


Abstract
We have cloned the human ionotropic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor GluR3 flip splice variant (hGluR3i) and developed a stable cell line expressing this receptor in HEK293 cells. Electrophysiological recordings demonstrated that glutamate-evoked currents desensitize rapidly, with a mean desensitization time constant of 5.4 ms. Robust glutamate-evoked increases in intracellular Ca++ ([Ca++]i) were observed in the presence of cyclothiazide, which attenuated receptor desensitization. [Ca++]i measurements were used to perform a detailed pharmacological characterization of hGluR3i with reference agonists and antagonists. The results of these studies showed that kainate and domoate were not fully efficacious agonists relative to glutamate. The binding affinities of agonists and competitive antagonists were determined in a [3H]AMPA competition binding assay. There was a good correlation between the functional data and the binding affinities obtained for competitive antagonists. However, the binding affinities of the agonists did not correlate with their functional EC50 values from [Ca++]i data, possibly because the binding assay predominantly measures the desensitized high-affinity state of the receptor. [3H]AMPA binding also was performed on membranes prepared from rat forebrain, and comparison of the data from HEK293 cells expressing hGluR3i and rat forebrain suggest that nearly all of the reference compounds show similar binding activities between the two membrane preparations, with the exception of fluoro-willardiine, kainate and 6-nitro-7-sulfamoylbenzo(f)quinoxaline-2–3-dione (NBQX). These data suggest that cells stably expressing recombinant hGluR3irepresent pharmacologically valid experimental systems to study human AMPA receptors.
Ionotropic glutamate receptors typically are classified into NMDA and non-NMDA receptor families. Molecular biological studies have identified nine non-NMDA receptor genes (reviewed by Bettler and Mulle, 1995), separated into the AMPA-preferring receptors (GluR1–4 or GluRA-D) and the kainate-preferring receptors (GluR5–7, KA1 and KA2). Studies with recombinant receptors have demonstrated that AMPA receptor subunits can assemble as homomeric and heteromeric receptors with distinct functional properties (Boulter et al., 1990; Keinänenet al., 1990). In addition to subunit composition, the properties of AMPA receptors also are affected by RNA editing events. All four members of the AMPA receptor family can undergo alternative splicing involving 38 amino acids at the flip/flop region (Sommeret al., 1990); in addition, GluR2, GluR3 and GluR4 can undergo an R/G edit near the flip/flop region (Lomeli et al., 1994); and finally, GluR2 can be edited at the Q/R site (Sommer et al., 1991), which dramatically impacts the Ca++ permeability of the receptor. Native AMPA receptors exhibit various degrees of Ca++permeability, depending on the neuronal preparations examined. For example, activation of AMPA receptors in Type II-hippocampal, neocortical and hypothalamic neurons, Purkinje cells and Bergmann glia show appreciable Ca++ permeability (reviewed byFletcher and Lodge, 1996). However, brain cell types expressing GluR2(R), such as CA3 pyramidal neurons, dentate gyrus granule cells and Hilar mossy cells, tend to have much lower Ca++ permeability following AMPA receptor stimulation (Geiger et al., 1995), consistent with the low Ca++ permeability observed for recombinant heteromeric AMPA receptors containing GluR2(R) (Hollmann et al., 1991; Burnashev et al., 1992).
AMPA receptors are thought to mediate the majority of fast synaptic transmissions in the mammalian central nervous system, and therefore play an important role in normal physiological functions. However, interest in the role of ionotropic receptors in pathophysiology such as ischemic damage has shifted the focus from NMDA receptors to AMPA receptors, subsequent to the findings that, in rodent models of global ischemia, AMPA receptor-selective antagonists are more effective neuroprotectants than NMDA receptor antagonists (reviewed by Gill, 1994). Many of these in vivo studies have been performed with the quinoxalinedione chemical series of AMPA receptor antagonists, such as NBQX, DNQX and CNQX. These compounds have adverse effects on memory, motor activity and autonomic functions, and poor solubility leading to nephrotoxicity. It is unclear whether inhibition of certain AMPA receptor subtypes mediates these adverse effects, or if they are caused by the non-AMPA receptor-mediated effects of quinoxalinediones. Therefore, the identification of new chemical classes of AMPA receptor antagonists, especially those that exhibit AMPA receptor subtype selectivity, is needed to discriminate between these possibilities.
The cloning of human genes encoding GluR1, GluR2 and GluR3 has been reported (Puckett et al., 1991; Sun et al., 1994;Rampersad et al., 1994). Functional data have only been presented for human GluR1 and GluR2 (Sun et al., 1994), but these studies have not been performed in sufficient detail to compare the properties of the human receptor with those of other species. Detailed characterization of human AMPA receptors is necessary, because even homologous receptors between species that have only minor amino acid changes may result in different pharmacological properties (Hallet al., 1994). In this study we have cloned the human AMPA receptor subtype GluR3-flip (hGluR3i) and have developed a stable cell line that expresses this receptor in HEK293 cells. We have investigated the biochemical, electrophysiological and pharmacological properties of recombinant hGluR3iand compared the pharmacology of reference agonists and antagonists by use of radioligand binding, intracellular Ca++([Ca++]i) measurements and whole-cell recordings.
Methods
cDNA cloning and construction of full-length human GluR3i cDNA.
A recombinant cDNA library was prepared with poly(A)+ RNA from a human hippocampus (Ellis et al., 1988) and probed with the cDNA encoding a full-length rat GluR3i (supplied by Dr. Stephen Heinemann-Salk Institute, San Diego, CA). One of the cDNAs (EAA8) encoded the hGluR3i sequence spanning from nt −97 to 1718 and from nt 1797 to 2923, but lacked a 78-bp exon (nt 1719–1796) and contained a 27-bp intron. An 868-bp fragment was amplified in a polymerase chain reaction from a human fetal brain cDNA library with oligonucleotide primers which flanked the improperly spliced region. The 778-bp fragment resulting fromClaI/BclI digestion was subcloned into the EAA8 cDNA to make the full-length human construct in the mammalian expression vector pCMV-T7–3-(-SD/SA)-hGluR3i. The pCMV-T7–3 expression vector was modified from the pCMV-β (Clontech, Palo Alto, CA) vector as described in Daggett et al. (1995).
Stable transfection of hGluR3i in HEK293 cells.
One day before transfection, 106HEK293 cells were plated in Dulbecco’s Modified Eagle’s Medium containing 6% bovine calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin. The cells were transfected with 20 μg of pCMV-T7–3-(-SD/SA)-hGluR3i and 2 μg of pSV2neo by the calcium phosphate precipitation method (Kingston, 1996). Two days later, the cells were placed under antibiotic selection by adding 0.5 mg/ml G418.
Transient transfection of pCMV-hGluR3i in HEK293 cells.
For transient transfections, 2 × 106 HEK293 cells were transfected with 5 μg of pCMV-hGluR3i, 2 μg pCMVβ-gal (Clontech, Palo Alto, CA) and 13 μg of pUC19 by the calcium phosphate method (Kingston, 1996). Transfection efficiency was determined by histochemical staining of β-galactosidase activity and was usually between 50 and 80%.
Measurement of hGluR3 immunoreactivity.
Approximately 0.75 mg of membranes from HEK293 cells stably or transiently transfected with hGluR3i were solubilized in 500 μl RIPA buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.5% deoxycholate, 1% NP-40, 0.1% sodium dodecyl sulfate) containing 100 μM phenylmethylsulfonyl fluoride, 1 μM calpeptin, 1 μM leupeptin and 1 μM pepstatin (Sheng et al., 1994; Varney et al., 1996). The particulate matter was removed by centrifugation at 13,000 × g. Membrane proteins were separated by electrophoresis through an 8 to 16% Tris-glycine polyacrylamide gel (Novex, San Diego, CA), and electrophoretically transferred to nitrocellulose membranes. The membranes were incubated with a 1:100 dilution of the anti-rat GluR2/3 antibody (Chemicon, Temecula, CA) followed by a 1:1000 dilution of horseradish peroxidase-conjugated donkey anti-rabbit IgG (Amersham, Arlington Heights, IL) and visualized by use of an enhanced chemiluminescence system (KPL, Gaithersburg, MD).
Intracellular [Ca++]imeasurements.
Cells were plated in 96-well plates at approximately 1.0 to 2.0 × 105 cells per well. Cells were loaded for 1 hr at 20°C with 10 μM fluo-3/AM or 3 μM fura-2/AM in HEPES-buffered saline (HBS, composition, in mM: NaCl, 125; KCl, 5; MgSO4, 0.62; CaCl2, 1.8; HEPES, 20; glucose, 6, pH 7.4) as described previously (Varney et al., 1996). Unincorporated dye was washed from the cells, and [Ca++]i measurements were performed in the presence of 100 μM cyclothiazide. For experiments with fluo-3, fluorescence levels were measured by a 96-well plate-reading fluorimeter (Cambridge Technical Instruments, Inc., Watertown, MA). Ten basal fluorescence readings were performed before the agonist was added in a one-tenth volume directly in the well with a pipette or a Digiflex automatic injector (ICN Flow), and a further 190 fluorescence readings were taken. In the antagonist studies, 30 μM glutamate (an approximate EC75 concentration) was added 2 to 5 min after the antagonist. Calibrated [Ca++]i levels were calculated for each well from Fmax andFmin values determined as described previously (Varney et al., 1996). For [Ca++]i measurements with fura-2, 350:385 nm fluorescence ratios were determined in a 96-channel fluorimeter (SIBIA-SAIC, La Jolla, CA), and [Ca++]i levels were calculated according to Grynkiewicz et al. (1985) with predetermined values for Rmin andRmax.
Electrophysiological recordings.
Currents were recorded with the whole-cell mode of the patch-clamp technique. Pipettes with resistances of 1.1 to 2.7 megohm were filled with (in mM): CsCl, 135; MgCl2, 1.0; EGTA, 10; HEPES, 10; pH 7.4. For current-voltage relationships, 100 μM spermine was also included in the pipette buffer. The external solution was mammalian Ringer’s, which contained (in mM): NaCl, 160; KCl, 5.0; MgCl2, 1.0; CaCl2, 2.0; HEPES, 5.0; glucose, 11; MgATP, 4.0; pH 7.3. Currents were measured with an Axopatch-1C amplifier, filtered at 1 KHz and sampled at 2.5 KHz with PClamp software and hardware (Axon Instruments, Foster City, CA). Agonists were applied using a 12–1 quartz array manifold (ALA, Inc., Westbury, NY) under manual control, positioned within 50 μm of the cell. For analysis of current-voltage properties, cells were held at −60 mV, and the membrane potential was stepped in 10-mV increments from −90 to 60 mV during repeated rapid applications of 1 mM glutamate by a piezo-electric application device.
Binding of [3H]AMPA to rat forebrain and HEK293 cells expressing hGluR3i.
Membranes were prepared essentially according to the methods described by Chazot et al. (1992), with minor modifications in buffer composition. Cells were removed from plates or flasks, resuspended in 50 mM Tris-citrate buffer, pH 7.0 and homogenized by use of a glass/Teflon homogenizer. The homogenate was centrifuged at 90,000 × g for 30 min at 4°C, and the pellet was resuspended in the same buffer by brief bursts with a Polytron homogenizer. This final membrane suspension was stored at −70°C. Rat forebrains were homogenized in 0.32 M sucrose and centrifuged at 800 ×g at 4°C for 20 min. The supernatant fraction was centrifuged at 54,000 × g for 20 min at 4°C, and the pellet was resuspended in cold (4°C) nanopure water and recentrifuged at 54,000 × g for 20 min at 4°C. The pellet was suspended in 50 mM Tris-citrate, pH 7.0, and centrifuged again at 54,000 × g for 20 min at 4°C, and the final pellet was resuspended in Tris-citrate buffer and stored at −70°C.
On the day of the binding assay, membranes were thawed and washed in assay buffer, which contained (in mM): HEPES, 50; EDTA, 1.0; potassium thiocyanate, 100; pH 7.2. The final washed pellet was resuspended at a protein concentration of 2 mg/ml. Protein concentrations were determined by the method of Lowry et al. (1951) with bovine serum albumin as the protein standard.
The binding assay was performed in deep-well 96-well plates with 300 μg membrane protein, 5 nM [3H]-(S)-AMPA and test compounds in a total volume of 500 μl for 75 min at 4°C. Nonspecific binding was determined in the presence of 1 mM glutamate. The experiment was terminated by filtration through Whatman GF/C filters with a 48-well Brandell cell harvester, followed by rapid washing three times. The filters were transferred to scintillation vials, 5 ml of scintillation fluid was added and the bound radioactivity in each sample was determined.
Data analysis and statistics.
EC50 and IC50 values were calculated from a best fit of the responses to a variable Hill slope with Prism software (version 2), and mean values were calculated with log-transformed data (geometric mean) with the lower and upper standard error (S.E.) values. For functional antagonist studies, IC50 values generated for competitive antagonists were expressed as the dissociation constants (Kb values) derived from the Leff-Dougall (Leff and Dougall, 1993) variant of the Cheng-Prusoff equation: Kb = IC50/(2 + ([A]/[A50])n)1/n− 1, where A is the agonist used, andA50 is the EC50 value for the agonist. For competition binding studies, IC50 values were converted toKi values by use of the Cheng-Prusoff equation. Statistics were performed on log-transformed data and subjected to a non-paired Student’s t test, or ANOVA and Student-Newman-Keuls test.
Materials.
[3H]-(S)-AMPA (specific activity, 45 Ci/mmol), (S)-AMPA, CNQX, (S)-5-fluorowillardiine, NBQX, NMDA, domoate and GAMS were obtained from Tocris Cookson (Bristol, U.K.). Glutamate, kainic acid, quisqualate, cyclothiazide, DNQX, GYKI-52466, NS102, NS257 and AMOA were obtained from Research Biochemicals International (Natick, MA). Fluo-3/AM and fura-2/AM were from Molecular Probes, Inc. (Eugene, OR). All other chemicals were reagent grade.
Results
Cloning and the Deduced Amino Acid Sequence of hGluR3i
A full-length hGluR3i cDNA was constructed with a human cDNA isolated from an adult hippocampal cDNA library and a cDNA fragment amplified from a human fetal brain cDNA library by polymerase chain reaction. The full-length hGluR3i cDNA encodes 894 amino acids, and the overall deduced amino acid sequence identity between the human and rat GluR3i is 99.4% (five amino acid changes;Keinänen et al., 1990; Boulter et al., 1990; Nakanishi et al., 1990). The putative human signal peptide is six amino acids longer than the rat sequence. The deduced amino acid sequence of the mature protein for the hGluR3i reported here is 99.7% identical with the hGluR3i sequence reported by Rampersadet al. (1994), with the following differences (Rampersadet al. sequence → our sequence): Leu525→Phe,Gly775→Arg, andSer854→Phe. (The underlined residues are identical with those reported for the rat sequence.) TheGly775→Arg difference between the two human sequences is the result of the R/G RNA editing event that occurs in GluR2, GluR3 and GluR4 cDNAs (Lomeli et al., 1994). Adopting the membrane topography proposed for rat GluR3 (Bennett and Dingledine 1995), all five putativeN-glycosylation sites and all four putative protein kinase C phosphorylation sites are conserved between rat and human proteins.
Establishment of Stable Cell Lines Expressing hGluR3i
Clones expressing hGluR3i were identified by the magnitude of the glutamate-evoked [Ca++]i signals in the presence of cyclothiazide. Responding clones were subcloned by limiting dilution and selected using the same procedure. Clone hGluR3i/HEK69–8 was chosen for further characterization based on the magnitude of glutamate-evoked [Ca++]i signals, typically in the range of 400 to 800 nM, and the stability of the glutamate-evoked responses over at least 40 cell passages, equivalent to approximately 20 weeks in culture (data not shown).
The stable expression of hGluR3i also was confirmed by immunoblotting experiments. A 110-kdalton immunoreactive species was detected in membranes prepared from the hGluR3i/HEK69–8 clone (fig.1, lane 1) and from cells transiently transfected with hGluR3i (lane 2) with an anti-rat GluR2/3 antibody. This immunoreactive species was not detected in membrane protein isolated from cells transfected with the control plasmid pCMV(−SD/SA) (lane 3, “control vector”). The expected size for an unglycosylated, monomeric hGluR3ipolypeptide is 102 kdaltons, and therefore the 110-kdalton immunoreactive species probably represents a glycosylated form of the recombinant hGluR3i receptor (Blackstone et al., 1992).

Immunochemical analysis of recombinant hGluR3i stably expressed in HEK69–8 cells (lane 1) and transiently expressed in HEK293 cells (lane 2). HEK293 cells also were transiently transfected with control vector (lane 3). Cell membranes were immunoprecipitated with an anti-GluR2/3 antibody, immunoblotted and probed with the same antibody, and detected by enhanced chemiluminescence.
Measurement of [Ca++]i
Agonist pharmacology of hGluR3i.
In the presence of 10 μM or 100 μM cyclothiazide (CTZ), 1 mM glutamate evoked a robust and rapid increase in [Ca++]i in hGluR3i/HEK69–8 cells (fig.2A). However, in the same cells we were unable to detect a glutamate-evoked change in [Ca++]i in the absence of CTZ (fig. 2A), most likely because of the rapid desensitization of the hGluR3i receptor. CTZ potentiated the AMPA- and kainate-evoked increases in [Ca++]i with similar potencies (fig. 2B), with EC50 values of 9.0 μM (7.8, 10.3; n = 3) and 6.1 μM (4.9, 7.5;n = 3), respectively [mean (lower, upper S.E.)]. These results are consistent with the reported potency of CTZ on a recombinant rat GluR1i splice variant (Partinet al., 1994). The glutamate-evoked [Ca++]i responses were unaffected by pretreatment with 1 μM nimodipine (data not shown), which suggests that the increase in [Ca++]i was caused by Ca++ entry through the hGluR3i receptor rather than indirectly through activation of endogenous voltage-gated Ca++channels that may be expressed in HEK293 cells (Berjukow et al., 1996).

[Ca++]i measurements in hGluR3i/HEK69–8 cells. (A) Kinetic traces of glutamate-evoked Ca++ signals in fluo-3-loaded HEK69–8 cells. The presence of 1 mM glutamate is shown by the striped bar. Cyclothiazide was added 2 min before assay. Data are from a single experiment and represent the mean from four replicate wells. (B) Concentration-response curves to cyclothiazide, obtained with 1 mM AMPA- and 1 mM kainate-evoked Ca++ responses in fluo-3-loaded hGluR3i/HEK69–8 cells. Cyclothiazide was added 2 min before assay. Data points represent the mean ± S.E. from three separate experiments, and are normalized in each experiment to the response obtained by 1 mM glutamate.
We used [Ca++]imeasurements to characterize the pharmacological properties of hGluR3i in the presence of 100 μM CTZ. Glutamate, AMPA, kainate, quisqualate, domoate and fluorowillardiine evoked concentration-dependent increases in [Ca++]i, albeit with different absolute efficacies (fig. 3A). The efficacies and EC50 values of agonists at hGluR3i are summarized in table1. Comparison of the geometric means by ANOVA and Student-Newman-Keuls test revealed the following rank order of potency: quisqualate > domoate ≥ kainate ≥ fluorowillardiine ≥ AMPA ≥ glutamate. The maximal [Ca++] signals (efficacy) evoked by AMPA, fluorowillardiine and quisqualate were equal to those evoked by glutamate; however, the efficacies of [Ca++] signals in response to domoate and kainate were lower (P < .05) than those signals obtained with glutamate (table 1; fig. 3A).

Pharmacological characterization of hGluR3i/HEK69–8 cells, determined by [Ca++]i measurements in fura-2-loaded cells. (A) Agonist pharmacology. Data points represent the mean ± S.E. from four to nine experiments, each performed in quadruplicate. Data are normalized to the maximal response obtained by 1 mM glutamate determined in each experiment. (B) Antagonist pharmacology. Data points represent the mean ± S.E. from three to eight separate experiments, each performed in quadruplicate. Data are normalized in each experiment to the response obtained by 30 μM glutamate. All Ca++ measurements were performed in the presence of 100 μM cyclothiazide, added 2 min before assay.
Agonist pharmacology of hGluR3i determined by [Ca++]i measurements in HEK69-8 cells
Antagonist pharmacology of hGluR3i.
We also investigated the antagonist pharmacology of hGluR3i by the [Ca++]i assay (table2; fig. 3B). The quinoxalinedione series of AMPA receptor antagonists (NBQX, CNQX, DNQX and YM-90K) and the noncompetitive antagonist GYKI-52466 (Donevan and Rogawski, 1993) completely inhibited [Ca++]i signals evoked by a submaximal concentration of glutamate (30 μM). The IC50 values generated for the competitive antagonists are also expressed as the dissociation constants (Kb values) estimated with the Leff-Dougall variant of the Cheng-Prusoff equation (Leff and Dougall, 1993). The putative AMPA receptor antagonist NS257 (Nielsen et al., 1995) inhibited only 76 ± 5% of the glutamate-evoked [Ca++]i signals, whereas the AMPA receptor antagonist AMOA (Wahl et al., 1992) showed no antagonist activity at concentrations up to 300 μM. In addition, the putative kainate receptor selective antagonist NS102 (Verdoornet al., 1994) did not show significant inhibition of glutamate-evoked [Ca++]i responses at concentrations up to 300 μM. GAMS, proposed to have AMPA/kainate receptor antagonist activity (Davies and Watkins, 1985), inhibited glutamate-evoked responses, albeit with a low potency (table 2).
Antagonist pharmacology of hGluR3i determined by [Ca++]i measurements in HEK69-8 cells
Comparison of the geometric mean Kb values by ANOVA and Student-Newman-Keuls test revealed the following rank order of potency: NBQX > DNQX = CNQX = YM-90K > NS257 > GYKI-52466 > GAMS.
Electrophysiological whole-cell recordings.
The electrophysiological properties of hGluR3i/HEK69–8 cells were investigated by whole-cell recording. In the absence of CTZ, the AMPA- and glutamate-induced currents were substantially smaller and showed greater desensitization than in the presence of CTZ. To resolve the peak current in the absence of CTZ, we used a rapid agonist application with a piezo element to achieve open-pipette switching times of approximately 470 μs. Under these conditions, cells responded to 10 mM glutamate with a mean 10 to 90% current rise time of 5.4 ± 2.6 ms, mean peak current of 75 ± 32 pA and mean desensitization time constant of 5.4 ± 1.1 ms (mean ± S.D. from three cells). This compares well with the desensitization time constant value of 4.8 ms obtained from outside-out patches from Xenopusoocytes expressing rat GluR3i (Mosbacher et al., 1994). Representative currents evoked by rapid application of 1 mM glutamate (in the absence of CTZ) are shown in figure4A, and the current-voltage relationship for the same cell is shown in figure 4B. The current-voltage relationship for hGluR3i is inwardly rectifying, as has been observed for rat GluR3 (Nakanishi et al., 1990), and no outward current was observed at positive potentials up to 50 mV.
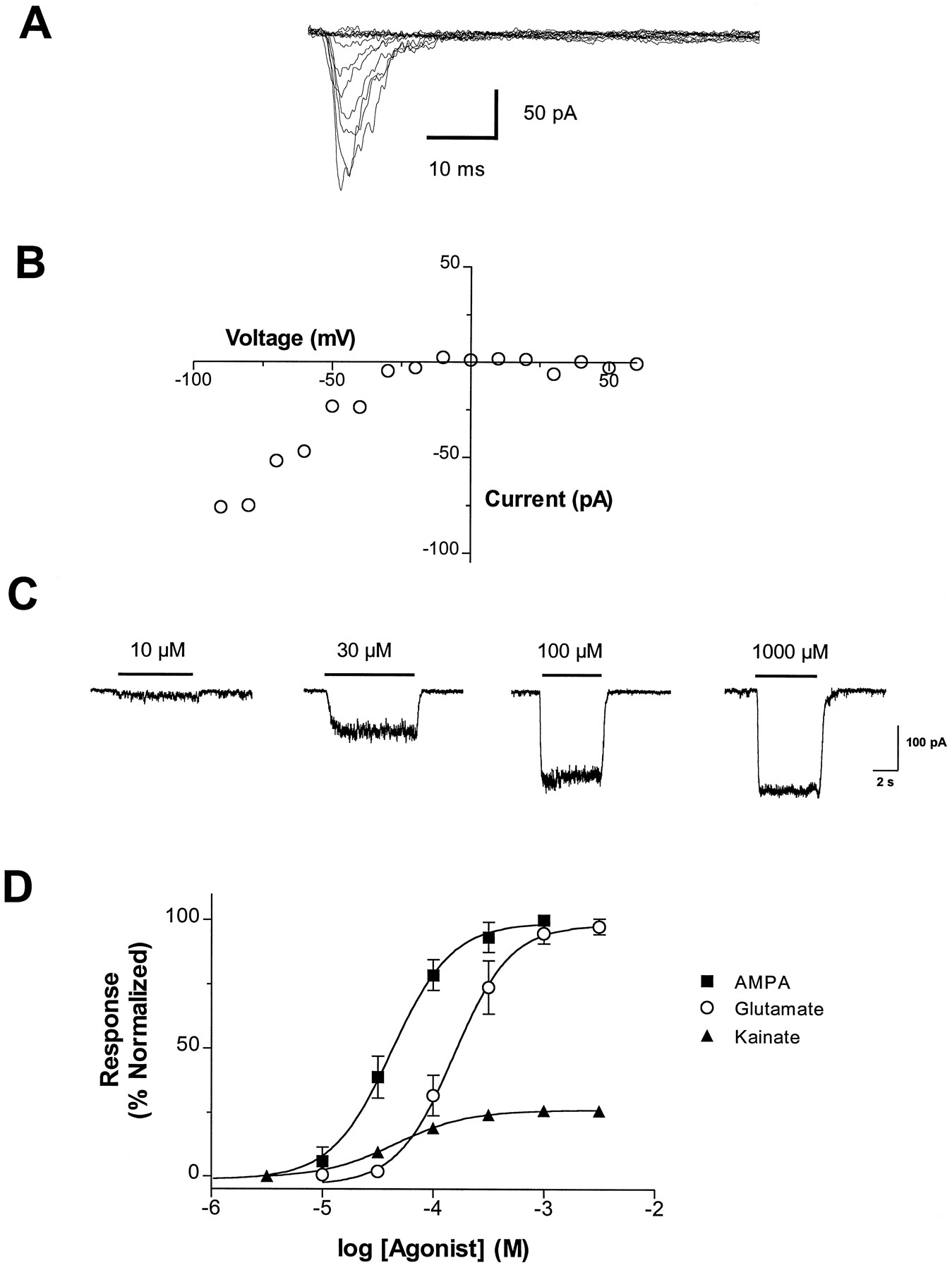
Whole-cell patch-clamp recordings of hGluR3i/HEK69–8 cells. (A) Representative currents evoked by rapid application of 1 mM glutamate in a cell held at −60 mV. The membrane potential was stepped in 10-mV increments from −90 to 60 mV during repeated applications of 1 mM glutamate. (B) Current-voltage relationship for the peak current from the cell shown in panel A is plotted. (C) Representative AMPA-evoked currents in the presence of 100 μM cyclothiazide. The cell was held at a membrane potential of −60 mV. (D) Agonist pharmacology of hGluR3i determined by whole-cell recording. Cells were held at a membrane potential of −60 mV. Data represent the mean ± S.D. from four to five cells. Data are normalized to the maximal current obtained from 3 mM glutamate.
In hGluR3i/HEK69–8 cells held at −60 mV, application of 1 mM AMPA in the presence of 100 μM CTZ resulted in a peak current of 347 ± 279 pA (mean ± S.D.,n = 58 cells). Dose-dependent AMPA-evoked currents are shown in figure 4C, and concentration-response curves to glutamate, AMPA and kainate (each in the presence of 100 μM CTZ) are shown in figure 4D. Estimated EC50 values (summarized in table 3) give a rank order of potency of kainate = AMPA > glutamate (P < .05). The EC50 values for these agonists are 6- to 7-fold higher than the values determined in the [Ca++]i assay. As in the [Ca++]i assay, we observed that glutamate and AMPA were equally efficacious, whereas kainate elicited approximately 22% of the current evoked by glutamate.
Pharmacological properties of hGluR3i determined by whole-cell recordings in HEK69-8 cells
[3H]AMPA binding.
We performed competitive radioligand binding studies with [3H]AMPA to further characterize the pharmacological properties of hGluR3i/HEK69–8 cells, and compared these results with those obtained for rat forebrain membranes. Under our assay conditions, the specific binding of [3H]AMPA to membranes prepared from rat forebrain and hGluR3i/HEK69–8 was greatly potentiated (data not shown) in the presence of the chaotropic agent potassium thiocyanate (Honoré and Drejer, 1988). All binding assays were performed, therefore, in the presence of 100 mM potassium thiocyanate.
A saturation analysis and Scatchard transformation of specific [3H]AMPA binding to hGluR3i/HEK69–8 cells (fig.5A) indicated a single class of binding sites, with a binding constant (Kd) of 12.3 (11.0, 13.7) nM [geometric mean (lower, upper S.E.)], and a binding capacity (Bmax) of 187 ± 27 fmol/mg protein (mean ± S.E., n = 7), corresponding to approximately 8,000 binding sites per cell. Under the same experimental conditions with rat brain membranes, [3H]AMPA bound with aKd of 16.7 (15.6, 17.9) nM (n = 3) and a Bmax of 711 ± 63 fmol/mg protein (n = 3, data not shown). The Kd value for rat brain is significantly different from hGluR3i/HEK69–8 cells (P < .001).

[3H]AMPA competition binding experiments in membranes prepared from hGluR3i/HEK69–8 cells. (A) Displacement of [3H]AMPA by unlabeled AMPA in the absence and presence of 100 μM cyclothiazide. Data points are the mean ± S.D. of triplicates from a single experiment, and representative of two further experiments. (B) Scatchard transformation of the data in panel A. Analysis of the saturation curves indicated that the data were best fit to a single binding site (P > .05,F-test). (C, D) Competition binding curves for AMPA (•), quisqualate (○), glutamate (□), CNQX (▪) and kainate (▵) in the absence (C) and presence (D) of 100 μM cyclothiazide. Data points are normalized to the amount of specific binding in the absence of unlabeled ligand, and represent the mean ± S.E. from three experiments.
Competitive binding studies with hGluR3iand rat forebrain.
A detailed pharmacological comparison between hGluR3i/HEK69–8 cells and rat forebrain was undertaken by comparing 15 AMPA receptor ligands for their potencies in displacing bound [3H]AMPA (table4). For most ligands, the calculatedKi values were similar in both membrane preparations (table 4). The largest difference was observed with fluorowillardiine, which was 10-fold more potent on rat forebrain than at hGluR3i. Conversely, kainate and NBQX were approximately 2-fold more potent on hGluR3i than rat forebrain. Some compounds showed weak or no displacement of [3H]AMPA in either membrane preparation The noncompetitive AMPA receptor antagonist GYKI-52466 and the putative kainate receptor antagonist NS102 showed no displacement at either preparation at concentrations up to 300 μM. Weak affinities for the two receptor preparations were observed for the putative AMPA receptor antagonists AMOA and GAMS, which showed Kivalues of greater than 100 μM.
[3H]AMPA binding to hGluR3i/HEK69-8 and rat forebrain
Effect of cyclothiazide on [3H]AMPA binding to hGluR3i.
Because the functional characterization of hGluR3i/HEK69–8 cells was performed in the presence of 100 μM CTZ to prevent receptor desensitization, and because [3H]AMPA likely is binding to the desensitized state of the receptor, we investigated the effect of CTZ on the affinity of ligands at the hGluR3i receptor in the [3H]AMPA binding assay. A maximum of approximately 50% reduction in [3H]AMPA binding was obtained with 100 μM CTZ (fig. 5A). Saturation binding experiments revealed that CTZ decreased specific [3H]AMPA binding by significantly reducing the affinity of AMPA for its receptor, because theKd was increased to 28.5 (22.3, 36.3) nM (different from control, P < .05, Student’s t test), without a significant change in the number of binding sites (Bmax = 253 ± 57 fmol/mg protein,n = 4; fig. 5B). These results are consistent with the previously reported effect of CTZ on [3H] AMPA binding in rat brain (Hall et al., 1993; Kessler et al., 1996).
The pharmacological properties of [3H]AMPA binding to hGluR3i/HEK69–8 cell membranes for five ligands were determined in the absence and presence of 100 μM CTZ (fig. 5, C and D). The IC50 values for these compounds were corrected for the difference in [3H]AMPA affinity in the two assay conditions by converting them to Ki values (table5). The Kivalues for these ligands were not significantly different from those determined in the absence of CTZ (table 5), with the exception of kainate, which had a 2-fold higher affinity in the presence of CTZ. The Hill coefficients for quisqualate and kainate were lower than unity in the presence of CTZ.
[3H]AMPA binding to hGluR3i/HEK69-8: Effect of cyclothiazide
Previous reports have suggested that CTZ and GYKI-52466 interact at overlapping binding sites on the AMPA receptor (Palmer and Lodge, 1993;Zorumski et al., 1993). Because the affinity of AMPA for hGluR3i was reduced in the presence of 100 μM CTZ, we tested the ability of GYKI-52466 to reverse this CTZ-mediated attenuation of [3H]AMPA binding in hGluR3i/HEK69–8 cell membranes. GYKI-52466, at concentrations up to 100 μM, did not alter [3H]AMPA binding in the absence or presence of CTZ (fig. 6), consistent with previous results from rat brain (Kessler et al., 1996), which indicates that CTZ and GYKI-52466 do not interact at the same binding site on hGluR3i (Partin and Mayer, 1996).
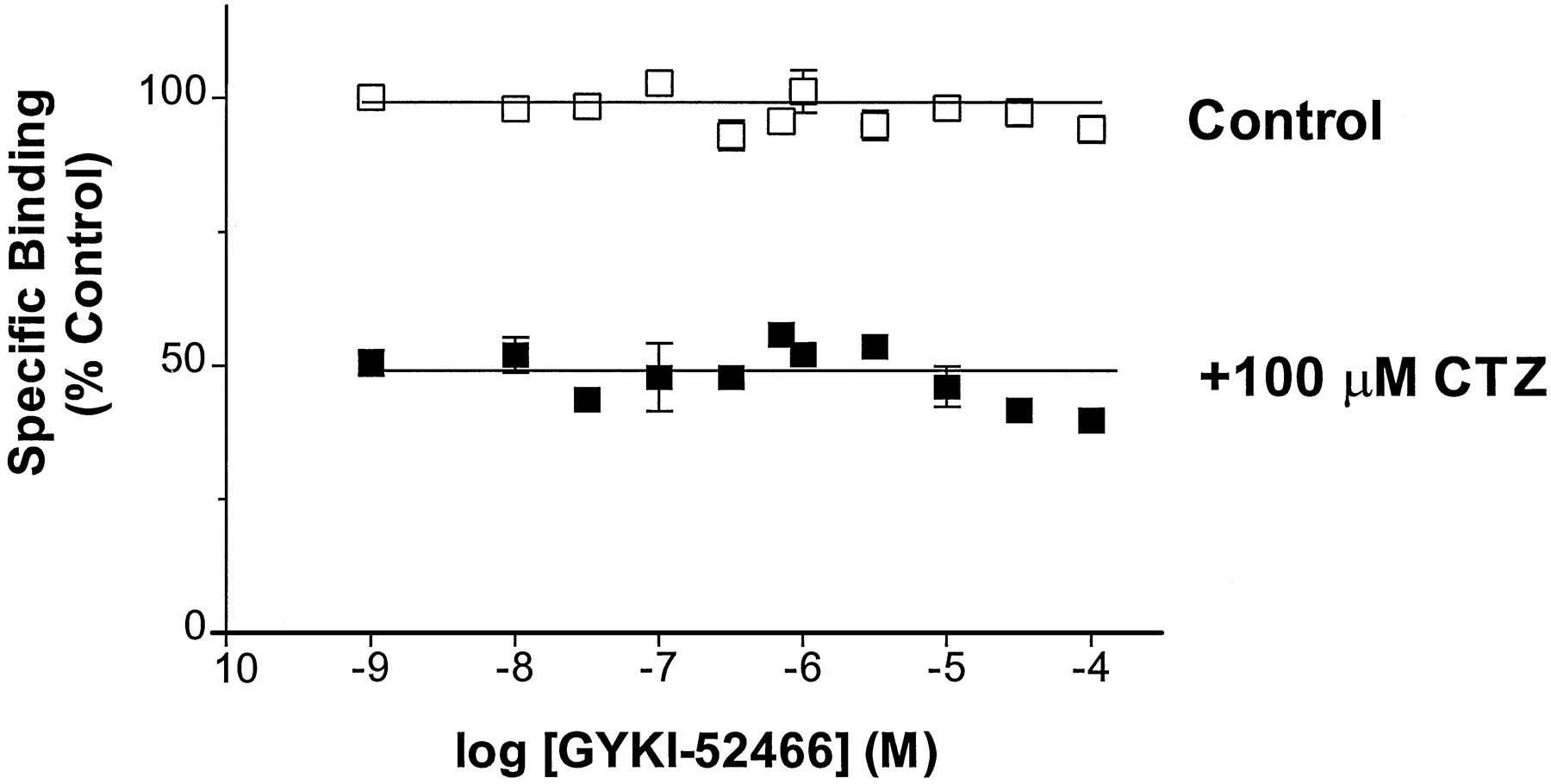
The effect of GYKI 52466 on specific [3H]AMPA binding to membranes prepared from hGluR3i/HEK69–8 cells in the absence (□) or presence of 100 μM cyclothiazide (▪). Data points are normalized to the amount of specific binding in the absence of cyclothiazide and GYKI 52466, and represent the mean ± S.E. from three experiments.
Discussion
Here we describe the characterization of an HEK293 cell line stably expressing the human AMPA receptor subtype GluR3i. This cell line has allowed us to investigate the biochemical, pharmacological and electrophysiological characteristics of hGluR3i under consistently high levels of receptor expression. This type of detailed characterization of a recombinant receptor should facilitate our understanding of native receptors in situ.
The sequence of the hGluR3-flip isoform described here differs from that identified by Rampersad et al. (1994) by three amino acids. One of these changes is an Arg/Gly edit, which is an RNA codon switching event that occurs in rat GluR2, GluR3 and GluR4 (Lomeliet al., 1994) at a site immediately preceding the flip and flop regions. The rat homolog of our clone, the nonedited GluR3i(Arg), has a slower rate of recovery from desensitization than the edited GluR3i(Gly) (Lomeli et al., 1994).
We confirmed the expression of the hGluR3iprotein by immunoblot analysis with an anti-rat hGluR2/3 antibody. The 110-kdalton immunoreactive species was larger than the 102-kdalton size predicted for a monomeric hGluR3i protein, which suggests that the GluR3 receptor, like other AMPA receptors, may beN-glycosylated (Hullebroeck and Hampson, 1992; Hunter and Wenthold, 1992). The role of glycosylation in AMPA receptor structure and function is not well understood (Hollmann et al., 1994;Kawamoto et al., 1995; Arvola and Keinänen, 1996).
Like all AMPA receptors, the hGluR3i receptor desensitizes rapidly. Our estimate of the desensitization time constant of 5.4 ms for hGluR3i determined with whole-cell recording is consistent with a time constant of 4.8 ms reported for rat GluR3i expressed in Xenopus oocytes with outside-out patches (Mosbacher et al., 1994). The use of outside-out patches rather than whole cells would allow us to make a more accurate determination of the time constant.
The rapid rate of receptor desensitization prevented us from detecting agonist-evoked [Ca++]ichanges in the absence of CTZ. However, robust [Ca++]i signals were detected in these cells in the presence of CTZ, which allowed us to perform a thorough characterization of the receptor pharmacology with this functional assay. The EC50 for the CTZ-mediated potentiation was approximately 10 μM, and was not dependent on the agonist used in the study. The potency of CTZ in the Ca++ assay is consistent with previous reports in the literature, generated from recombinant flip isoforms of AMPA receptors expressed in Xenopus oocytes (Partin et al., 1994), and native neuronal receptors (Patneau et al., 1993, Yamada and Tang, 1993; Rammes et al., 1996;Hoyt et al., 1995). CTZ differentially affects the flip/flop splice variants of AMPA receptors, potentiating the agonist responses of the flip isoform with a greater affinity and efficacy than the flop isoform (Partin et al., 1994). Furthermore, CTZ strongly attenuates desensitization of the flip isoform, whereas the desensitization of the flop isoform remains pronounced even in the presence of CTZ (Partin et al., 1994). Consistent with these reports, in initial experiments with hGluR3-flop, we were unable to detect robust increases in [Ca++]i, even in the presence of CTZ (data not shown).
Under our [3H]AMPA binding assay conditions, we detected a single class of [3H]AMPA binding sites to membranes prepared from hGluR3i/HEK69–8 cells, with a Kd value consistent with values previously reported for recombinant rat AMPA receptors (Keinänen et al., 1990; Kawamoto et al., 1995; Andersen et al., 1996). TheKd value that we obtained for rat forebrain is consistent with the high-affinity site reported by others (Honoré et al., 1982; Murphy et al., 1987;Cha et al., 1992; Hall et al., 1993). Likewise, for rat forebrain, the Ki values and rank order of potencies that we obtained for the various ligands from competitive binding experiments are consistent with previous reports from rat brain (Honoré et al., 1989; Nielsen et al., 1990; Giberti et al., 1991; Hawkins et al., 1995a), and they were similar to the binding results for hGluR3i. However, we observed a 10-fold difference between rat forebrain and hGluR3i/HEK69–8 cells for fluorowillardiine, and smaller differences for AMPA, NBQX and kainate. Because [3H]AMPA binding to rat forebrain presumably reflects multiple AMPA receptor subtypes, the differences in ligand potencies between the two preparations may indicate that these compounds exhibit a degree of receptor subtype selectivity. This is supported further by the observation that we were unable to detect significant [3H]fluorowillardiine binding to hGluR3i/HEK69–8 cells, yet we readily detected binding to rat forebrain (data not shown). Based on these results, therefore, one should be cautious in assuming that the binding of [3H]fluorowillardiine to rat brain detects all subtypes of AMPA receptors equally (Hawkins et al., 1995b), unlike [3H]AMPA which does not show marked selectivity at recombinant AMPA receptors (Keinänen et al., 1990; Andersen et al., 1996; Varney M and Rao S, unpublished observation).
The data from functional [Ca++]i measurements and radioligand binding experiments demonstrate that for some agonists the concentrations required to activate hGluR3i are several orders of magnitude higher than their bindingKd values. This suggests that [3H]AMPA binds to a desensitized form of the receptor, as has been observed for some other ionotropic receptors including nicotinic (Romano and Goldstein, 1980) and γ-aminobutyric acidA (Bristow and Martin, 1989) receptors. For hGluR3i-expressing cells, there is no significant correlation (r2 = 0.03, P = .76) between Ki values determined for [3H]AMPA binding and EC50values from functional Ca++ data for agonists (fig. 7A). For example, AMPA, glutamate and kainate are 920-, 86- and 3.1-fold less potent in the Ca++ assay than the [3H]AMPA binding assay. These differences are unlikely to be caused by the presence of CTZ, which was present in the functional assay, because inclusion of the same concentration of CTZ in the binding assays did not dramatically affect theKi values. The affinity of agonists in the binding assay is measured by their ability to compete with [3H]AMPA at the higher affinity desensitized conformational state of the receptor. Therefore, agonists such as kainate, which induce less desensitization than glutamate or AMPA at rat GluR3i (Partin et al., 1994), would be expected to have a lower affinity for the desensitized state of the receptor. The relatively lower affinity of kainate for the desensitized state of the receptor would be consistent with the model proposed by Patneau et al. (1992) to accommodate for differences in agonist-evoked desensitization of AMPA receptors by willardiine derivatives. In their model, simulated responses for a weakly desensitizing agonist (such as kainate in our studies) were reproduced successfully by decreasing the ratio of the affinities of the agonist for the active over the desensitized states of the receptor, which resulted in a reduced affinity of the agonist for the desensitized receptor (Patneau et al., 1992).

Correlation of potency values obtained from [3H]AMPA competition binding experiments and functional [Ca++]i measurements for agonists (A) and antagonists (B). Potency values are expressed as mean log EC50 (pEC50) or log IC50(pIC50) values. Correlations were determined by linear regression, and are shown by the solid lines, with the 95% confidence intervals shown by the dashed lines. For panel A,r2 = 0.03 and the slope = 0.40; for panel B, r2 = 0.97 and the slope = 1.06.
In contrast to agonist data, there was an excellent correlation (r2 = 0.97, P < .0001) in the affinities of competitive antagonists determined by binding and functional studies (fig. 7B). These data suggest that competitive antagonists recognize, with similar affinity, the desensitized and active states of the AMPA receptor. Furthermore, they indicate that for competitive antagonists, binding affinities can predict antagonism in an in vitro functional assay, whereas binding data cannot be used to predict functional potencies for agonists. The lack of displacement of [3H]AMPA binding by the GYKI-52466 also demonstrates the limitations of binding assays, because noncompetitive antagonists can only be detected reliably in functional assays.
The mechanism of action of CTZ in attenuating AMPA receptor desensitization is not yet clear, but three models have been proposed based on recent experimental results: CTZ destabilizes a desensitized state of the receptor, or it stabilizes the closed nondesensitized state of the receptor, or both (Partin et al., 1996). These models take into account the apparent increase in receptor affinity measured at hippocampal neurons where CTZ increased the potency of quisqualate by 300-fold (Yamada and Tang, 1993) and that of kainate by 3-fold (Patneau et al., 1993). Radioligand binding data from rat brain also support these models. Because CTZ prevents agonist-induced desensitization of hGluR3i, a reduction in the rate of receptor desensitization by CTZ would be expected to reduce the affinity of the receptor for [3H]AMPA, because [3H]AMPA binding predominantly measures the desensitized states of the receptor. This is consistent with our and other (Hall et al., 1993) experimental observations, in which [3H]AMPA displayed a lower affinity for the receptor in the presence of CTZ. The affinity of [3H]CNQX for AMPA receptors in rat brain was also reduced by CTZ (Kessler et al., 1996). Because [3H]CNQX binds with equal affinity to different desensitized states of the receptor (Honoré et al., 1988), the effect of CTZ cannot be caused simply by an interconversion of the nondesensitized and desensitized receptor states. However, these results would be consistent with CTZ stabilizing the agonist-bound nondesensitized closed state of the receptor by increasing the agonist affinity and decreasing the rate of onset of desensitization (Kessleret al., 1996; Partin et al., 1996). [3H]AMPA will bind to both states of the receptor whether CTZ is present or not. Because CTZ shifts the distribution of the receptors, [3H]AMPA predominantly binds to the nondesensitized (closed) state of the receptor in the presence of CTZ. The pharmacology of [3H]AMPA binding to hGluR3i in the presence of CTZ is similar to that in the absence of CTZ when the IC50 values are corrected for the reduced affinity of [3H]AMPA by calculating Ki values. However, kainate exhibited a 2-fold higher affinity for hGluR3i in the presence of CTZ, and reduced Hill coefficients were observed for quisqualate and kainate. This suggests that in the presence of CTZ, [3H]AMPA binds to more than one conformational state of the receptor with similar affinities, but kainate and quisqualate exhibit different affinities for these conformational states.
Although activation of AMPA receptors favors Na+entry through the channel, approximately 3.5% of the inward current at −60 mV is carried by Ca++, which is less than the fractional Ca++ current of 11% measured for NMDA receptors (Burnashev et al., 1995). Although the fractional Ca++ current in hGluR3i-expressing cells is lower than for NMDA receptors, it was detected easily by fluorescent Ca++-sensitive dyes after agonist stimulation in the presence of CTZ. The time course of the Ca++signal does not reflect the responses observed by electrophysiology, with the [Ca++]i signals having a slower onset compared with the patch-clamp response. There are several possible explanations for this. First, we are measuring [Ca++]i in a population of cells, and not in single cells. Second, the overall increase in [Ca++]i results from the combined contributions of Ca++ influx, Ca++ release from internal stores, sequestration into organelles, Ca++ efflux and cellular Ca++ buffering mechanisms, which slow the onset of the [Ca++]i signals (Neher and Augustine, 1992). Furthermore, unlike in electrophysiological recordings, the addition of the agonist is not performed by a rapid exchange protocol.
The robust increases in [Ca++]i allowed us to perform and report, for the first time, an extensive characterization of recombinant AMPA receptor pharmacology with [Ca++]i measurements. The agonist pharmacology determined with this functional assay is consistent with that obtained by electrophysiological recordings for rat recombinant receptors in the presence of CTZ. For example, for rat GluR3 receptors expressed in Xenopus oocytes, EC50 values for AMPA, glutamate, quisqualate and kainate of 16, 58, 0.8 and 73 μM, respectively, were obtained in the presence of 100 μM CTZ (Stern-Bach et al., 1994). In our electrophysiological studies, agonists were about 7-fold less potent than in [Ca++]imeasurements. Because both assays were performed in the presence of CTZ and during a similar time course of agonist application, we should be measuring the same receptor states. These differences in potency suggest that the Ca++ assay is saturating at lower concentrations of agonist than whole-cell recordings. One explanation for these observations is that both responses depend on the membrane potential, and in one case it is fixed (whole-cell recording), and in the other (the Ca++ assay) it is not. All functional receptors would be detected in whole-cell recordings because of the linear relationship of channel opening events and whole-cell currents. However, in the Ca++ assay, because activation of only a small proportion of the total receptors may be sufficient to maximally depolarize the cell, not all functional receptors may be detected. This level of depolarization in hGluR3i-expressing cells would be dictated by the inwardly rectifying properties of the receptors, which would become nonconducting at a membrane potential of −10 or −20 mV. Once this threshold is reached, activating additional receptors does not result in a further increase in [Ca++]i, and therefore these additional receptors would behave as “spare receptors” in the Ca++ assay. The 7-fold leftward shift in the dose-response curves obtained in the Ca++ assay suggest that activation of approximately 30 to 40% of the functional receptors suffices to evoke a maximal Ca++signal.
In both [Ca++]i and whole-cell recording measurements, kainate was less efficacious than glutamate or AMPA. This is similar to data obtained in hippocampal neurons, where kainate was only about 45% as efficacious as glutamate in the presence of CTZ (Patneau et al., 1993). From our [Ca++]i measurements, we noticed that domoate, a closely related analog of kainate, and also a weakly desensitizing agonist, was not as efficacious as glutamate. Our results do not provide an explanation for the lower efficacy of kainate and domoate, but it may involve partial agonist effects, or agonist-evoked differences in single-channel conductance states of the hGluR3i receptor. In this regard, in recombinant GluR4i, kainate exhibits a lower single-channel conductance than those obtained for AMPA or glutamate (Swanson et al., 1997).
The most potent AMPA receptor antagonists characterized in this study were from the quinoxalinedione-like series (CNQX, DNQX, NBQX and YM-90K), which completely inhibited glutamate-evoked [Ca++]i withKb values in the range 0.3 to 2.1 μM. NS257 was the most potent antagonist of the nonquinoxalinediones, although it failed to completely inhibit glutamate-evoked [Ca++]i signals, and AMOA and NS102 both failed to inhibit hGluR3i, even at 300 μM. The noncompetitive AMPA-selective antagonist GYKI-52466 inhibited hGluR3i with a slightly lower potency than those reported from electrophysiological studies on AMPA receptors in the absence of CTZ (Donevan and Rogawski, 1993; Zorumski et al., 1993). Because the antagonist affinities were measured in the presence of CTZ, the question arises as to what the true affinities of these compounds are in the absence of CTZ. From electrophysiological studies on AMPA receptors expressed in Xenopus oocytes, CTZ allosterically modulates the affinity of the competitive antagonists NBQX and YM-90K, resulting in a slightly lower apparent affinity of the antagonists for the AMPA receptor (Okada et al., 1996). This is consistent with the CTZ-mediated reduction in binding affinity of [3H]CNQX for AMPA receptors (Kessler et al., 1996). However, the antagonist affinity of noncompetitive antagonists such as GYKI-52466 may be affected by CTZ to a greater degree depending on the receptor subtypes (Palmer and Lodge, 1993;Zorumski et al., 1993; Johansen et al., 1995;Rammes et al., 1996). For example, the antagonist activity of GYKI-52466 is reduced 6-fold by 50 μM CTZ at rat GluR2i/4i heteromers, but unaffected by CTZ at rat GluR1i and GluR4i homomers (Johansen et al., 1995). In our studies GYKI-52466 failed to reverse the CTZ-mediated attenuation of [3H]AMPA binding to hGluR3i, which suggests that the two binding sites are nonoverlapping. However, it is conceivable that CTZ could allosterically modify the GYKI-52466 binding site, without being modified significantly itself by GYKI-52466 (Rammes et al., 1996). Therefore, the Ca++ assay likely provides a good estimate of the affinity of competitive antagonists, but may underestimate noncompetitive antagonist activities whose affinity is affected by CTZ, or the desensitization state of the receptor.
There is some evidence for the involvement of GluR3 in pathophysiology. The chromosomal localization of the human GluR3 gene has been mapped to the X chromosome (q25–26), and linkage analysis studies suggest that this overlaps the regions involved in oculocerebral-renal syndrome of Lowe, and a syndrome characterized by congenital nerve deafness and albinism (McNamara et al., 1992). In addition, Rogerset al. (1994) have reported that sera from patients with Rasmussen’s encephalitis (an intractable pediatric disease) react with GluR3, and may result in GluR3 receptor activation (Twyman et al., 1995). Rabbits immunized with a portion of the GluR3 receptor also exhibited seizure-like behaviors, and consistent with the human data, these rabbit antisera also activated AMPA receptors. Furthermore, after status epilepticus, selective up-regulation of GluR3 mRNA levels has been reported in the dentate gyrus (Condorelli et al., 1994), which implicates a potential role for GluR3 in some forms of epilepsy.
In conclusion, these studies demonstrate the stable expression of recombinant hGluR3i in a human host cell line, with biophysical, biochemical and pharmacological characteristics similar to those reported for native tissues. The functional [Ca++]i assay will allow us to use the hGluR3i/HEK69–8 cell line as a high-throughput screening target to facilitate the discovery of novel, selective ligands for AMPA receptors.
Acknowledgments
We thank Dr. Y. Auberson, Novartis, Switzerland, for supplying YM-90K. The excellent technical assistance of K. Lariosa, C. Liaw and B. Siegel, and the editorial assistance of K. Payne are greatly appreciated. We thank Drs. Sandy Madigan and Michael Harpold from SIBIA, and Drs. Hans Allgeier, Rainer Kuhn and Dirk Sauer from Novartis, Switzerland for providing critical input into this manuscript.
Footnotes
-
Send reprint requests to: Dr. Mark Varney, SIBIA Neurosciences, Inc., 505 Coast Blvd. South, La Jolla, CA 92037.
- Abbreviations:
- AMOA
- (±)-2-amino-3-[3-(carboxymethoxy)-5-methyl-isoxazol-4-yl]propionic acid
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CNQX
- 6-cyano-7-nitroquinoxaline-2,3-dione
- GAMS
- γ-d-glutamylaminomethylsulfonic acid
- DNQX
- 6,7-dinitroquinoxaline-2,3-dione
- YM-90K
- 6-(1H-imadazol-1-yl)-7-nitro-2,3-(1H, 4H)-quinoxalinedione
- GYKI-52466
- 1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine hydrochloride
- HEPES
- N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- HBS
- HEPES-buffered saline
- hGluR3i
- human AMPA receptor subtype 3-flip
- NBQX
- 6-nitro-7-sulfamoylbenzo(f)quinoxaline-2–3-dione
- NS 102
- 6,7,8,9-tetrahydro-5-nitro-1H-benz[g]indole-2,3-dione 3-oxime
- NS 257
- 1,2,3,6,7,8-hexahydro-3-(hydroxyimino)-N,N,7-trimethyl-2-oxo-benzo[2,1-b:3,4-c′]dipyrrole-5-sulfonamide hydrochloride
- NMDA
- N-methyl-d-aspartate
- fluo-3/AM
- fluo-3-acetoxymethyl ester
- fura-2/AM
- fura-2-acetoxymethyl ester
- CTZ
- cyclothiazide
- nt
- nucleotide
- ANOVA
- analysis of variance
- Received August 1, 1997.
- Accepted December 29, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}