


Abstract
Cell lines expressing the human metabotropic glutamate receptor subtype 5a (hmGluR5a) and hmGluR1b were used as targets in an automated high-throughput screening (HTS) system that measures changes in intracellular Ca2+ ([Ca2+]i) using fluorescence detection. This functional screen was used to identify the mGluR5-selective antagonist, SIB-1757 [6-methyl-2-(phenylazo)-3-pyridinol], which inhibited the glutamate-induced [Ca2+]i responses at hmGluR5 with an IC50 of 0.37 μM compared with an IC50 of >100 μM at hmGluR1. Schild analysis demonstrated a noncompetitive mechanism of inhibition. Pharmacophore mapping was used to identify an additional compound, SIB-1893 [(E)-2-methyl-6-(2-phenylethenyl)pyridine], which was also shown to block glutamate-induced increases in [Ca2+]i at hmGluR5 with an IC50of 0.29 μM compared with an IC50 of >100 μM at hmGluR1. SIB-1757 and SIB-1893 showed little or no activity when tested for agonist and antagonist activity at the other recombinant human mGluR subtypes, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, kainate, and N-methyl-d-aspartate receptors. In rat neonatal brain slices, SIB-1757 and SIB-1893 inhibited (S)-3,5-dihydroxyphenylglycine (DHPG)-evoked inositol phosphate accumulation in hippocampus and striatum by 60% to 80%, with a potency similar to that observed on recombinant mGluR5. However, in the cerebellum, a brain region with low mGluR5 expression, SIB-1757 failed to inhibit DHPG-evoked inositol phosphate accumulation. In cultured rat cortical neurons, SIB-1757 and SIB-1893 largely inhibited DHPG-evoked [Ca2+]i signals, revealing a population of neurons that were less sensitive to SIB-1757 and SIB-1893. This is the first description of highly selective, noncompetitive mGluR5 antagonists. These compounds will be useful tools in evaluating the role of mGluR5 in normal physiology and in animal models of disease.
Glutamate is the principal excitatory transmitter in the central nervous system acting through ionotropic glutamate receptors; however, it also plays a major role in activating modulatory pathways through the metabotropic glutamate receptors (mGluRs). Because of their presynaptic, postsynaptic, or perisynaptic localization (Baude et al., 1993;Petralia et al., 1996; Shigemoto et al., 1997), the activation of mGluRs is typically involved in modulating synaptic transmission or neuronal signaling. Only mGluR6, localized to the ON-bipolar cells, postsynaptic to photoreceptors, has been shown to directly mediate synaptic transmission (Masu et al., 1995).
To date, eight mGluRs have been cloned and functionally expressed. These have been classified into three groups based on their amino acid sequence homology (reviewed in Conn and Pin, 1997). Group I mGluRs include mGluR1 and mGluR5 and have been shown to be coupled to stimulation of phospholipase C, resulting in phosphoinositide hydrolysis and elevation of intracellular Ca2+levels ([Ca2+]i). Group I mGluRs have also been shown to modulate ion channels, such as K+ channels (Ikeda et al., 1995), Ca2+ channels (McCool et al., 1998), and nonselective cation channels (Zhou and Hablitz, 1997). Group II (mGluR2 and mGluR3) and group III (mGluR4, mGluR6, mGluR7, and mGluR8) mGluRs have been shown to couple to inhibition of cAMP formation when heterologously expressed in mammalian cells (reviewed in Pin and Duvoisin, 1995). Group II and III mGluRs have also been shown to couple to G protein-activated inward rectifying potassium channels inXenopus oocytes and in unipolar brush cells in the cerebellum (Saugstad et al., 1996; Knoflach and Kemp, 1998). Besides mGluR6, which essentially is expressed only in the retina, the mGluRs are widely expressed throughout the central nervous system.
The two group I mGluR members have somewhat complementary regional expression patterns. mGluR1 is highly expressed in the cerebellum, olfactory bulb, hippocampus, lateral septum, thalamus, globus pallidus, entopeduncular nucleus, ventral pallidum, and substantia nigra (Shigemoto et al., 1992; Petralia et al., 1997). In contrast, mGluR5 shows very little expression in the cerebellum but shows markedly higher levels of expression in the striatum and cortex (Romano et al., 1995). In the hippocampus, mGluR5 is widely and diffusely expressed, whereas mGluR1 in the CA1 is expressed in the stratum oriens and more diffusely in the CA3 and dentate (Blumcke et al., 1996; Lin et al., 1997; Shigemoto et al., 1997). Mutant mice lacking mGluR1 were found to have impaired motor learning as well as deficient long-term depression in cerebellar Purkinje synapses; the defects in the mutant animals are likely related to the absent expression of mGluR1 in the cerebellum (Aiba et al., 1994). In contrast, mutant mice lacking mGluR5 were deficient in spatial learning and in long-term potentiation in the CA1, whereas LTP was normal in the CA3 region (Lu et al., 1997). Excessive activation of group I mGluRs has been implicated in many diseases, and selective group I mGluR antagonists may be of therapeutic benefit in indications such as epilepsy, cerebral ischemia, chronic neurodegeneration, pain, and psychiatric disorders (reviewed inKnöpfel et al., 1995).
Localization and mutant mouse studies have helped in understanding the role of group I mGluRs in the central nervous system; however, without potent and selective antagonists of the group I mGluRs, further understanding of their functional roles is limited. In addition, existing antagonists are competitive inhibitors and have low potency and limited selectivity. We used cell lines that stably express human mGluR1b (hmGluR) and mGluR5a in a high-throughput screening (HTS) system to identify a novel series of potent compounds that are selective for mGluR5 and antagonize the receptor in a noncompetitive manner. These compounds have little or no detectable activity on other mGluRs or ionotropic glutamate receptors. We also demonstrate that they antagonize the (S)-3,5-dihydroxyphenylglycine (DHPG)-evoked inositol phosphate (InsP) turnover in rat brain slices and inhibit DHPG-evoked [Ca2+]isignals in rat cortical neurons. This class of mGluR5-selective antagonists should be valuable tools for understanding the role of mGluR5 in the nervous system.
Materials and Methods
Generation of Stable Cell Lines Expressing Recombinant Glutamate Receptors.
We previously described the generation and characterization of Ltk− cells expressing recombinant hmGluR1b (Lin et al., 1997) and hmGluR5a (Daggett et al., 1995), Chinese hamster ovary cells expressing hmGluR2 (Flor et al., 1995a), hmGluR4 (Flor et al., 1995b), hmGluR6 (Laurie et al., 1997), and hmGluR7 (Flor et al., 1997) and human embryonic kidney 293 cells expressing hNMDAR1A/2A, hNMDAR1A/2B (Varney et al., 1999), and hGluR3i (Varney et al., 1998). In addition, we generated stable cell lines expressing hmGluR3 and hmGluR8; the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors hGluR1-flip (hGluR1i), hGluR2i(Q), hGluR1i/2i(R), and hGluR4i (M. A. Varney, S. D. Hess, and E. C. Johnson, in preparation); and the kainate receptors hGluR5 and hGluR6(I, Y, Q).
HTS Against hmGluR5a/L38-20 Cells.
The activity of compounds was examined at a final concentration of 25 μM against the hmGluR5a receptor stably expressed in mouse fibroblast Ltk− cells (the hmGluR5a/L38-20 cell line). Receptor activity was detected by changes in [Ca2+]i, measured using the fluorescent Ca2+-sensitive dye Fura-2. Briefly, hmGluR5a/L38-20 cells were plated on 96-well plates and loaded with 3 μM Fura-2 for 1 h. Unincorporated dye was washed from the cells, and the cell plate was transferred to a custom-built 96-channel fluorimeter (SIBIA-SAIC, La Jolla, CA), which was integrated into a fully automated plate handling and liquid delivery system. Cells were excited at 350 and 385 nm with a xenon source combined with optical filters. Emitted light was collected from the sample through a dichroic mirror and a 510-nm interference filter and directed into a cooled CCD camera (Princeton Instruments). Image pairs were captured approximately every 1 s, and ratio images were generated after background subtraction. After a basal read of 20 s, an EC80 concentration of glutamate (10 μM) was added to the well, and the response was evaluated for an additional 60 s. The glutamate-evoked increase in [Ca2+]i in the presence of the screening compound was compared with the response of glutamate alone (the positive control) and expressed as a percent of the positive control.
Second Messenger Assays.
InsP assays were performed basically as described previously (Daggett et al., 1995). For native receptor studies, tissue slices were prepared from 7-day-old (neonatal) Sprague-Dawley rats, as described previously (Sacaan et al., 1998).
Measurements of cAMP accumulation in cell lines expressing recombinant mGluRs were performed as described previously (Flor et al., 1995b). For antagonist tests, the following submaximal concentrations of agonist were used: 30 μM (1S,3R)-ACPD for hmGluR2; 1 μM l-AP4 for hmGluR4a, hmGluR6, and hmGluR8a; and 500 μM l-AP4 for hmGluR7b.
[35S]Guanosine 5′-O-(3-thio)triphosphate ([35S]GTPγS) binding assays were performed on membranes prepared from recombinant cells using the methods described by Lazareno et al. (1993). Agonist tests were performed with 50 μg of membrane protein, 5 μM GDP, and test compound. For antagonist tests, EC80 values of glutamate were used (30 μM for hmGluR2, 0.3 μM for hmGluR3, and 5 μM for hmGluR4a).
Primary Cortical Neuronal Cultures.
Cortices were isolated from the brains of embryonic day 16 (E16) Sprague-Dawley rats. Cortical neurons were isolated using the Worthington Papain Dissociation System (Worthington Biochemical Corp., Freehold, NJ) with modification. Briefly, cortices were dissociated in 5 ml of papain dissociation solution by trituration, followed by agitation for 20 min at 37°C. The cell suspension was centrifuged at 300g for 5 min, and the pellet was resuspended in a Deact solution (albumin-ovomucoid inhibitor/DNase solution). This was layered on top of a discontinuous density gradient of albumin-ovomucoid inhibitor mixture and then centrifuged at 70g for 5 min. The cell pellet was resuspended in growth medium [Neurobasal medium (GIBCO, Grand Island, NY) supplemented with 2 mM glutamine, B27 supplement, 100 U/ml penicillin, and 100 μg/ml streptomycin]. Cells were grown on 35-mm coverslips in 6-well plates containing growth medium and fed every 3 to 4 days. Cortical neurons were used for experiments between 15 and 28 days in vitro.
In single-cell imaging measurements, the cortical neurons were assayed on 35-mm coverslips at a density of approximately 8.5 × 105 cells per coverslip, as described previously (Daggett et al., 1998). Imaging data were analyzed in each run by measuring the peak response to the agonist from each cell and scoring the cell as a responder if the maximal signal was more than 3 S.D. above the average signal evoked by perfusion of buffer alone.
Data Analysis and Statistics.
IC50values were calculated from a best fit of the responses to a variable Hill slope using Prism software (GraphPAD, San Diego, CA), and mean values were calculated using log-transformed data (geometric mean) with the lower and upper S.E. values. Statistical differences were determined by a one-way ANOVA followed by Student-Newman-Keuls test. For competitive antagonist studies, the dissociation constant,Kb, was calculated using the Leff-Dougall variant of the Cheng-Prusoff equation (Leff and Dougall, 1993).
Results
Discovery of SIB-1757 [6-Methyl-2-(phenylazo)-3-pyrindol].
A random, small-molecule library was screened against cell lines expressing hmGluR5a (hmGluR5a/L38-20; Daggett et al., 1995) and hmGluR1b (hmGluR1b/L13-23-7; Lin et al., 1997). We previously described the development and validation of the screening assay that, in the case of these cells, uses agonist-induced increases in [Ca2+]i (Veliçelebi et al., 1998). We designed the screening assay to allow for discovery of both agonists and antagonists. Identified agonists (compounds that increased [Ca2+]i) and antagonists (compounds that did not elicit a detectable agonist response but inhibited the response to an EC80concentration of glutamate) were then tested at six concentrations on both hmGluR5a/L38-20 and hmGluR1b/L13-23-7 cells to evaluate potency, efficacy, and subtype selectivity. Subtype-selective and potent compounds were evaluated further against other glutamate receptors expressed in stable cell lines (see Tables1 and 2).
Activity of SIB-1757 and SIB-1893 on recombinant mGluRs
Activity of SIB-1757 and SIB-1893 on recombinant ionotropic glutamate receptors
Using this approach, SIB-1757 (Fig. 1A) was identified as a potent and highly selective inhibitor of the hmGluR5a subtype (Fig. 1B). SIB-1757 inhibited the glutamate-induced [Ca2+]i response in hmGluR5a/L38-20 cells with an IC50 value of 0.37 μM [0.28, 0.50 μM (geometric mean: lower, upper S.E.;n = 4)] compared with a minimal inhibition of hmGluR1b/L13-23-7 cells at concentrations up to 100 μM (i.e., a >270-fold selectivity). The selective inhibition of the [Ca2+]i response in the recombinant hmGluR5a/L38-20 cells by SIB-1757 was validated by measurement of the inhibition of agonist-induced InsP accumulation. SIB-1757 inhibited the quisqualate-induced InsP accumulation in hmGluR5a/L38-20 cells with an IC50 value of 3.1 μM (2.5, 3.8 μM) (Fig. 1C) and gave minimal inhibition of hmGluR1b/L13-23-7 cells at concentrations up to 100 μM, consistent with the results of [Ca2+]i measurements.

SIB-1757 is a mGluR5-selective antagonist of glutamate-stimulated responses identified from high-throughput random screening. A, chemical structure of SIB-1757. B, SIB-1757 selectively antagonized glutamate-induced [Ca2+]iresponses in Fura-2-loaded hmGluR5a/L38-20 cells (○) but not in hmGluR1b/L13-23-7 cells (▪). Glutamate was used at its ∼EC80 value (10 μM for hmGluR5 and 100 μM for hmGluR1b). C, SIB-1757 selectively antagonized the quisqualate-induced InsP accumulation in a dose-dependent manner in hmGluR5a/L38-20 cells (○) but not in hmGluR1b/L13-23-7 cells (▪). Quisqualate was used at its ∼EC80 value (0.3 μM for hmGluR5 and 20 μM for hmGluR1b). Data points represent the mean ± S.E. from three to five experiments and are expressed relative to the response obtained by the reference agonist.
Mechanism of Action of SIB-1757.
We investigated whether SIB-1757 inhibited mGluR5 by a competitive or noncompetitive mechanism by performing a Schild analysis. We generated concentration-response curves to glutamate in the presence of several concentrations of SIB-1757 using [Ca2+]imeasurements in hmGluR5a/L38-20 cells. The results, shown in Fig.2, indicate that the maximal response to glutamate was reduced by the presence of SIB-1757 in a concentration-dependent manner without changing the EC50 value of glutamate. For example, the EC50 value of glutamate alone was 1.5 μM compared with 2.3, 2.0, and 1.3 μM in the presence of 50 nM, 250 nM, and 1 μM SIB-1757, respectively. Thus, SIB-1757 inhibits hmGluR5a by a noncompetitive mechanism.

SIB-1757 antagonized glutamate-stimulated [Ca2+]i responses in hmGluR5a/L38-20 cells in a noncompetitive manner. A Schild analysis, examining the potency of glutamate alone (○), and in the presence of 50 (▴), 250 (▪), and 1000 (♦) nM SIB-1757, was performed to determine the mechanism of antagonism of glutamate-stimulated [Ca2+]iresponses in hmGluR5a/L38-20 cells. The efficacy of glutamate was reduced with increasing concentrations of the SIB-1757, whereas the EC50 value for glutamate was unchanged. Data points represent the mean ± S.D. from a single experiment, performed in quadruplicate and representative of a further experiment. Data points are expressed relative to the [Ca2+]iresponse obtained by 1 mM glutamate in the absence of antagonist.
SAR Studies: Identification of SIB-1893 [(E)-2-Methyl-6-(2-phenylethsenyl)pyridine].
Similarity mapping analyses of virtual libraries of small molecules were performed using UNITY software (Tripos, Inc.). Using specific two- and three-dimensional pharmacophore mapping parameters, we identified analogs that satisfied the spatial and stereoelectronic requirements of the pyridyl and phenyl rings in SIB-1757 but that constrained them within an identical molecular volume. In this manner, a number of analogs were discovered, including the potent and selective hmGluR5 antagonist SIB-1893 (Fig. 3A). SIB-1893 inhibited the glutamate-induced [Ca2+]i response in hmGluR5a/L38-20 cells with an IC50 value of 0.29 μM (0.19, 0.43 μM), with minimal inhibition of hmGluR1b/L13-23-7 cells at concentrations up to 100 μM (i.e., >340-fold selectivity). SIB-1893 inhibited the quisqualate-induced InsP accumulation in hmGluR5a/L38-20 cells with an IC50 value of 2.3 μM (2.1, 2.6 μM) (Fig. 3C), with minimal inhibition at hmGluR1b/L13-23-7 cells at concentrations up to 100 μM.

SIB-1893, a stilbene analog of SIB-1757, is an mGluR5-selective antagonist of glutamate-stimulated responses. A, chemical structure of SIB-1893. B, SIB-1893 selectively antagonized the glutamate-induced [Ca2+]i signals in hmGluR5a/L38-20 cells (○) but not in hmGluR1b/L13-23-7 cells (▪). C, SIB-1893 selectively antagonized the quisqualate-induced InsP accumulation in a dose-dependent manner in hmGluR5a/L38-20 cells (○) with no marked inhibition in hmGluR1b/L13-23-7 cells (▪). Agonists were used at the same concentrations as described in Fig. 1. Data points represent the mean ± S.E. of three or four experiments and are expressed relative to the response obtained by the reference agonist.
Activity of SIB-1757 and SIB-1893 on Recombinant Metabotropic and Ionotropic Glutamate Receptors.
To determine their selectivity, we examined the agonist and antagonist activities of SIB-1757 and SIB-1893 on recombinant glutamate receptors stably expressed in cell lines (results summarized in Tables 1 and 2). The activities of SIB-1757 and SIB-1893 on group II and III mGluRs were determined using forskolin-stimulated cAMP measurements (Fig.4) or [35S]GTPγS binding (Fig.5). At 100 μM, SIB-1757 had minimal or no agonist activity over the forskolin-stimulated cAMP levels. However, SIB-1757 showed general elevations in the forskolin-stimulated cAMP levels in hmGluR4a, hmGluR7b, and hmGluR8a (Fig. 4A). In the [35S]GTPγS binding assay, no agonist activity of SIB-1757 was seen at hmGluR2, hmGluR3, or hmGluR4a (Fig. 5A).

SIB-1757 and its analog, SIB-1893, have minimal activity against group II and III mGluRs examined using forskolin-stimulated cAMP measurements. A, at 100 μM, SIB-1757 and SIB-1893 have minimal agonist activity at group II and III mGluRs, as determined by the lack of attenuation of forskolin-stimulated cAMP levels, except for a weak agonist effect of SIB-1893 at mGluR4a (B). No antagonist activity of SIB-1757 (C) or SIB-1893 (D) was observed on group II or group III mGluRs. For the antagonist test, reference agonists were used at their corresponding ∼EC80 values (see Materials and Methods). Data points were normalized to the potentiation obtained by forskolin alone and represent the mean ± S.D. for three to seven experiments performed in triplicate. Dashed lines show the control cAMP levels, which have been normalized to 100%. Reference antagonists were tested against some of these cell lines and displayed the following activities: at 100 μM, (2S)-α-ethylglutamate gave a 38 ± 4% inhibition of the response to ACPD at hmGluR2, and 100 μM (R,S)-α-methylserine-O-phosphate gave a 64 ± 6% inhibition of the response to l-AP4 at hmGluR4 and 44 ± 5% at hmGluR7.

SIB-1757 and SIB-1893 have minimal agonist (A and C, ■, ▿, and ○) or antagonist (B and D, ▪, ▾, and ●) activity against mGluR2 (■ and ▪), mGluR3 (▿ and ▾), or mGluR4a (○ and ●), determined by glutamate-stimulated [35S]GTPγS binding. For antagonist tests, glutamate was used at the EC80 value for the corresponding receptor (seeMaterials and Methods). Data points were normalized to the potentiation obtained by either an EC100 value (for agonist tests) or EC80 value (for antagonist tests) of glutamate alone and represent the mean ± S.E.M. from three to five experiments performed in triplicate. Reference antagonists were tested against these cell lines and displayed the following activities: At 100 μM, MCPG gave a 26 ± 3% inhibition of the glutamate response at hmGluR2 and 52 ± 5% at hmGluR3, and 100 μM (S)-2-amino-2-methyl-4-phosphonobutanoic acid gave a 58 ± 6% inhibition of the glutamate response at hmGluR4.
SIB-1893, at 100 μM, did not show agonist activity at hmGluR2, hmGluR6, hmGluR7b, or hmGluR8a, but did exhibit some agonist activity at hmGluR4a (Fig. 4, A and B) in the cAMP assay. A modest agonist effect of SIB-1893 was seen in cAMP measurements, with an EC50 value of 26.4 μM (20.7, 33.5 μM) and an efficacy of around 50% of that seen with l-AP4. However, this agonist effect was not seen in the [35S]GTPγS binding assay at concentrations up to 100 μM (Fig. 5C), nor was any agonist activity of SIB-1893 seen in this assay at hmGluR2 or hmGluR3.
The antagonist activity of SIB-1757 and SIB-1893 on group II and III mGluRs was tested against submaximal concentrations of agonists (seeMaterials and Methods). SIB-1757 showed no antagonist activities on any of the group II or group III mGluR members at concentrations up to 100 μM in the cAMP (Fig. 4C) or [35S]GTPγS binding assays (Fig. 5B). Similarly, SIB-1893 showed no antagonist activities at group II or group III mGluRs in either assay (Figs. 4D and 5D). The activity of reference antagonists in these assays is summarized in the legends of Figs. 4 and 5.
The activity of SIB-1757 and SIB-1893 was also examined at recombinant ionotropic glutamate receptors using Ca2+measurements (Table 2). No agonist or antagonist activity of 30 μM SIB-1757 or 30 μM SIB-1893 was seen at the recombinant AMPA (hGluR1, hGluR2, hGluR3, hGluR4, hGluR1/2), kainate (hGluR5, hGluR6), orN-methyl-d-aspartate (NMDA) (hNMDAR1A/2A, 1A/2B) receptor subtypes (Table 2). The activity of reference compounds at recombinant ionotropic glutamate receptors is also summarized in Table 2.
Activity of mGluR5 Antagonists on DHPG-Evoked InsP Accumulation in Rat Brain Regions.
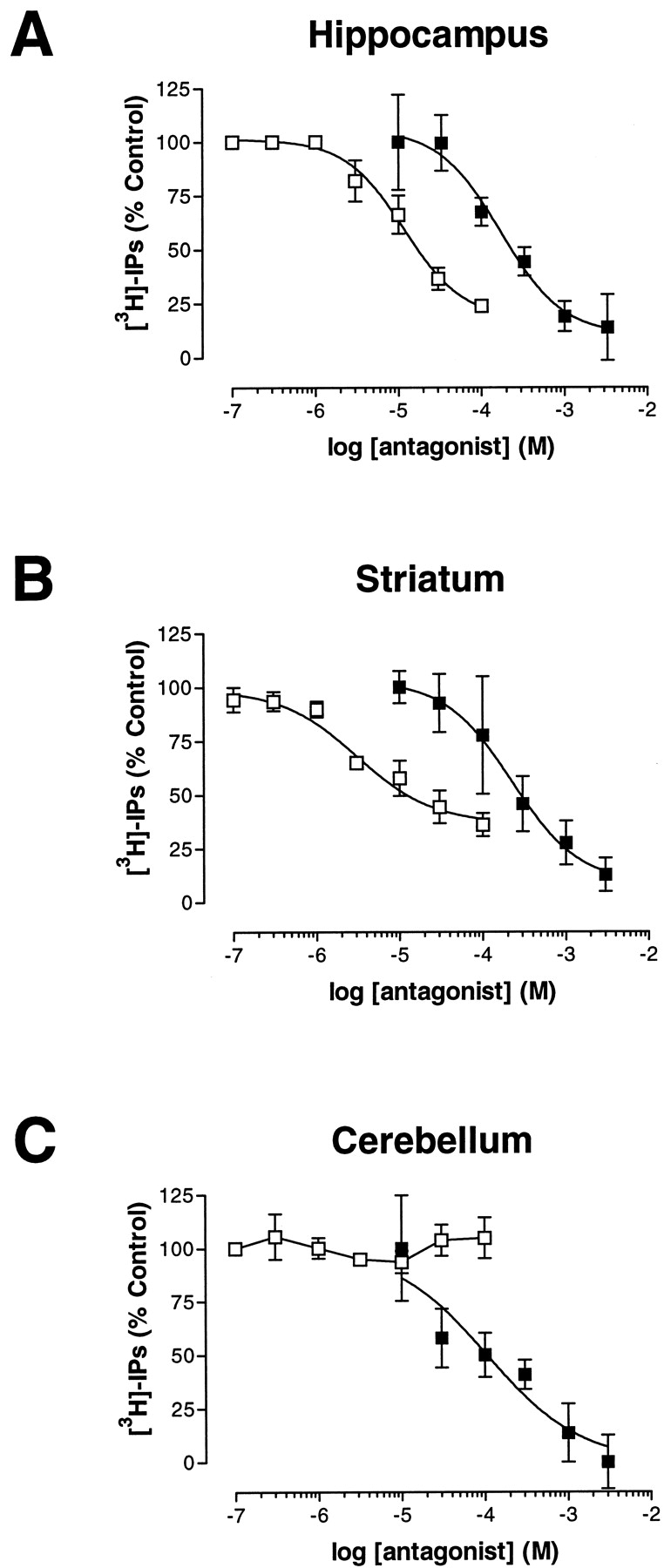
To determine the action of the mGluR5-selective antagonists on native receptors, we investigated the activity of SIB-1757 and SIB-1893 on native mGluRs expressed in the striatum, hippocampus, and cerebellum of the neonatal rat brain. We previously reported the profiling of several metabotropic ligands on the InsP accumulation in these brain regions (Sacaan et al., 1998). SIB-1757 inhibited the stimulation of InsP accumulation by the group I mGluR agonist DHPG in hippocampal and striatal slices with IC50 values of 5.2 μM (2.9, 9.4 μM) and 10.1 μM (4.7, 15.9 μM), respectively. However, the extent of inhibition was not complete because SIB-1757 gave a maximal inhibition of 78.2 ± 1.1% in the hippocampus and 64.3 ± 5.5% in the striatum (Fig. 6, A and B, summarized in Table 3). In contrast to these two regions, SIB-1757 did not significantly inhibit DHPG-stimulated InsP accumulation in cerebellar slices (Fig. 6C, Table 3). We compared the inhibition of DHPG-evoked InsP accumulation in these brain regions by SIB-1757 with the inhibition seen with the nonselective mGluR antagonist (S)-α-methyl-4-carboxyphenylglycine (MCPG; Fig.6, Table 3). MCPG inhibited DHPG-evoked responses to a greater extent than that achieved by SIB-1757, especially in the cerebellum, where SIB-1757 was ineffective (Fig. 6).

SIB-1757 selectively inhibits DHPG-induced InsP accumulation in rat brain regions. Inhibition curves for antagonism of DHPG (10 μM)-induced InsP accumulation by SIB-1757 (■) or MCPG (▪) in slices of neonatal rat hippocampus (A), striatum (B), and cerebellum (C). Data points represent the mean ± S.E. of three independent experiments performed in triplicate. IC50values for SIB-1757 and Kb values for MCPG, together with the extent of inhibition, are reported in Table 3.
Inhibition of DHPG-evoked InsP accumulation in rat brain regions
We also evaluated the potency of SIB-1893 in neonatal brain regions. SIB-1893 produced a concentration-dependent inhibition of DHPG-induced InsP in the hippocampus and striatum, with IC50values of 3.18 (2.44, 4.14) and 2.73 μM (1.92, 3.88 μM), respectively (Fig. 7). Consistent with the results from SIB-1757, SIB-1893 did not completely inhibit DHPG-evoked InsPs in the striatum and hippocampus and failed to inhibit the DHPG-evoked response in the cerebellum at concentrations up to 100 μM (Fig. 7; results summarized in Table 3).

Inhibition of DHPG-induced InsP accumulation in rat brain regions. Inhibition curves for SIB-1893 in the presence of 10 μM DHPG in neonatal rat hippocampus (▪), striatum (○), and cerebellum (▴). Data points represent the mean ± S.D. of three independent experiments run in triplicate. IC50 values for SIB-1893, together with the extent of inhibition, are reported in Table3.
Activity of mGluR5 Antagonists on DHPG-Induced [Ca2+]i Signals in Cultured Rat Cortical Neurons.
To examine the effects of mGluR5-selective antagonists on cortical neurons, we first verified the expression of group I mGluRs in cultured rat cortical neurons by looking at DHPG-evoked [Ca2+]i signals in individual neurons by single-cell Ca2+ imaging. DHPG evoked rapid, robust, and transient increases in [Ca2+]i in primary cultures. Reproducible DHPG-evoked responses were obtained after the application of a 30-s pulse of 100 μM DHPG followed by 3-min washing (Fig. 8A). We investigated the antagonist sensitivity of DHPG-evoked [Ca2+]i signals and found that 10 μM SIB-1757 and 10 μM SIB-1893 inhibited the combined [Ca2+]i signals by 88.0 ± 8.2% and 70.0 ± 5.5%, respectively (Fig. 8, B and C). Furthermore, the inhibition by SIB-1757 and SIB-1893 was fully and rapidly reversible because DHPG-evoked responses were obtained after washout of the antagonist that were comparable to control DHPG-evoked responses (Fig. 8). The responses to DHPG were completely sensitive to MCPG because only 1 of 81 cells responded to 100 μM DHPG in the presence of 3 mM MCPG (data not shown).
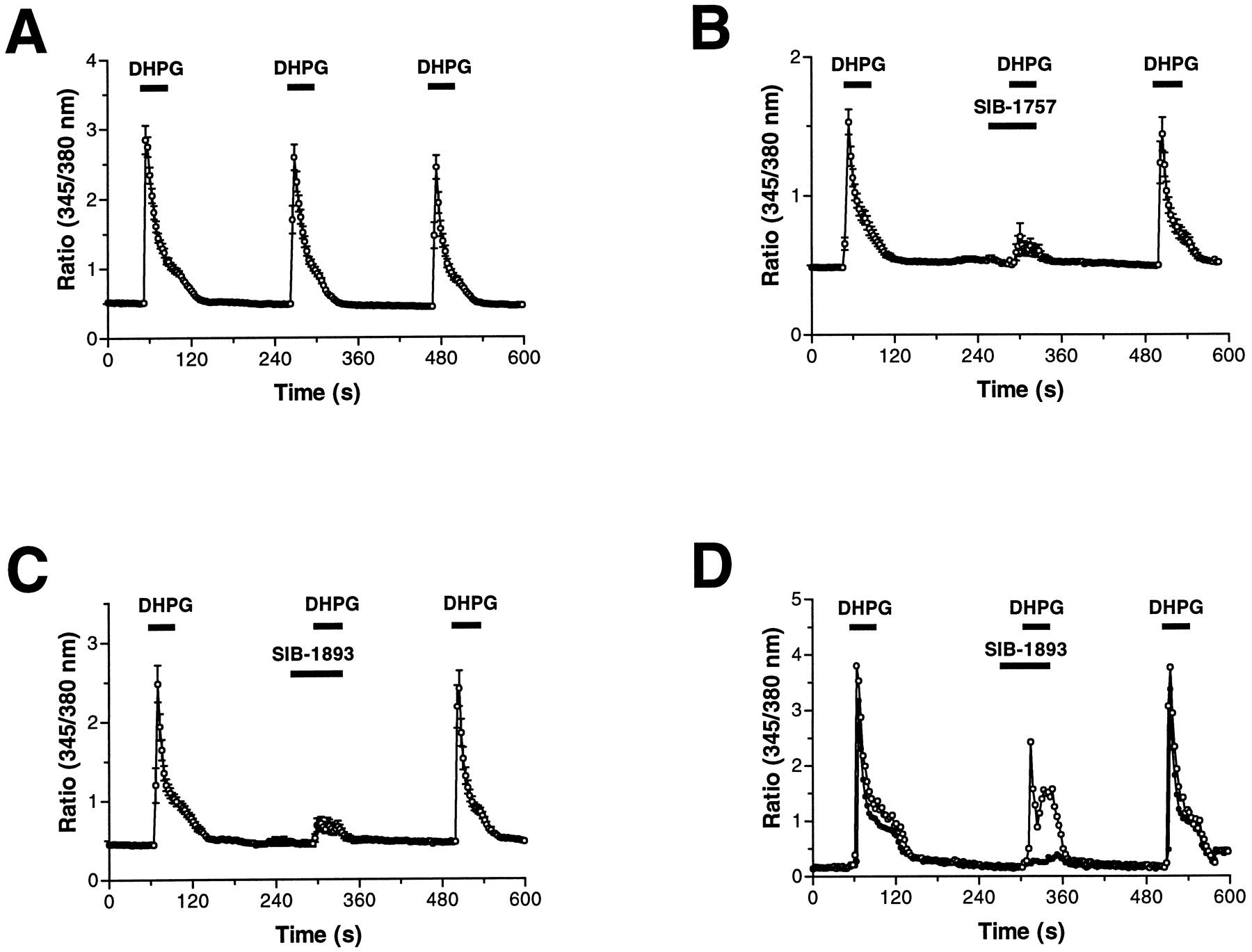
Effect of SIB-1757 and SIB-1893 on DHPG-induced [Ca2+]i responses from individual cultured cortical neurons. [Ca2+]i signals, expressed as an increase in the Fura-2 emission ratio (345/380 nm), are shown in response to a 30-s application of 100 μM DHPG. Either 10 μM SIB-1757 (B) or 10 μM SIB-1893 (C) was added 30 s before DHPG. The data points are mean ± S.D. values from 32 (A), 36 (B), and 22 (C) cells. D, traces from two individual neurons from C, showing the different antagonist sensitivities of neurons in the population.
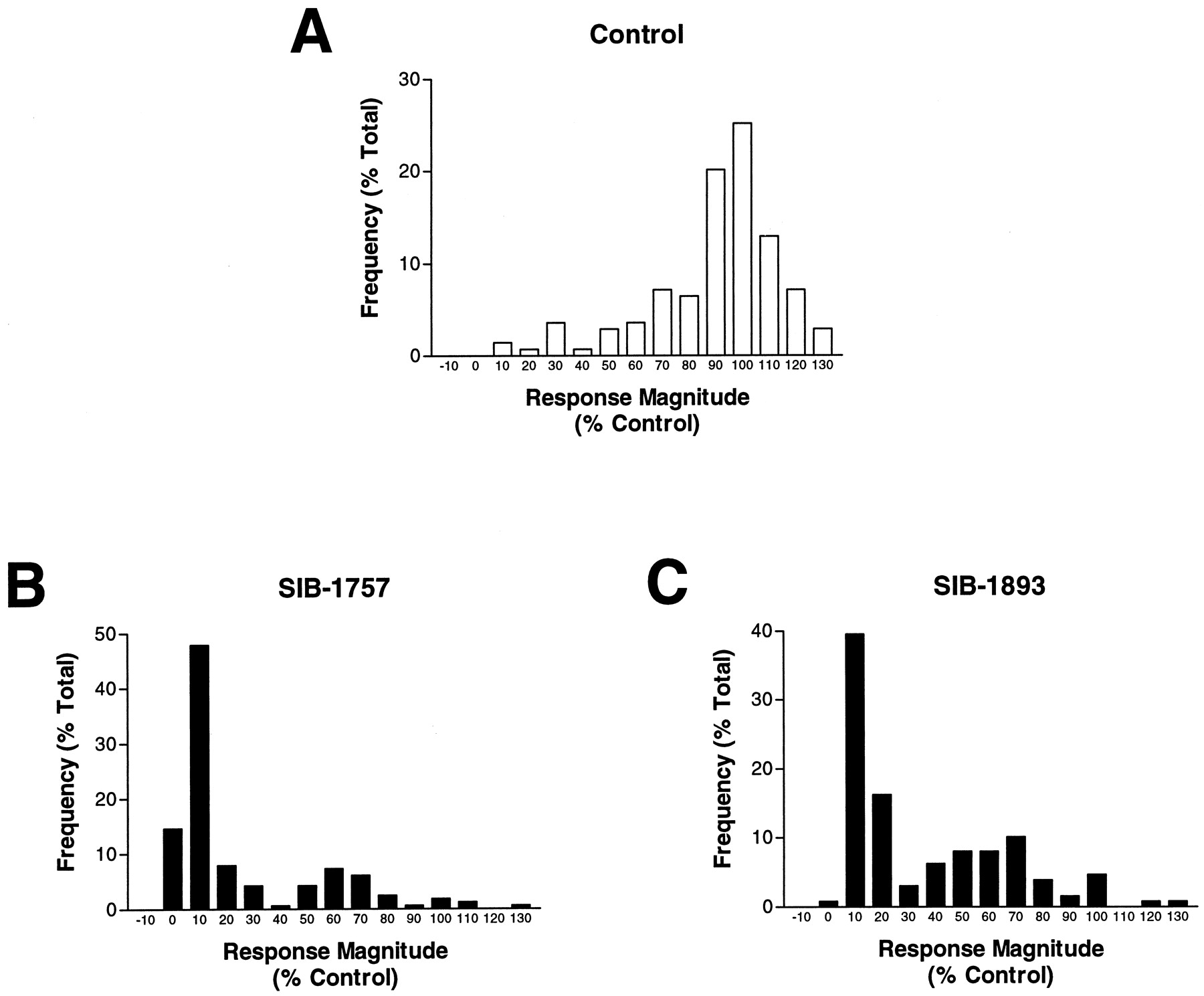
We performed additional analysis of the imaging data from these experiments at a single-cell level (Table4). The application of DHPG alone evoked [Ca2+]i responses (seeMaterials and Methods for definition of responders) in 100% of neurons. Similarly, a second exposure of DHPG evoked responses in 98% of neurons with a magnitude that was 80 ± 37% of the initial response compared on a cell-by-cell basis (Table 4). However, the application of a second DHPG pulse in the presence of SIB-1757 or SIB-1893 reduced not only the number of cells responding to DHPG but also the magnitude of the responses from those cells that still elicited a response to DHPG. We compared the response magnitudes for the second agonist application, expressed as a percent of the first agonist response for each individual neuron, to construct a frequency distribution plot (Fig. 9). The DHPG1/DHPG2 responses were distributed around 100%, indicating the second response was similar in magnitude to the first response. However, DHPG/SIB-1893 frequency distributions were centered around 10%, with a second, smaller population around 50% of the control response. The frequency distribution for the DHPG/SIB-1757 was similar to the DHPG/SIB-1893 distribution (Fig. 9) and suggests the presence of two populations of neurons in the culture. Examples of these two cell populations are shown from the experiment summarized in Fig. 8D, where one neuron is completely sensitive to SIB-1893, and the another in the same microscope field is considerably less sensitive to SIB-1893. Neurons that showed a reduced sensitivity to SIB-1893 were completely inhibited by 3 mM MCPG (data not shown).
Summary of antagonist activities of SIB-1757 and SIB-1893 on cultured cortical neurons

Frequency histograms of DHPG-evoked [Ca2+]i signals in rat cultured cortical neurons demonstrate a major population of neurons are fully antagonized by the mGluR5-selective antagonists SIB-1757 or SIB-1893. Responses to 100 μM DHPG were measured in each neuron alone, and after 3-min washing, the neurons were rechallenged with DHPG in the absence (A) or presence of SIB-1757 (B) or SIB-1893 (C). The data are expressed as the percent of the second response magnitude, relative to the first DHPG response. The frequency histograms use a 10% bin range. The total number of neurons used for the analysis was 123 (A), 128 (B), and 147 (C).
Discussion
The most characterized series of compounds that discriminate between the eight mGluR subtypes are the phenylglycine derivatives. This class of compounds possesses a wide spectrum of activity on mGluRs, from agonist to antagonist activity (reviewed by Watkins and Collingridge, 1994). More recently, this series of compounds is starting to yield moderately potent ligands, some of which are able to discriminate among mGluR group members. As yet, there are no known competitive antagonists that are able to discriminate between individual mGluR members.
We used an HTS system to search for potential novel hmGluR5-selective compounds. Our approach was first to clone the hmGluR5 gene, stably express the cDNA in a recombinant cell line (Daggett et al., 1995), and set up a functional assay to monitor receptor activity. The group I mGluRs couple robustly to phospholipase C, resulting in the generation of InsPs and an increase in [Ca2+]i. We developed a fluorescence detection HTS system that is capable of simultaneously measuring [Ca2+]i from all 96 wells of a microtiter plate (Veliçelebi et al., 1998). This system is rapid, it evaluates the agonist and antagonist activity of compounds in the same assay, and it is a functional assay, measuring receptor activation rather than ligand affinity, as traditionally measured in ligand binding. We can therefore detect competitive and noncompetitive interactions, unlike radioligand binding assays.
A random, small molecule library was tested using the HTS assay, leading to the identification of SIB-1757. This relatively simple, low-molecular-mass (MW = 213) compound potently inhibited glutamate-evoked [Ca2+]isignals in hmGluR5-expressing cells with remarkable selectivity over hmGluR1. Measurement of glutamate-stimulated InsP accumulation in the same cell lines confirmed the high degree of selectivity for SIB-1757 at hmGluR5 over hmGluR1. However, the potencies of SIB-1757 and SIB-1893 in the InsP assay were about 10-fold lower than those in the Ca2+ assay. The reason for these differences in antagonist potency is not clear, but they may be due to assay differences. Nevertheless, we observed the same potency in the InsP measurements between recombinant hmGluR5 and hippocampal and striatal rat brain slices.
We examined the selectivity of SIB-1757 and SIB-1893 at members of the mGluR family, in addition to many of the ionotropic glutamate receptors. Both SIB-1757 and SIB-1893 showed essentially no cross-reactivity at the AMPA, kainate, and NMDA ionotropic glutamate receptors. Furthermore, SIB-1757 and SIB-1893 exhibited minimal agonist or antagonist effects at group II and III mGluRs. SIB-1893 exhibited weak agonist activity at mGluR4 in cAMP measurements, in terms of both efficacy and potency. However, further examination of SIB-1893 in a second functional assay, the [35S]GTPγS binding assay, did not confirm this activity. We are unclear why these two assays might detect different activities of SIB-1893; the potency and efficacy of all reference agonists and antagonists do not differ between these two assays. Nevertheless, both mGluR5 antagonists show a high degree of selectivity for mGluR5 over all glutamate receptor subtypes examined so far. This is in contrast to the current series of competitive mGluR antagonists based on the phenylglycine backbone, some members of which cross-react with AMPA and NMDA receptors (Contractor et al., 1998).
Schild analysis indicated that the mechanism of inhibition of hmGluR5 by SIB-1757 was noncompetitive. The concentration-response curves to glutamate were not shifted in the presence of the antagonist, but the maximal responses to glutamate were reduced. This is the second reported example of a noncompetitive, selective inhibitor of mGluRs (Litschig et al., 1999). At present, we are unclear of the exact location of the binding site for SIB-1757/SIB-1893, although a feasible approach might involve the use of chimeric hmGluR1/5 receptors to identify the residues involved in this antagonism. The identification of a modulatory site on mGluR5 raises the question of whether there are endogenous regulatory molecules that interact with this site. There is some precedence for this type of modulation with other receptors: the 5-hydroxytryptamine1B/1D G protein-coupled receptor is noncompetitively antagonized by the endogenous peptide 5-hydroxytryptamine moduline (Massot et al., 1996), and the nicotinic acetylcholine receptor channel is noncompetitively antagonized by steroids, 5-hydroxytryptamine, and substance P (reviewed by Arias, 1998).
The stable cell lines that we have established express the human recombinant forms of mGluR5. Although human and rat sequences are very similar (95.4% from the deduced amino acid sequences; Daggett et al., 1995), work on other receptor families has indicated species homolog differences even with this level of sequence similarity (Hall et al., 1993). For example, the selective NK-1 antagonist CP96345 demonstrates a 100-fold higher affinity at the human NK-1 receptor compared with its affinity for the rat NK-1 receptor, despite the 94% sequence homology between the two receptors (Fong et al., 1992). Because of the potential species homolog differences, we examined the activity of SIB-1757 and SIB-1893 in two tissue preparations: neonate rat brain slices taken from the hippocampus, striatum, and cerebellum, in which receptor activation was measured by InsP accumulation, and cortical neurons prepared from rat embryonic tissue, where receptor activity was determined by single-cell Ca2+ imaging.
In the InsP assay, SIB-1757 and SIB-1893 inhibited DHPG-evoked InsP accumulation in hippocampal and striatal slices with potencies similar to those observed at the human mGluR5, confirming the antagonist activity of these compounds on rat mGluRs. Two pieces of evidence suggest that the potency and selectivity of SIB-1757 and SIB-1893 for mGluR5 over mGluR1 are retained at rat receptors. First, because the DHPG-evoked InsP accumulation in rat brain is due to activation of mGluR1 and mGluR5, the incomplete inhibition of DHPG-induced InsP accumulation in these two regions likely reflects a selective inhibition of mGluR5, leaving an mGluR1-insensitive component. In addition, the potency of the inhibition of InsP accumulation in the striatum and the hippocampus is similar to that observed in hmGluR5a/L38-20 cells. Consistent with this, MCPG, a nonselective mGluR antagonist, completely inhibited DHPG-evoked InsP accumulation. Furthermore, SIB-1757 and SIB-1893 failed to inhibit DHPG-induced InsP accumulation in the cerebellum, which was completely inhibited by MCPG. These results are consistent with the known expression pattern of mGluR5 and mGluR1. In neonatal and adult rat brain regions, results from quantitative Western blotting and immunocytochemistry indicate that mGluR5 is highly expressed in the striatum and hippocampus, with much lower levels expressed in the cerebellum (Romano et al., 1995). Conversely, the expression of mGluR1, determined by in situ hybridization (Shigemoto et al., 1992) and immunocytochemistry (Martin et al., 1992), confirms a high level of mGluR1 expression in the cerebellum and lower levels of expression in the hippocampus and striatum. Therefore, the activities of SIB-1757 and SIB-1893 on native rat mGluRs are supportive of their mGluR5 selectivity and indicate that these antagonists possess a similar potency at rat and human recombinant mGluR5.
Additional studies were performed with SIB-1757 and SIB-1893 on cultured rat cortical neurons. In single-cell Ca2+ imaging, DHPG evoked a rapid and robust increase in [Ca2+]i in almost all neurons. The repeated application of DHPG, interspersed with washing, gave reproducible Ca2+ signals. SIB-1757 and SIB-1893 inhibited the DHPG-evoked [Ca2+]i signals to a large degree, suggesting that these DHPG-evoked responses were largely mediated by mGluR5. However, further investigation of the single-cell Ca2+ imaging data indicated that there was a differential sensitivity of the neurons to the antagonists. Frequency histograms, generated from the sensitivity to SIB-1757 and SIB-1893, suggested the presence of two neuronal populations: in one population, the DHPG-evoked responses were inhibited by around 90%, and the second population was inhibited by around 50%. DHPG-evoked responses in both populations were completely inhibited by MCPG. These findings suggest a differential expression of mGluR5 and mGluR1 in these two neuronal populations and are consistent with the high level of mGluR5 and low level of mGluR1 expression in rat cortical neurons reported by others (Bruno et al., 1995). These single-cell Ca2+imaging studies also confirmed the reversibility of SIB-1757 and SIB-1893 because a brief washing period was sufficient to fully restore the DHPG-evoked Ca2+ responses to control levels.
In summary, we identified a series of compounds, represented by SIB-1757 and SIB-1893, that have a selectivity unprecedented for mGluRs; this degree of selectivity may be due, at least in part, to its noncompetitive mechanism. These compounds are likely acting outside of the ligand-binding domain in regions of high sequence divergence relative to other mGluRs. In contrast, competitive compounds act at the highly conserved ligand domain and thus have a lower likelihood of the high selectivity observed with the present compound series. Because these antagonists are noncompetitive, they have the advantage of antagonizing the receptor even in the presence of high levels of glutamate that may be present in the diseased state. The excessive activation of mGluR5 has been implicated in many diseases, and a selective antagonist may be of therapeutic benefit in indications such as epilepsy, cerebral ischemia, chronic neurodegeneration, pain, and psychiatric disorders (reviewed in Knöpfel et al., 1995). SIB-1757 and SIB-1893 will be important tools in determining the role of mGluR5 in animal models of these disorders.
Acknowledgments
We thank K. Lariosa, M. Akong, and R. Siegel for technical assistance. We also acknowledge K. Stauderman, M. Harpold, I. McDonald, W. Comer, and K. Lloyd for valuable input throughout this research.
Footnotes
-
Send reprint requests to: Mark Varney, Ph.D., SIBIA Neurosciences, Inc., 505 Coast Boulevard South, Suite 300, La Jolla, CA 92037. E-mail:mvarney{at}sibia.com
- Abbreviations:
- mGluR
- metabotropic glutamate receptors
- [Ca2+]i
- intracellular Ca2+
- GTPγS
- guanosine-5′-O-(3-thio)triphosphate
- HTS
- high-throughput screening
- DHPG
- (S)-3,5-dihydroxyphenylglycine
- InsP
- inositol phosphate
- hmGluR
- human metabotropic glutamate receptor
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- MCPG
- (S)-α-methyl-4-carboxyphenylglycine
- SIB-1757
- 6-methyl-2-(phenylazo)-3-pyridinol
- SIB-1893
- (E)-2-methyl-6-(2-phenylethenyl)pyridine
- Received January 26, 1999.
- Accepted March 18, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}