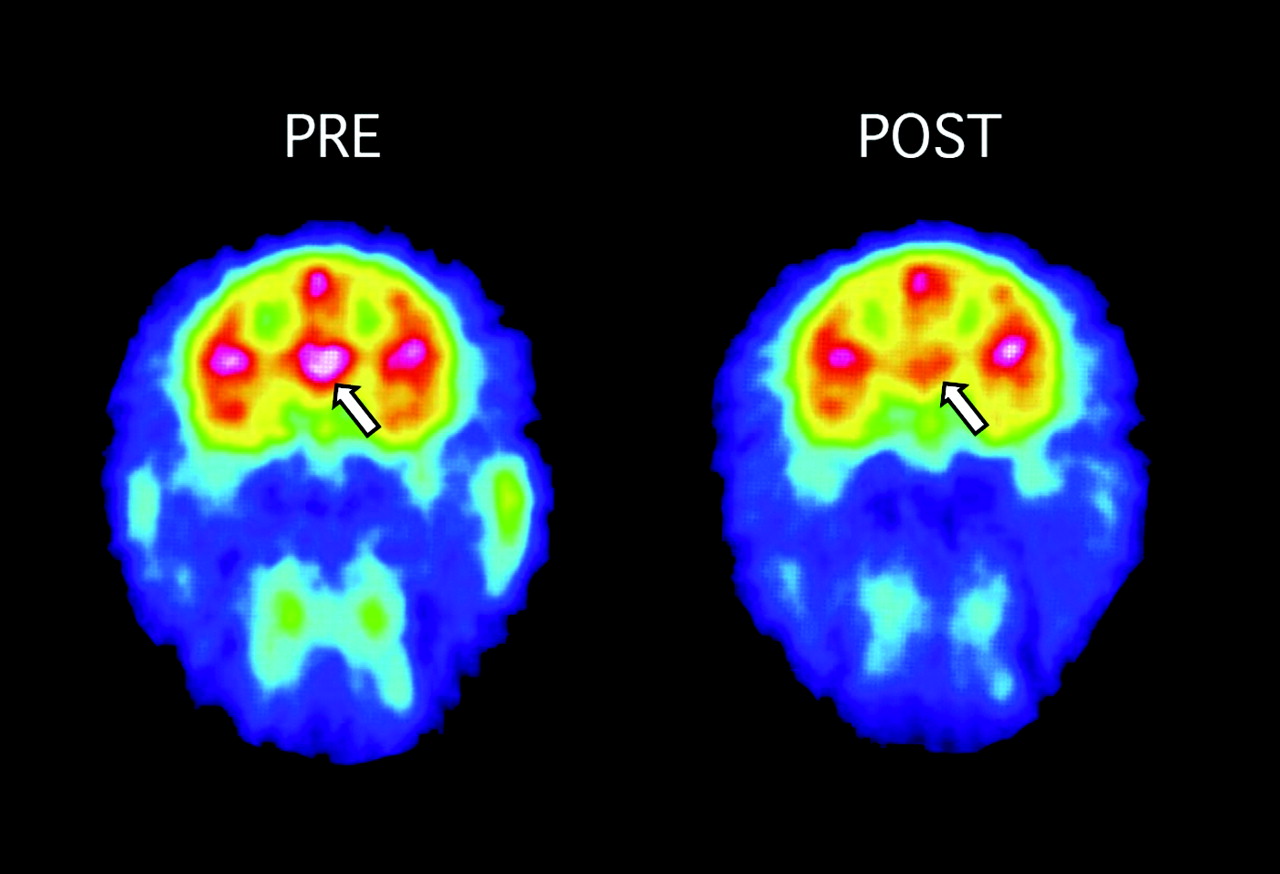
Modafinil (8 mg/kg) occupancy by the NET in thalamus, as detected by PET imaging of the NET with [11C]MeNER. Left, an adult rhesus monkey was injected with [11C]MeNER and scanned over 60 min to develop baseline measures of NET binding potential in the thalamus. Images were color-transformed to display occupancy of the NET by [11C]MeNER, with high levels detected in the thalamus (white-red), as designated by the arrow, and lowest levels in blue-purple. Regions of interest are drawn over the thalamus. Right, after decay of [11C]MeNER radioactivity, modafinil was injected i.v. and 1 h later, [11C]MeNER was injected. [11C]MeNER accumulation was significantly lower compared with baseline levels of accumulation. Data are summarized in Table 1 and Fig. 1.
Modafinil Inhibition of DAT, NET, and SERT. We measured modafinil affinity for the DAT in radioligand binding assays and for DAT, NET, and SERT in transport assays. Because modafinil is weakly soluble in aqueous media, we were cognizant of the potential confound of solvent interference with transport binding or activity assays. Pilot radioligand binding assays demonstrated that at certain concentrations, solvents (dimethyl sulfoxide or ethanol/HCl) significantly affected DAT and NET binding assays, and assignment of the relative contributions of solvent and modafinil to the data were not possible. Solvent levels were adjusted accordingly to prevent this confound. In competition studies with [3H]CFT (DAT), modafinil affinity was relatively modest (IC50 = 4360 nM; Table 2) and corresponded to its potency for inhibiting [3H]dopamine transport (IC50 = 6390 ± 2050 nM; Fig. 4). Modafinil was a weak inhibitor of [3H]norepinephrine or [3H]dopamine transport by the NET (Table 2; Fig. 5). Modafinil potency for blocking [3H]serotonin transport was low, with an IC50 value >500 μM (Table 2). With the exception of bupropion, modafinil potency was at least 10-fold lower in comparison with other therapeutic drugs that elicit pharmacological responses partly via the DAT (Table 2). Compared with modafinil, methylphenidate was 100-fold more potent at blocking [3H]dopamine transport by the DAT, and benztropine was 30- and 10-fold more potent.
Modafinil affinities for DAT and NET and comparison with other clinically relevant DAT inhibitors, at 25°C
Modafinil Effects on TA1 Alone or with DAT, NET, or SERT. We investigated the effects of modafinil on TA1 because modafinil blocks [3H]dopamine transport, and DAT-mediated dopamine release is facilitated by TA1 (Sotnikova et al., 2004). We initially monitored whether modafinil modulated CRE-luc expression mediated by TA1. In HEK293 cells transfected with the reporter constructs, there was no dose-response effect of modafinil with or without TA1, with only extremely small elevations in CRE-luc activation detected in the presence of modafinil (Fig. 6A).
PEA is an agonist at TA1 and is an effective substrate for DAT (Miller et al., 2005; B. K. Madras, A. K. Jassen, Z. Lin, H. Panas, T. J. Spencer, G. M. Miller, and A. J. Fischman, unpublished data). We determine whether modafinil alters PEA activation of TA1 indirectly, by blocking DAT, NET, or SERT function. We postulated this may occur as a population of TA1 is localized intracellularly, based on immunohistochemical localization of the receptor in vitro (Bunzow et al., 2001; Miller et al., 2005). Indirect evidence also points to an intracellular localization of TA1 in vitro, because TA1 activation by PEA is markedly enhanced in DAT-TAR1-expressing cells (Miller et al., 2005). To determine whether modafinil could modify TA1 activity via monoamine transporters, albeit in vitro, various concentrations of modafinil were incubated with cells coexpressing DAT-TA1, NET-TA1, or SERT-TA1. Modafinil augmented TA1 activity in the presence of DAT only at 100 μM, but not NET or SERT (Fig. 6B). Because the three cell lines share an identical HEK293 background, the response is mediated by the DAT. Accordingly, modafinil does not directly activate TA1 receptors at doses below 100 μM.

Modafinil inhibition of [3H]dopamine transport by the DAT. Various concentrations of modafinil were preincubated with HEK cells stably transfected with the human DAT. [3H]Dopamine (4 nM, approximately) was added, and cells were incubated for 10 min at 25°C, as described under Materials and Methods.

Modafinil inhibition of [3H]norepinephrine transport by the NET. Various concentrations of modafinil were preincubated with HEK cells stably transfected with the human NET. [3H]Norepinephrine (4 nM, approximately) was added, and cells were incubated for 10 min at 25°C, as described under Materials and Methods.
Modafinil Effects on PEA Activation of TA1 Alone or with DAT, NET, or SERT. PEA activation of TA1 is dose-dependent and is robustly enhanced by either DAT, NET, or SERT coexpression in vitro (Fig. 7). The enhancement of PEA activation of TA1 in the presence of DAT, NET, or SERT is blocked by pretreatment with the transporter inhibitors methylphenidate, desipramine, and citalopram, respectively, at a dose of 10 μM (Fig. 7). In the absence of monoamine transporters, modafinil (10–9–10–4 mol/l) had no effect on PEA activation of TA1 (Fig. 8A, left top). We tested a single dose of modafinil, 10 μM, and found that it was able to augment the enhancement of PEA activation of TA1 in the presence of DAT (Fig. 8B, right top) or NET (Fig. 8C, left bottom), but there was no augmentation of the enhancement of PEA activation of TA1 in the presence of SERT.
Discussion
Modafinil is widely used to treat narcolepsy (Banerjee et al., 2004), and it is undergoing clinical trials as a therapeutic for a wide range of psychiatric, neurological, and medical illnesses. These include cocaine addiction (Dackis and O'Brien, 2003; Vocci and Ling, 2005), Parkinson's disease (Serrano and Garcia-Borreguero, 2004), attention deficit hyperactivity disorder (Biederman et al., 2005), depression (Price and Taylor, 2005), cancer, and multiple sclerosis-related fatigue (Morrow et al., 2005) and opioid-induced sedation in chronic pain (Reissig and Rybarczyk, 2005). The rapid expansion of these presumptive clinical indications for modafinil is empirically driven, because no broad consensus exists on the underlying mechanisms of modafinil pharmacology. We now provide compelling evidence that modafinil occupies the DAT and NET in living brain of rhesus monkeys at doses that are presumably clinically relevant. Occupancy was comparable with DAT occupancy by the attention deficit hyperactivity disorder medication methylphenidate, also administered at a therapeutic dose. The clinical significance of this discovery is supported by in vitro data, because modafinil blocked 50% of DAT-mediated dopamine transport in vitro at a lower concentration than the plasma concentrations of modafinil estimated in human subjects within 2 h after a modest dose (200 mg) of the drug (Wong et al., 1999). Accordingly, clinically relevant doses (400–600 mg) of modafinil conceivably achieve brain concentrations sufficient to bind to DAT and NET, notwithstanding the caveats of 1) extrapolating modafinil plasma levels to brain extracellular concentrations, 2) extrapolating in vitro to in vivo transporter affinities, 3) potential differences in the pharmacokinetic and metabolic properties of modafinil in humans and nonhuman primates, and 4) a similarity in plasma levels that results from i.v. modafinil in rhesus monkeys compared with oral modafinil in humans. If our data are confirmed in human subjects by PET imaging, we surmise that modafinil is a modulator of DAT and/or NET function in various brain regions, a process that conceivably contributes to enhanced wakefulness and possibly other therapeutic effects of the drug.

Effects of modafinil on rhesus monkey TA1-mediated increases in CRE-luciferase expression, a measure of cAMP production. TA1 was transiently transfected with a CRE-luc reporter system alone (a measure of cAMP production) into HEK293 cells or into cells expressing the human DAT, NET, and SERT. Cells were incubated for 18 h. A, modafinil effects on HEK cells transfected with luciferase reporters (open circles) or additionally with TA1 (closed circles). Results are expressed as percentage of increase in RLU above baseline levels. B, dose-response effects of modafinil in DAT + TA1 (black circles), NET + TA1 (dark gray circles), or SERT + TA1 (light gray circles) cells. With the exception of 100 μM modafinil, which produced a statistically significant increase in RLU in DAT-TA1 cells (p < 0.02), no other significant changes were detected.
Although initial observations of modafinil pharmacology were consistent with drug-induced enhancement of dopamine activity, the contribution of dopamine neurotransmission to modafinil pharmacology has been repeatedly questioned, on the basis of nuances of neurochemical or behavioral data and in vitro DAT affinity. In particular, dopaminergic contributions were mitigated by evidence of contrasting amphetamine and modafinil pharmacology, brain activation patterns, and the failure of dopamine receptor antagonists to attenuate modafinil-mediated behavioral effects (Duteil et al., 1990; Akaoka et al., 1991; Mignot et al., 1994; Simon et al., 1995; Lin et al., 1996; Ferraro et al., 1997a,b; Engber et al., 1998). Although these differences may be attributable to contrasting doses, potencies, and the spectrum of molecular targets and affinities for various receptors and transporters in brain, they do not exclude dopaminergic mechanisms for both drugs.
A functional interaction between DAT and modafinil has been discounted, because of low modafinil affinity for DAT (Mignot et al., 1994). This is an insufficient reason to disregard the DAT, because relatively high doses of modafinil ranging from 200 to 600 mg (e.g., Schwartz et al., 2005) are administered clinically. The doses we used to detect DAT occupancy were 2 to 8 times lower than oral doses used to promote wakefulness in monkeys (Hermant et al., 1991). At a 200-mg dose, initial plasma modafinil levels range from 15 to 19 μM (Wong et al., 1999), a concentration 3 times higher than the 6 μM modafinil concentration needed to inhibit 50% of DAT sites in vitro. Other DAT modulators (e.g., amphetamine or methylphenidate) have much higher DAT affinity, and accordingly, are given at far lower doses (approximately 5–20 mg).
Our finding of modafinil occupancy of striatal DAT sites provides evidence that DAT modulation by modafinil may contribute to modafinil pharmacology. This hypothesis is supported by the capacity of 5 mg/kg modafinil to rapidly double striatal efflux of dopamine in dog brain and the failure of modafinil to promote wakefulness in DAT null mutant mice (Wisor et al., 2001). Intriguingly, the latter study demonstrated that both modafinil and amphetamine produced similar levels of dopamine release at equipotent wake-promoting doses. DAT occupancy was similar with two different DAT probes and comparable with occupancy levels by methylphenidate, an attention deficit hyperactivity disorder medication (Spencer et al., 2006). At this level of occupancy, extracellular dopamine is augmented in brain by modafinil.

Dose-response effects of PEA in TA1-transfected HEK cells (TA1) compared with TA1-transfected DAT (DAT-TA1), NET (NET-TA1), or SERT (SERT-TA1) cells. Pretreatment of DAT-TA1, NET-TA1, or SERT-TA1 cells with 10 μM methylphenidate, 10 μM desipramine, or 10 μM citalopram, respectively, reduces the transporter-dependent enhancement of TA1 activation.
Modafinil also occupied NET sites in the thalamus. We monitored this brain region because the thalamus yielded the highest NET signal-to-noise ratio combined with high density of NET in a previous screening of NET binding sites in various regions of primate brain (B. K. Madras, unpublished data). Given at equal doses (5 mg/kg), modafinil occupancy of the NET was approximately 50% lower than DAT. Nevertheless, occupancy was higher than anticipated, because modafinil is 7 times less potent at blocking NET than DAT function in vitro. Other factors, in addition to in vitro affinity, conceivably dictate the extent of DAT or NET occupancy, including ligand dissociation rates, relative affinities of the two PET probes, and relative DAT or NET densities in these brain regions. Furthermore, levels of endogenous substrates (neurotransmitters and modulators) in various brain regions are likely to affect modafinil interaction with DAT and NET. Modafinil is a relatively weak inhibitor of catecholamine transporters, and its capacity to block transporter function will be sensitive to local concentrations of competing endogenous neurotransmitters, to a greater extent than other DAT or NET inhibitors, with affinities 10 to 100 times higher (e.g., cocaine or methylphenidate). Although we did not monitor SERT occupancy, given the exceedingly low affinity of modafinil for the SERT, it is unlikely that therapeutic doses of the drug will interact with SERT to affect SERT function directly, but indirect effects cannot be ruled out.
Based on the in vivo and in vitro data, it is reasonable to surmise that modafinil could elevate dopamine and possibly norepinephrine, and perhaps also trace amine levels in select brain regions, and thereby indirectly activate dopamine, norepinephrine, and/or trace amine receptor subtypes. Elevation of dopamine levels has not conventionally been implicated in sleep-wake regulation, but the present study and that of Wisor et al. (2001) implicate the DAT as a potential mediator of modafinil-induced wakefulness. Potent DAT inhibitors, such as difluoropine or indatraline, promoted sleep fragmentation and increased nighttime wakefulness (Madras et al., 2006). The role of norepinephrine is less certain, because chemical ablation of noradrenergic neurons did not block modafinil-induced wakefulness (Wisor and Eriksson, 2005). It has been postulated that modafinil increases wakefulness by promoting glutamate release and inhibiting GABA release (Duteil et al., 1990; Ferraro et al., 1997a,b, 1999; Wisor et al., 2001). In parallel with modafinil, the DAT inhibitor cocaine also promotes glutamate release and attenuates GABA release in select brain regions (Cameron and Williams, 1994; Reid et al., 1997).
We found that the monoamine transporter inhibitors methylphenidate, desipramine, and citalopram at 10 μM almost entirely block DAT-, NET- or SERT-dependent enhancement of TA1 activation by PEA. The enhanced response of TA1 to monoamines observed with the CRE-luc assay may be due to the ability of monoamine transporters to deliver agonists intracellularly, where TA1 receptors are observed to be sequestered in vitro (Bunzow et al., 2001; Miller et al., 2005). However, it may also be that during the long period of agonist treatment (18 h) required to maximize the signal-to-noise ratio in the CRE-luc assay, TA1 receptors may traffic to the membrane, and that this trafficking is somehow facilitated by transporter function, although this has yet to be investigated. We also tested modafinil at the same dose (10 μM), which we show does not directly activate TA1. But unlike the other inhibitors, which have 15 to 100 times higher potency, 10 μM modafinil is near its IC50 value for the DAT. At this dose, modafinil augmented the enhancement of TA1 activity by PEA in DAT- and NET-expressing cells, but not SERT-expressing cells. This finding gives an initial indication that modafinil could affect, albeit indirectly, PEA activation of the TA1 receptor in dopamine and norepinephrine neurons. TA1 receptor mRNA is present in the major monoaminergic cell groups (Borowsky et al., 2001), and we have used double-label DAT and TA1 immunocytochemistry to demonstrate that a subset of dopamine neurons express TA1 receptors in both rhesus monkey and mouse substantia nigra (Xie et al., 2005). Regardless, the augmentation of enhancement of PEA activation of TA1 receptors by modafinil in DAT- and NET-expressing cells, but not SERT cells, further supports the hypothesis that modafinil interacts with the DAT and NET.

Effects of 10 μM modafinil on PEA activation of TA1 activity in HEK cells alone or in HEK cells also expressing the human DAT, NET, or SERT. A, In HEK cells transfected with TA1 alone (with reporter constructs), PEA dose-dependently increased TA1 activity (gray bars). Modafinil (10 μM; black bars) did not affect PEA dose-response activity on TA1 activity. B, dose-response effects of PEA in HEK-DAT cells transfected with TA1 (gray bars). At concentrations of 10–8 and higher, PEA tested, modafinil (10 μM; black bars) enhanced PEA activation of TA1 (*, p < 0.05). C, dose-response effects of PEA in HEK-NET cells transfected with TA1, in absence (gray bars) and presence (black bars) of 10 μM modafinil. Modafinil (10 μM; black bars) enhanced PEA activation of TA1 (*, p < 0.05, 10–6 and higher; **, p < 0.01, at 10–7 and 10–8). D, in HEK cells transfected with TA1 and SERT, PEA dose dependently increased TA1 activity (gray bars). Modafinil (10 μM; black bars) did not enhance PEA activation of TAR1, but it seemed to slightly attenuate OEA activation of TAR1, albeit not significantly.
The interaction of modafinil with the DAT may account for some of its preliminary positive clinical results in cocaine addicts (Vocci and Ling, 2005). Although the underlying mechanisms are unknown for this indication, the low DAT affinity of modafinil may be advantageous as a cocaine medication. In this regard, it is likely that extracellular dopamine inundation that ensues from potent DAT inhibitor may lead to undesirable receptor adaptations, arising from the inability of pre- or postsynaptic receptors to recycle appropriately, particularly if dopamine levels achieve a concentration that is likely to bind to dopamine receptors in the low-affinity state.
In summary, the present data raise the possibility that modafinil affects wakefulness by interacting with catecholamine transporters in brain. Although modafinil is likely to mediate effects via several mechanisms, the present study supports the view that the DAT and NET warrant further scrutiny as an important molecular target for this intriguing drug.
Acknowledgments
We thank Dr. Bryan Roth for helpful discussions and Jennifer Carter for excellent assistance in manuscript preparation.
Footnotes
-
This study was supported by National Institutes of Health Grants DA11558 (to B.K.M.), DA06303 (to B.K.M.), DA15305 (to B.K.M.), DA16606 (to G.M.M.), and RR00168 (to NEPRC).
-
This study was reported in abstract form. Madras BK (2006) Modafinil potentiates trace amine receptor activity via the dopamine transporter in vitro and partially occupies the dopamine transporter in vivo, The Annual Meeting of the Society for Nuclear Medicine; 2006 Jun 3–7, San Diego, CA, Society for Nuclear Medicine, Reston, VA.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.106583.
-
ABBREVIATIONS: DAT, dopamine transporter; NET, norepinephrine transporter; PET, positron emission tomography; CFT (WIN 35,428), 2β-carbomethoxy-3β-4-(fluorophenyl)tropane; MeNER, (S,S)-2-(α-(2-methoxyphenoxy)benzyl)morpholine; PEA, phenethylamine; TA1, trace amine receptor 1; HEK, human embryonic kidney; SERT, serotonin transporter; DA, dopamine; CRE, cAMP response element; RLU, relative light unit; TAR1, rhesus monkey trace amine receptor 1.
- Received April 21, 2006.
- Accepted July 26, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}