


Abstract
Anthracyclines and many other antitumor drugs induce cardiotoxicity that occurs “on treatment” or long after completing chemotherapy. Dose reductions limit the incidence of early cardiac events but not that of delayed sequelae, possibly indicating that any dose level of antitumor drugs would prime the heart to damage from sequential stressors. Drugs targeted at tumor-specific moieties raised hope for improving the cardiovascular safety of antitumor therapies; unfortunately, however, many such drugs proved unable to spare the heart, aggravated cardiotoxicity induced by anthracyclines, or were safe in selected patients of clinical trials but not in the general population. Cardio-oncology is the discipline aimed at monitoring the cardiovascular safety of antitumor therapies. Although popularly perceived as a clinical discipline that brings oncologists and cardiologists working together, cardio-oncology is in fact a pharmacology-oriented translational discipline. The cardiovascular performance of survivors of cancer will only improve if clinicians joined pharmacologists in the search for new predictive models of cardiotoxicity or mechanistic approaches to explain how a given drug might switch from causing systolic failure to inducing ischemia. The lifetime risk of cardiotoxicity from antitumor drugs needs to be reconciled with the identification of long-lasting pharmacological signatures that overlap with comorbidities. Research on targeted drugs should be reshaped to appreciate that the terminal ballistics of new “magic bullets” might involve cardiomyocytes as innocent bystanders. Finally, the concepts of prevention and treatment need to be tailored to the notion that late-onset cardiotoxicity builds on early asymptomatic cardiotoxicity. The heart of cardio-oncology rests with such pharmacological foundations.
Cardiotoxicity of Antitumor Drugs: Old Aspects and New Issues
Antitumor drugs have long been known to induce untoward but manageable cardiovascular effects such as transient blood pressure disorders, ischemia, arrhythmias, systolic dysfunction, and pericardial effusions. Hormonal treatment of hormone-responsive tumors also introduces a risk of cardiovascular events. There are cases where the cardiac sequelae of antitumor therapies may be life-threatening; for example, cumulative doses of antibiotics, such as anthracyclines, mitomycin, or mitoxantrone, induce dilative cardiomyopathy and congestive heart failure (CHF) (Minotti et al., 2004; Menna et al., 2008).
Almost all of the approved antineoplastics have been shown to induce some type of cardiotoxicity (see Table 1 for a representative list of cardiotoxic drugs). With that said, the clinical manifestations of cardiotoxicity seem to have changed in the last few years. Reducing the cumulative dose of anthracyclines (topoisomerase II inhibitors and DNA intercalators) was of help in diminishing acute or subacute cardiotoxicity but not chronic cardiotoxicity that occurred 5 or more years after completing chemotherapy; moreover, some survivors of cancer developed CHF, whereas others developed ischemic disease and myocardial infarction (MI) (Carver et al., 2007; Swerdlow et al., 2007). Nonanthracycline chemotherapeutics (antimetabolites, alkylators, tubulin-active agents, or other topoisomerase II inhibitors such as etoposide) were known to induce systolic dysfunction but also, if not primarily, ischemia that occurred within hours or days of treatment (Menna et al., 2008); however, currently available evidence shows that nonanthracycline chemotherapeutics also introduce a lifetime risk of cardiotoxicity (Carver et al., 2007). The importance of age of first treatment has been reappraised. Children-adolescents and the elderly had long been considered to be more vulnerable by anthracyclines; however, recent studies show that late-onset cardiotoxicity could occur independent of age of first treatment (Swerdlow et al., 2007).
Antitumor drugs with known, probable, or presumed cardiotoxicity
Cardiotoxicity is induced by antibodies or small molecules targeted at growth factors, or their receptors, or receptor-associated kinases. Receptors thought to be crucial to tumor cells (e.g., the epidermal growth factor receptor 2, HER-2) are also expressed in cardiomyocytes and relay a number of survival signals (Cheng and Force, 2010). Therefore, blocking HER-2 with the antibody trastuzumab precipitates cardiotoxicity from concomitant anthracyclines or causes dysfunction in patients with prior exposure to anthracyclines (Menna et al., 2008). Antibodies or small tyrosine kinase inhibitors (TKIs) targeted at the vascular endothelial growth factor (VEGF) or its receptors (VEGFr) may cause hypertension, contractile dysfunction, and ischemia (Schmidinger et al., 2008). Imatinib and other TKIs of Bcr-Abl and C-kit of leukemic or gastrointestinal sarcoma cells are also suspected to cause cardiotoxicity (Cheng and Force, 2010).
What Is Cardio-Oncology?
Concerns about cardiotoxicity from antitumor drugs should be weighed against survival or curability benefits; for example, anthracyclines caused lifesaving effects that outweighed the risk of cardiac-related death at 10 or 20 years of follow-up (Gianni et al., 2008). Less is known about the recently developed targeted drugs. Studies with a short follow-up demonstrate that commencing trastuzumab after chemotherapy improved the event-free survival of women with HER-2+ breast cancer, antiangiogenic drugs were active in many advanced tumors, imatinib was both lifesaving in chronic myeloid leukemia and active in otherwise untreatable gastrointestinal sarcomas (Suter et al., 2007; Albini et al., 2010; Cheng and Force, 2010). These facts show that patients with cancer should not be denied the benefits from drugs with known or suspected cardiotoxicity; similar concepts apply to the elderly as well (Carver et al., 2008). With that said, cardiotoxicity compromises the quality of life and calls for costly medical assistance. Therefore, oncologists and cardiologists were asked to jointly assess the risk/benefit of antitumor therapies and identify the best possible cardiac surveillance or preventative measures that improved the therapeutic index of antitumor therapies. This is the clinical dimension of cardio-oncology (Cardinale, 1996; Gianni et al., 2008), sometimes referred to as onco-cardiology (Albini et al., 2010).
Confining cardio-oncology to the merging of cardiology with oncology would be an unforgivable oversight. Cardio-oncology is a much broader discipline that goes from bench to bedside and builds clinical initiatives on pharmacological reasoning. In brief, we highlight some contemporary issues that illustrate the pharmacological foundations of cardio-oncology.
Lifetime Risk of Cardiotoxicity: A Matter of Long-Lasting Pharmacological Signatures
There is an unmet need for deciphering the lifetime risk of cardiotoxicity; this would be much important for drugs, such as the anthracyclines, that are cleared from the heart quite rapidly and decrease to below levels of toxic concern (Salvatorelli et al., 2009).
In rats, acute treatment with the anthracycline doxorubicin impaired cardiac oxidative phosphorylation; this was followed by formation of reactive oxygen species that caused both quantitative and qualitative alterations of mitochondrial DNA and its encoded respiratory chain subunits. Inasmuch as mitochondrial dysfunction persisted and worsened after completing anthracycline treatment, these studies suggested that the lifetime risk of cardiotoxicity could be caused by a mitochondriopathy that was self-maintained in the absence of continued exposure to doxorubicin (Lebrecht and Walker, 2007). In humans, however, mitochondriopathy did not correlate with cardiotoxicity induced by other anthracyclines. Post-mortem studies of hearts from patients with cancer showed that mitochondriopathy was caused by doxorubicin but not by its analog epirubicin (Lebrecht and Walker, 2007); in spite of that, epirubicin retained ≥60% of the cardiotoxic potential of doxorubicin (Menna et al., 2008).
The lifetime risk of cardiotoxicity from anthracyclines could be better explained by their conversion to secondary alcohol metabolites. Being more polar than their parent drugs, such metabolites are poorly cleared from cardiomyocytes and accumulate to become a long-lasting anthracycline signature in the heart (Minotti et al., 2004; Menna et al., 2008); moreover, secondary alcohol metabolites are many times more potent than their parent anthracyclines at inactivating Ca2+-handling proteins of the contraction-relaxation cycle (Minotti et al., 2004) or key regulators of energy metabolism and redox balance, such as cytoplasmic aconitase (Minotti et al., 1998; Cairo et al., 2002). Therefore, secondary alcohol metabolites cause cardiotoxicity both during and well after the course of chemotherapy. The levels of alcohol metabolite formation correlated with clinical cardiotoxicity from different anthracyclines. Experiments with ex vivo human myocardial samples exposed to anthracyclines in vitro showed that both doxorubicin and epirubicin were converted to secondary alcohol metabolites; however, epirubicin formed approximately 50% less alcohol metabolite than did doxorubicin (Salvatorelli et al., 2006, 2007, 2009).
Anthracyclines are not the only drugs that release long-lasting signatures of cardiotoxicity. Survivors of testicular cancer with a history of treatment with the alkylating agent cisplatin carry an increased risk for MI (Carver et al., 2007). In these subjects, the plasma levels of cisplatin remain measurable up to 20 years after completion of therapy, causing cumulative dysfunction of endothelial cells that eventually detach from the intima of vessels (Vaughn et al., 2008). Therefore, a long-lasting pharmacological signature (circulating cisplatin) correlates with molecular mechanisms of damage (endothelial dysfunction) and clinical facts (risk of MI).
Other chemotherapeutics seem to lack long-lasting signatures in spite of their proven or suspected implication in late-onset cardiotoxicity. This calls for ad hoc studies and changing strategies in drug development; new chemical entities should be routinely scrutinized for long-lasting pharmacological signatures that primed survivors of cancer to cardiotoxicity.
Drugs Are Not Alone: The Multiple-Hit Hypothesis
Roughly half of all childhood survivors of cancers with a history of anthracycline treatment showed asymptomatic cardiac dysfunction at noninvasive testing; however, symptomatic events only occurred in approximately 5% of them (Mulrooney et al., 2009). Similar concepts apply to survivors of adult-onset cancer with a history of anthracycline treatment (Hequet et al., 2004). On the other hand, circulating cisplatin would be seen in the vast majority of survivors of testicular cancer, but MI only occurred in ≤6% of them (van den Belt-Dusebout et al., 2006). Although still consistent with higher hazard ratios for cardiac events in survivors of cancer compared with age-matched siblings or other subjects from the general population, these figures denote that the incidence of cardiotoxicity might be influenced by interpatient variability.
Some patients might retain stronger pharmacological signatures of cardiotoxicity compared with others. Gain of function V244M polymorphism of carbonyl reductases 3, one of the many reductases that could convert anthracyclines to secondary alcohol metabolites, was retrospectively associated with an increased risk of CHF in long-term survivors of childhood cancer (Blanco et al., 2008). However, it is noteworthy that experiments with ex vivo human myocardial samples exposed to anthracyclines in vitro showed that carbonyl reductases metabolized some anthracyclines (daunorubicin) but not others (doxorubicin, epirubicin); the latter anthracyclines were much better substrates of aldehyde reductases belonging to the superfamily of aldo-keto reductases (Salvatorelli et al., 2007; Menna et al., 2008). Thus, carbonyl reductase polymorphisms would be important in survivors of childhood cancer who had received daunorubicin for the treatment of acute lymphoblastic leukemias, for example, but not in survivors who had received doxorubicin for the treatment of sarcomas or other malignancies. For similar reasons, carbonyl reductase polymorphisms would not explain interpatient variability among survivors who received doxorubicin or epirubicin rather than daunorubicin for adult-onset breast cancer or lymphomas. With regard to cisplatin, pharmacokinetic studies showed that, for any given body surface-based dose level, there was a 3-fold interpatient variability in the plasma exposure to cisplatin (Peng et al., 1997). Patients who developed higher plasma cisplatin levels during chemotherapy might be considered to carry such higher levels also after completion of chemotherapy; yet, there is a paucity of studies that explored cause-and-effect relationships between acute or chronic levels of cisplatin and the risk of late-onset cardiotoxicity.
Absent universal markers for the lifetime risk of cardiotoxicity, one should look at cardiotoxicity from alternative but not mutually exclusive viewpoints. Pre-existing comorbidities (hypertension, diabetes, hyperlipidemia) or unfavorable lifestyle choices (reduced physical activity) have long been known to increase the risk of cardiotoxicity in patients scheduled to receive chemotherapy (Minotti et al., 2004). The available evidence suggests that this picture should be viewed the other way around; in comparison with age-matched controls, previously healthy survivors of cancer developed more comorbidities or tended to reduce physical activity (Jones et al., 2007; Vaughn et al., 2008; De Bruin et al., 2009). It follows that asymptomatic, potentially reversible cardiotoxicity from “safe doses” of anthracyclines or nonanthracycline chemotherapeutics may progress to symptomatic events by overlapping with risk factors that matured after completion of chemotherapy. This is the so-called multiple-hit hypothesis, according to which late-onset cardiotoxicity builds on pharmacologic and nonpharmacologic sequential injuries (Jones et al., 2007; Menna et al., 2008). These concepts call for new approaches in caring for survivors of cancer: in these subjects, comorbidities or unfavourable lifestyle choices should be prevented or treated more vigorously than in the general population.
From Antitumor Drugs to Diastolic Dysfunction and More
Asymptomatic diastolic dysfunction fingerprints many survivors of cancer (Gianni et al., 2008). Anthracyclines cause diastolic elevated [Ca2+]i and left ventricular (LV) wall stiffness (diastolic dysfunction) by a number of mechanisms (Minotti et al., 2004). Diastolic stiffness then increases interstitial pressure and diminishes coronary conductance, eventually inducing ischemia that further increases [Ca2+]i and stiffness by its own mechanisms. Nonanthracycline chemotherapeutics would trigger this vicious cycle through ischemia due to endothelial dysfunction and/or coronary spasm (Menna et al., 2008).
Reciprocal interactions between diastolic dysfunction and ischemia need to be brought center stage. Nonanthracycline chemotherapeutics that caused persistent endothelial dysfunction and, hence, ischemia would induce diastolic dysfunction that aggravated ischemia. Such reciprocal interactions may remain asymptomatic for many years; accordingly, diastolic dysfunction at noninvasive testing preceded MI in survivors of testicular cancer with a history of cisplatin-based therapy (Altena et al., 2009). Patients treated with anthracyclines and chemotherapeutics that only caused transient endothelial dysfunction or coronary spasm (such as the tubulin-active vinca alkaloid, vincristine, or the glycopeptide antibiotic bleomycin) would be exposed to a blending of diastolic dysfunction and ischemia that made diastolic stiffness and reduced coronary conductance develop stronger and more persistent; accordingly, the risk of MI in previously nonischemic survivors of Hodgkin's lymphoma correlated with their prior exposure to anthracyclines in combination with vincristine and/or bleomycin (Swerdlow et al., 2007). Diastolic dysfunction and reduced coronary conductance would render the heart more vulnerable by comorbidities that diminished coronary flow or increased oxygen demand, such as premature atherosclerosis or hypertension, for example. Radiotherapy applies in this context where mediastinal irradiation causes diastolic dysfunction and premature coronary artery disease in survivors of cancer (Carver et al., 2007).
Different mechanisms may govern the switching of diastolic dysfunction to systolic failure. Anthracyclines cause reduced expression and/or increased disorganization of myofibrils, induce apoptosis, and trigger LV hypertrophy that deteriorates to wall thinning and dilation because of chronically unbalanced afterload (Gianni et al., 2008). With no synergism of anthracyclines with factors promoting diastolic dysfunction, the equilibrium would be pushed toward systolic failure. HER-2 protects against myofibrillar disorganization and serves a number of antiapoptotic functions (Pentassuglia et al., 2009; Cheng and Force, 2010); it follows that blocking HER-2 would accelerate progression to systolic failure. Therefore, women with breast cancer who received trastuzumab in combination with “safe doses” of anthracyclines developed dilative cardiomyopathy and systolic failure rather than MI. Administering trastuzumab after anthracyclines diminished the severity of cardiotoxicity but not its clinical phenotype; systolic dysfunction, although reversible and manageable, prevailed over ischemic disease (Suter et al., 2007).
In clinical practice, patients with or survivors of cancer are monitored by serial measurements of left ventricular ejection fraction (LVEF). Pharmacological notions suggest that this would be inadequate to capture the blending of diastolic dysfunction with systolic failure or ischemia (Fig. 1). New imaging techniques should be adopted and tailored to the characteristics of patients and chemotherapy regimens.
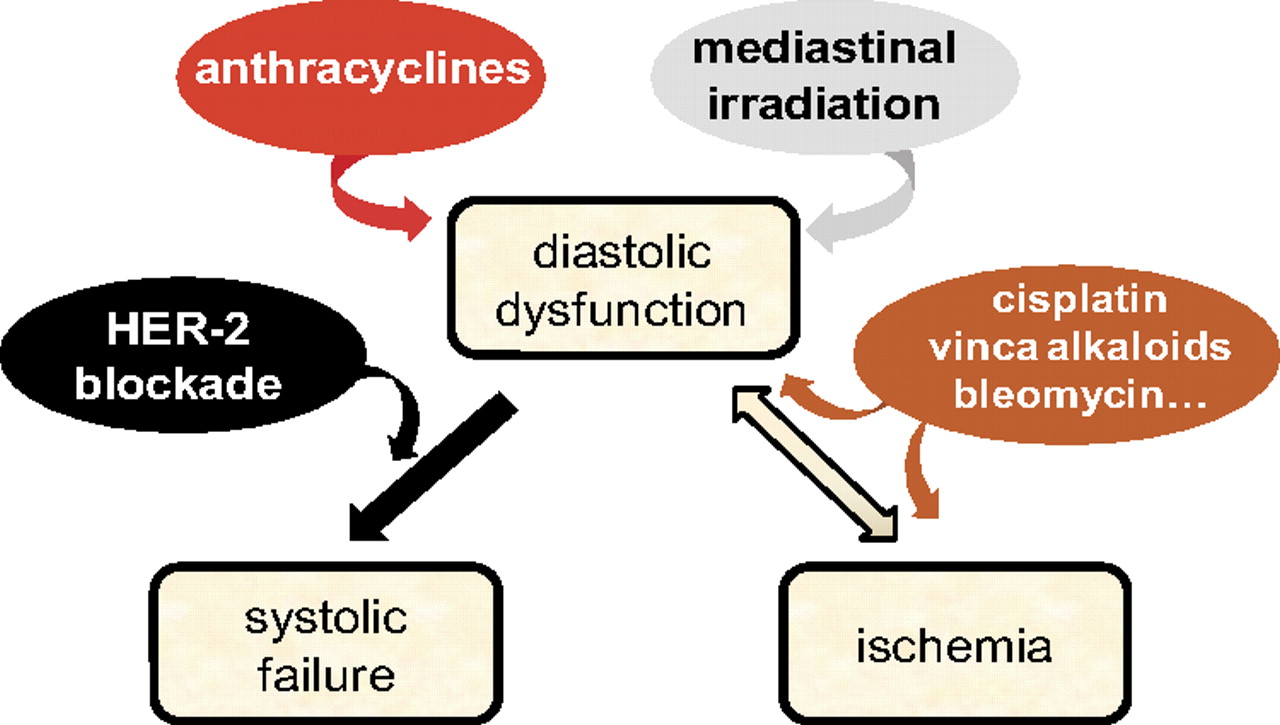
Diastolic dysfunction bridges anticancer therapies to ischemia or systolic failure. Anthracyclines or mediastinal irradiation cause diastolic dysfunction that induces ischemia or aggravates with ischemia induced by other chemotherapeutics. HER-2 blockade facilitates progression of diastolic dysfunction to systolic failure.
Targeted Drugs Are Not Magic Bullets
More than 1000 drugs are being developed by pharmaceutical companies to target tyrosine kinases or, to a lesser extent, serine-threonine kinases that are crucial to tumor growth; meanwhile, reports or concerns about cardiotoxicity from the approved targeted drugs accumulate steadily. Thus, both hopes and fears dominate the field of targeted therapies.
Cardiotoxicity from targeted drugs is often said to occur “unexpectedly” (De Keulenaer et al., 2010). Pharmacologists would perceive this as an oversimplification. As long as a molecular entity governed cardiac development or maintained expression levels in the postnatal heart, targeting that particular entity would cause cardiotoxicity quite predictably. Defective expression of VEGF caused embryonic death that was accompanied by prominent cardiac malformations (Bi et al., 1999); genetic deletion of HER-2 caused cardiac embryonic death (Lee et al., 1995). Either information was available before bevacizumab and trastuzumab were shown to cause cardiotoxicity in patients with cancer. Global development of drugs targeted at many other biological moieties will emerge over the next few years. Cardiotoxicity from new agents should be said to occur “predictably” if a given molecular target participated in one or more cardiac functions.
Research on targeted drugs is also biased by the assumption that modeling a TKI to a given kinase or an intended number of kinases would minimize the risk of targeting other kinases; hence, cardiac toxicity that was caused by any such designed TKI would be liable to interpretation and proper treatment. Pharmacologists would perceive this as a case of overt enthusiasm. Sunitinib was thoughtfully designed as a multikinase inhibitor that targeted VEGFr, platelet-derived growth factor α/β, colony-stimulating factor 1 receptor, FMS (Fibromyalgia Syndrome-like)-related tyrosine kinase 3, and few other kinases; however, the available evidence shows that, at therapeutic plasma levels, sunitinib would inhibit approximately 90 kinases. Inhibition of which kinase or combination of kinases caused cardiotoxicity from sunitinib would be very difficult to ever identify. Almost all of the approved TKIs bind to more kinases, albeit with different Kd values; cardiotoxicity from TKIs therefore correlated with such a lack of target specificity (Hasinoff, 2010).
As mentioned previously in text, trastuzumab worsened cardiotoxicity from concomitant anthracyclines; however, trastuzumab administered after anthracyclines caused reversible systolic dysfunction that only occasionally accompanied with ultrastructural changes typical of anthracycline cardiotoxicity. This and other factors provided a rationale for distinguishing type 1 cardiotoxicity (induced by anthracyclines) from type 2 cardiotoxicity (induced by trastuzumab) (Ewer and Lippman, 2005). Other targeted drugs might fit in type II cardiotoxicity. With that said, one cannot ignore that targeted therapies cause more cardiotoxicity in the general oncologic population than in selected patients of clinical trials. This occurs with angiogenesis inhibitors (Schmidinger et al., 2008), it probably occurs with trastuzumab to some extent (Menna et al., 2008), and it remains under scrutiny for imatinib and cogeners and dual TKIs of HER-1/HER-2 such as lapatinib (see also Table 1). Therefore, it seems that comorbidities influenced cardiotoxicity from targeted therapies similar to the way they influenced cardiotoxicity from old generation chemotherapeutics, presumably because elimination of growth and survival factors by the targeted drugs made the heart more vulnerable by hemodynamic or metabolic stress. Furthermore, early clinical experience showed that cardiotoxicity from targeted drugs may move beyond contractile dysfunction or ischemia; angiogenesis inhibitors and lapatinib cause QT prolongation that might put patients at risk for torsade de pointes and sudden death (Menna et al., 2008).
Targeted therapies have been around for few years only; the epidemiologic dimension of cardiotoxicity from these drugs, whether in isolation or in concert with comorbidities, will need to be defined at longer periods of follow-up. With these concerns in mind, it would be wise surrender to the concept that there are no magic bullets that cure a patient without introducing dangerous liaisons with the cardiovascular system or comorbidities. Pharmacologists would better serve patients with cancer and survivors by predicting the risk of cardiotoxicity or by elucidating means for preventing or treating it.
The Need for Better Preclinical Models and Clinical Trials
There is an unmet need for preclinical models that predict the risk of cardiotoxicity from antitumor drugs. Studies with ex vivo human myocardial samples exposed to anthracyclines in vitro helped to define the enzymology and pharmacokinetics of secondary alcohol metabolites (Salvatorelli et al., 2007, 2009); however, this model could not explore the pathobiology of the “multiple-hit hypothesis.” Animal models of chronic cardiotoxicity are limited by heterogeneous drug metabolism, such that the enzymology and net levels of anthracycline secondary alcohol metabolites would be quite different from those of human heart (Minotti et al., 2004); moreover, animal models require weeks of treatment and weeks or months of observation but in the absence of comorbidities only few animals would develop cardiac abnormalities (Gianni et al., 2008). The spontaneously hypertensive rat (SHR) shows a greater sensitivity to chronic anthracycline treatment and develops functional, histologic, and biochemical aspects of cardiotoxicity that more closely resemble those of patients with cancer (Herman et al., 1985); however, SHR models would be biased by the same metabolic pitfalls as those of normotensive rats, confounding an interpretation of the cause-and-effect relations between the cardiac levels of secondary alcohol metabolites and their toxic synergism with hypertension. An alternative but not mutually exclusive approach would be to treat SHR with both anthracyclines and nonanthracycline chemotherapeutics, thereby reproducing clinical conditions where diastolic dysfunction from one drug interacted with ischemia from another drug; unfortunately, there is a lack of studies in that direction.
Animal models do not always anticipate the risk of cardiotoxicity from targeted drugs. Trastuzumab did not recognize the ectodomain of HER2 in the murine heart, and hence, it lacked cardiotoxicity in preclinical studies (Pegram and Ngo, 2006). Mice treated with sunitinib developed cardiomyocyte apoptosis but this only occurred after inducing hypertension with a sympathicomimetic agents (Cheng and Force, 2010). This denotes that VEGF-VEGFr signaling regulates capillary density and helps the cardiovascular system to withstand hemodynamic load (Schmidinger et al., 2008). There is a lack of models that explored the capability of sunitinib of causing hypertension and then cardiotoxicity by its own.
Clinical trials may underestimate the risk of cardiotoxicity from antitumor drugs: phase III studies exclude or only marginally include patients with risk factors; cardiac surveillance almost invariably rests with LVEF measurements only; and follow-up may be too short to predict late-onset cardiotoxicity due to sequential stressors. Pharmacologic reasonings call for proof-of-concept trials in which patients are stratified by risk factors and probed for asymptomatic dysfunction(s) precursor to late-onset cardiotoxicity.
Reshaping Prevention and Treatment
For many years, dexrazoxane was synonymous with preventing anthracycline cardiotoxicity. Formally labeled as an iron chelator, dexrazoxane was thought to diminish the formation of anthracycline-iron complexes that generated reactive oxygen species in excess of the antioxidant defenses of cardiomyocytes. This mechanism has been questioned in recent years; perhaps more importantly, clinical use of dexrazoxane has been limited by unconfirmed concerns that it could also diminish anthracycline activity in tumors (Gianni et al., 2008; Menna et al., 2008). With the absence of regulatory or educational initiatives to resuscitate the popularity of dexrazoxane, anthracycline cardiotoxicity can be prevented by replacing rapid infusions with slow infusions or liposomal formulations that, by different mechanisms, diminish the cardiac uptake of anthracyclines (Minotti et al., 2004); unfortunately, many doctors perceive these procedures to be too expensive or laborious.
The concepts of prevention and treatment clearly need to be reshaped. The notion that late-onset cardiotoxicity develops over months or years through asymptomatic cardiac dysfunction suggests that drugs used “to treat” symptomatic events should be used earlier “to prevent” subclinical damage. In limited studies, prophylactic commencement of β blockers or angiotensin I-converting enzyme inhibitors (ACEI) prevented systolic dysfunction induced by cumulative doses of chemotherapeutics (Kalay et al., 2006; Cardinale et al., 2006), as one would expect from the known effects of such drugs on reducing heart rate and afterload. However, it is noteworthy that there may be other mechanisms for the preventative efficacy of β blockers and ACEI (or angiotensin II receptor blockers, ARB). Catecholamines and angiotensin II down-regulate endothelial expression of neuregulin 1, i.e., the ligand promoting heterodimerization of HER-2 with HER-4 and activation of salvage pathways downstream of HER-2 (Lemmens et al., 2006). By shielding endothelial cells from catecholamines or angiotensin II, both β blockers and ACEI (or ARB) would indirectly up-regulate neuregulin 1 expression, making cardiomyocytes more resistant to hemodynamic and chemical stress.
Prophylactic commencement of cardiovascular drugs is uncommon outside of exploratory studies; doctors believe that patients with cancer showing normal cardiac function should not be exposed to discomfort that can be caused by such drugs. These concerns should be weighed against the notion that delayed commencement of ACEI would only transiently affect the progression of chemotherapy-related cardiomyopathy (Gianni et al., 2008); moreover, experimental evidence suggests that progressive cardiomyopathy might be accompanied by functional uncoupling of HER-2 activation from downstream survival signals, making β blockers and ACEI (or ARB) less protective (Doggen et al., 2009). With the possible exception of conditions where oncologists may need a persistent blockade of HER-2 and microvasculature-derived signals for optimal control of tumor growth, the balance of evidence would favor the earliest possible commencement of cardiovascular medications.
The building of late-onset systolic failure or ischemia on earlier asymptomatic diastolic dysfunction calls for pharmacological interventions specific to diastolic LV stiffness. In patients with chronic diastolic dysfunction (i.e., heart failure with preserved LVEF), nitrates and diuretics are indicated for reducing fluid retention and cardiac filling pressure. The efficacy of β blockers is controversial; recent evidence suggests that nebivolol could be better than others, presumably because of its corollary effects on improving endothelial dysfunction through antioxidative properties and functional coupling of nitric-oxide synthase (Kamp et al., 2010). Research on drugs that specifically prevented or mitigated diastolic LV stiffness (lusitropic agents) should nonetheless be considered a priority in the years to come.
Pharmacology and the Future of Cardio-Oncology
We described how cardiotoxicity from antitumor drugs has changed in past years. Asymptomatic cardiotoxicity (contractile dysfunction or ischemia) is an increasingly frequent consequence of antitumor therapies. Without early cardiovascular medications, asymptomatic cardiotoxicity synergizes with multiple hits and progresses toward late-onset cardiac events (Fig. 2). New waves of antitumor drugs will make cardiotoxicity change even faster in the coming years. The vulnerability of the heart as a paracrine intercellular system anticipates the occurrence of new cardiotoxicity phenotypes caused by interruption of cross-talks between the cardiac microvasculature and myocytes, for example (De Keulenaer et al., 2010). In the next few years, research on cardiac progenitor cells that repopulate foci of myocyte damage will be significant. Progenitor cells succumb to anthracyclines (De Angelis et al., 2010); however, little is known regarding their vulnerability by targeted drugs. This calls for ad hoc studies because targeting progenitor cells would prime survivors of cancer to inefficient myocardial repopulation.

Early cardiovascular medications and cardiac safety versus multiple hits and late-onset cardiac events. Asymptomatic cardiotoxicity may be induced by anthracyclines, nonanthracycline chemotherapeutics, targeted drugs, in concert with polymorphisms, or mediastinal irradiation. Early commencement of cardiovascular medications pushes asymptomatic cardiotoxicity toward cardiac safety. Without such medications, asymptomatic cardiotoxicity synergizes with multiple hits (comorbidities, other stressors) and progresses toward late-onset events. See “text” for explanations.
Cardiologists and oncologists will be asked to manage new paradigms of cardiotoxicity; however, mechanistic insight and therapeutic measures will only come from pharmacological research. The heart of cardio-oncology will rest with pharmacological foundations.
Footnotes
This work was supported by Associazione Italiana Ricerca sul Cancro [IG 2004–2008] and University Campus Bio-Medico of Rome [Special Project 1057/08 “Cardio-Oncology”].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.165860.
-
ABBREVIATIONS:
- CHF
- congestive heart failure
- MI
- myocardial infarction
- HER-2
- epidermal growth factor receptor 2
- VEGF
- vascular endothelial growth factor
- VEGFr
- VEGF receptor
- TKI(s)
- small tyrosine kinase inhibitor(s)
- LV
- left ventricular
- LVEF
- left ventricular ejection fraction
- SHR
- spontaneously hypertensive rat
- ACEI
- angiotensin I-converting enzyme inhibitors
- ARB
- angiotensin II receptor blockers.
- Received March 3, 2010.
- Accepted March 23, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue




{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Cardiotoxicity of Antitumor Drugs: Old Aspects and New Issues
- What Is Cardio-Oncology?
- Lifetime Risk of Cardiotoxicity: A Matter of Long-Lasting Pharmacological Signatures
- Drugs Are Not Alone: The Multiple-Hit Hypothesis
- From Antitumor Drugs to Diastolic Dysfunction and More
- Targeted Drugs Are Not Magic Bullets
- The Need for Better Preclinical Models and Clinical Trials
- Reshaping Prevention and Treatment
- Pharmacology and the Future of Cardio-Oncology
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters