


Abstract
The antitumor anthracycline doxorubicin induces a dose-related cardiotoxicity that correlates with the myocardial levels of its secondary alcohol metabolite doxorubicinol. Combining doxorubicin with taxanes such as paclitaxel or docetaxel may aggravate cardiotoxicity, presumably because the taxanes cause an allosteric-like stimulation of cytoplasmic aldehyde reductases that convert doxorubicin to doxorubicinol in the heart. A less severe aggravation of cardiotoxicity was observed on combining taxanes with epirubicin, a closely related analog of doxorubicin; therefore, we characterized whether the cardiac tolerability of epirubicin-taxane therapies could be due to a defective taxane stimulation of the conversion of epirubicin to its secondary alcohol metabolite epirubicinol. Comparisons between doxorubicin and epirubicin in isolated human heart cytosol showed that epirubicin exhibited a lower Vmax/Km value for reaction with aldehyde reductases and a defective stimulation of epirubicinol formation by paclitaxel or docetaxel. A similar pattern occurred in the soluble fraction of human myocardial strips incubated in plasma with anthracyclines and paclitaxel or docetaxel, formulated in their clinical vehicles Cremophor EL or polysorbate 80. Doxorubicin, but not epirubicin, was also able to generate reactive oxygen species in the membrane fraction of myocardial strips; however, the levels of doxorubicin-derived reactive oxygen species were not further augmented by paclitaxel. These results support the notion that taxanes might aggravate the cardiotoxicity of doxorubicin through a specific stimulation of doxorubicinol formation. The failure of paclitaxel or docetaxel to stimulate epirubicinol formation therefore uncovers an important determinant of the improved cardiac tolerability of epirubicin-taxane combinations.
The anthracycline doxorubicin and the taxanes paclitaxel or docetaxel are highly active antitumor drugs. Doxorubicin-paclitaxel combinations were shown to induce high response rates in women with metastatic breast cancer, particularly when the two drugs were administered almost concomitantly; unfortunately, however, the clinical use of doxorubicin-paclitaxel combinations was limited by a higher than expected incidence of the cardiac toxicity known to be induced by doxorubicin. For example, doxorubicin immediately followed by paclitaxel caused congestive heart failure (CHF) at a cumulative dose of 420 to 480 mg of doxorubicin/m2, which was below a safety limit usually set at ∼500 to 550 mg of doxorubicin/m2 (Gianni et al., 1995). Paclitaxel alone had not been reported to induce CHF; therefore, the unexpected cardiotoxicity of doxorubicin-paclitaxel uncovered a cardiotoxic synergism between the anthracycline and the taxane and prompted studies that identified 360 mg of doxorubicin/m2 as the highest cumulative dose of anthracycline, which could be safely administered in combination with paclitaxel (Perotti et al., 2003).
A higher than expected incidence of cardiotoxicity was not observed in metastatic breast cancer patients treated with doxorubicin immediately followed by docetaxel, but the median cumulative dose of doxorubicin adopted in these studies was quite low (378 mg/m2) (Nabholtz et al., 2003). Moreover, studies of doxorubicin-taxane combinations as primary or adjuvant therapies of operable breast cancer cast doubt on the actual safety of doxorubicin-docetaxel versus doxorubicin-paclitaxel combinations and suggest that the cardiotoxicity of doxorubicin-taxane schedules would be determined primarily by the cumulative dose of doxorubicin associated with paclitaxel or docetaxel (Nabholtz et al., 2002; Gianni et al., 2005; Martin et al., 2005).
We demonstrated that both paclitaxel and docetaxel were able to stimulate an enzymatic conversion of doxorubicin to its secondary alcohol metabolite doxorubicinol (Salvatorelli et al., 2006b). The taxanes acted through an allosteric modulation of NADPH-dependent cytoplasmic reductases, and such an effect was characterized with translational models of human heart that obviated the different levels of expression and activity of the reductases in the heart of laboratory animals (Maessen et al., 1987; Minotti et al., 2004a). Inasmuch as doxorubicinol had been implicated to induce CHF, we proposed that the stimulation of doxorubicinol formation induced by paclitaxel or docetaxel could help to explain the higher than expected incidence of cardiotoxicity induced by doxorubicin-taxane therapies (Salvatorelli et al., 2006b).
Epirubicin is a closely related anthracycline that shows antiproliferative activity at doses ∼1.5 times higher than doxorubicin. This is caused by a well established pattern of epirubicin glucuronidation and improved elimination in bile and urine (Innocenti et al., 2001); interestingly, however, epirubicin induces CHF at doses ∼1.8 to 2 times higher than doxorubicin (Bonadonna et al., 1993), as if glucuronidation and body clearance were not the only factors that diminished its dose-related cardiotoxicity. Experiments with human myocardial samples suggested that the cardiotoxicity of epirubicin might be limited by its impaired conversion to the secondary alcohol metabolite epirubicinol (Salvatorelli et al., 2006a).
Clinical trials of epirubicin immediately followed by paclitaxel showed that the incidence of CHF was low up to 990 mg of epirubicin/m2, a dose level that formally exceeded a 2:1 ratio with the highest doxorubicin dose that proved safe in combination with paclitaxel. The incidence of CHF increased at a cumulative dose of 1080 mg of epirubicin/m2, but this usually occurred in patients with additional cardiac risk factors (Gennari et al., 1999). Epirubicin immediately followed by docetaxel showed a similar profile of cardiac tolerability; moreover, a severe but reversible decline of myocardial contractility only occurred in a patient who received a cumulative dose of 870 mg of epirubicin/m2, a dose level that remained >2 times higher than the maximal cumulative dose of doxorubicin recommended for use in combination with paclitaxel (Pagani et al., 2000). On balance, these results suggested that paclitaxel or docetaxel caused a less severe aggravation of the dose-related cardiotoxicity of epirubicin.
We hypothesized that paclitaxel and docetaxel failed to stimulate epirubicinol formation, and we designed experiments that probed such a possibility in translational models of human heart. In addressing this issue, we considered that anthracyclines might cause cardiotoxicity also by forming reactive oxygen species (ROS) (Gewirtz, 1999; Minotti et al., 2004a). We therefore characterized whether doxorubicin and epirubicin also exhibited a different taxane modulation of their metabolic conversion to ROS.
Materials and Methods
Chemicals. We used doxorubicin [7-(3-amino-2,3,6-trideoxy-α-l-lyxo-hexopyranosyl)doxorubicinone]; doxorubicinol [7-(3-amino-2,3,6-trideoxy-α-l-lyxo-hexopyranosyl)-13-dihydroxydoxorubicinone]; epirubicin [7-(3-amino-2,3,6-trideoxy-R-l-arabino-hexopyranosyl)-doxorubicinone]; epirubicinol [7-(3-amino-2,3,6-trideoxy-R-l-arabino-hexopyranosyl)-13-dihydroxydoxorubicinone] (Nerviano Medical Sciences, Milan, Italy); lyophilized paclitaxel [5β,20-epoxy-1,2α,4,7β,10β,13α-hexahydroxytax-11-en-9-one, 4,10-diacetate, 2-benzoate, 13-ester, with (2R,3S)-N-benzoyl-3-phenylisoserine] (Bristol Myers Squibb, Wallingford, CT); lyophilized docetaxel [(2R,3S)-N-carboxy-3-phenylisoserine,N-tert-butyl ester, 13-ester, with 5β-20-epoxy-1,2α,4,7β,10β,13α-hexahydroxytax-11-en-9-one, 4-acetate, 2-benzoate, trihydrate] (Rhône-Poulenc Rorer, Vitry-surSeine Cedex, France); ethyl-1-benzyl-3-hydroxy-2(5H)-oxopyrrole-4-carboxylate (EBPC) (Tocris Cookson Inc., Bristol, UK); 2,7-difluorospirofluorene-9,5′-imidazolidine-2′,4′-dione (AL1576) (Alcon Laboratories, Fort Worth, TX); and dichlorofluorescin-diacetate (DCFH-DA) and dichlorofluorescein (DCF) (Invitrogen, Carlsbad, CA). Doxorubicinone and doxorubicinolone (13-dihydroxydoxorubicinone) were prepared by us through the thermoacid hydrolysis of doxorubicin and doxorubicinol, respectively (Menna et al., 2002). Ethanol was from Carlo Erba (Milan, Italy); Cremophor EL (polyoxyethyleneglycerol triricinoleate 35), polysorbate 80 (polyoxyethylenesorbitan-20-mono-oleate), quercetin (3,3′,4′,5,7-pentahydroxyflavone), bafilomycin (type A1), and all other chemicals were obtained from Sigma-Aldrich (Milan, Italy).
Human Myocardial Samples. Small myocardial samples were obtained from 91 male and female patients (62 ± 6 years) undergoing aortocoronary bypass grafting. All of the specimens were derived from the lateral aspect of excluded right atrium, and they were routinely disposed of by the surgeons during cannulation procedures; therefore, the patients were not exposed to any unjustified loss of tissue (Salvatorelli et al., 2006b). Sampling procedures were in compliance with the guidelines of the Institutional Ethical Committee, and written informed consent was obtained from all of the patients.
Anthracycline Metabolism in Human Heart Cytosol. Pools of 10 to 15 myocardial specimens were processed for cytosol preparation by homogenization, ultracentrifugation, and 65% ammonium sulfate precipitation of 105,000g supernatants. Next, the cytosol was treated with 100 mM dithiothreitol, pH 8.9, and gel-filtered on homemade Sepharose 6B minicolumns to induce the disassembly of iron-sulfur clusters that otherwise oxidized secondary alcohol metabolites back to their parent anthracyclines and limited accurate kinetic measurements of metabolite formation (Minotti et al., 2004b). Anthracycline metabolism was reconstituted in 0.25-ml incubations that contained cytosol (0.15 mg of protein), 0.25 mM NADPH, and anthracyclines, in 0.3 M NaCl, at pH 7.0 and 37°C. Where indicated, taxanes or inhibitors were also included. The incubation time and the concentration of each test compound are specified under Results and in the legends for figures or tables. Because of their limited water solubility, doxorubicinone and taxanes or inhibitors were stored concentrated in ethanol and diluted as appropriate just before their use. To permit direct comparisons, all the incubations were adjusted to contain the same final volume of ethanol (5 μl, formally corresponding to 0.34 mM); under such defined conditions, ethanol did not precipitate cytosolic proteins during the course of the experiments.
Anthracycline Distribution and Metabolism in Human Myocardial Strips. We used a model that was developed and validated in our laboratory (Salvatorelli et al., 2006a,b). Thin strips (∼2 × 10 mm; <0.1 g) were carefully dissected from the ex vivo myocardial samples and incubated in 2 ml of fresh human plasma with anthracyclines in the absence or presence of taxanes; anthracyclines were used 10 μM, whereas taxanes were used 2 and 6 or 15 μM. Paclitaxel and docetaxel were formulated in their clinical vehicles Cremophor EL or polysorbate 80, respectively; both vehicles were mixed with equal volumes of ethanol to reproduce the clinical ethanol-vehicle cosolvent systems (see Results). After 4-h incubation at 37°C in a Dubnoff metabolic bath under an air atmosphere, the strips were washed with ice-cold 0.3 M NaCl, homogenized in a minimal volume of the same medium, and centrifuged for 90 min at 105,000g to separate soluble and whole-membrane fractions that were assayed for anthracyclines, secondary alcohol metabolites, and taxanes. Aliquots of plasma were also taken and assayed for anthracyclines and alcohol metabolites.
Where indicated, the experimental conditions were adapted to monitor the formation of basal or anthracycline-induced formation of ROS. We used a modification of the method based on the sequential uptake of DCFH-DA, its deacetylation to DCFH by cellular esterases, and the oxidation of DCFH to DCF by H2O2 and peroxidases or trace amounts of iron (Salvatorelli et al., 2006a). The strips were incubated in 4 ml of 50 mM phosphate buffer, 123 mM NaCl, and 5.6 mM glucose, pH 7.4, added with the membrane-permeable DCFH-DA. The latter was used 50 μM, a concentration that gave a homogeneous distribution of DCFH to both soluble and membrane fractions of the strips (Salvatorelli et al., 2006a). After 1 h in the dark, the strips were washed with 0.3 M NaCl and incubated for 4 h in human plasma added with anthracyclines and taxanes; where indicated the strips were also exposed to bafilomycin A1 (300 or 90 nM during the loading with DCFH-DA or the subsequent incubation in plasma, respectively). At the end of the incubations the strips were homogenized in 2 ml of ice-cold 123 mM NaCl that was added with 1 mM 4-hydroxytempol, an antioxidant, to prevent further oxidation of DCFH to DCF during tissue disruption and homogenization. Soluble and membrane fractions were eventually assayed for DCF.
Myocardial Efflux of Anthracyclines and Secondary Alcohol Metabolites. In selected experiments, the strips were incubated in plasma with anthracyclines for 4 h, quickly washed with 0.3 M NaCl, pH 7.0, and reincubated for an additional 15 min in 2 ml of anthracycline-free fresh plasma. Aliquots of plasma were taken at 5 or 15 min and assayed for anthracyclines or secondary alcohol metabolites. To permit direct comparisons between strips with different contents of anthracyclines or metabolites the efflux of a given analyte was calculated as the ratio [analyte in plasma]/[analyte in plasma + analyte in the strips].
High-Performance Liquid Chromatography Assays for Anthracyclines and Taxanes. Plasma, isolated cytosol, and soluble or membrane fractions of myocardial strips were extracted with a 4-fold volume of CHCl3/CH3OH (1:1), and the organic phases were combined to obtain a total extract (Salvatorelli et al., 2006b). Twenty microliters of the extracts was assayed for anthracyclines and taxanes by reversed phase high-performance liquid chromatography (HPLC) in a Hewlett Packard 1100 system (Hewlett Packard Palo Alto, CA). The extracts of samples with doxorubicin and taxanes were loaded onto a Nucleosil C18 column (100 × 4.6 mm, 5 μm; Supelco, Bellefonte, PA) operated at 25°C and eluted at the flow rate of 1 ml/min for a total 25-min run time [15-min linear gradient from 100% 50 mM NaH2PO4, pH 4.0, to (50–50%) CH3CN-50 mM NaH2PO4, followed by a 10-min isocratic elution with (50–50%) CH3CN-50 mM NaH2PO4]. Retention times were 11.7 and 12.4 min for doxorubicinol and doxorubicin, respectively, and 18.9 and 20.6 min for docetaxel and paclitaxel, respectively. The extracts of samples with epirubicin or doxorubicinone and taxanes were eluted over the same run time, but the linear gradient was completed in 5 min; the retention times were 5.9 and 6.3 min for epirubicinol and epirubicin, 6.7 and 7.2 min for doxorubicinolone and doxorubicinone, and 10.6 and 11.9 min for docetaxel and paclitaxel, respectively. Withinday and between-day coefficients of variation were ∼2 and ∼7%. Anthracyclines were detected fluorimetrically (excitation at 480 nm, emission at 560 nm), whereas paclitaxel and docetaxel were detected by UV absorbance (λmax = 230 nm) (Grasselli et al., 2001). Due to differences in the fluorescence yield of alcohol metabolites versus parent anthracyclines, we did not use a fixed internal standard such as daunorubicin; instead, all analytes were carefully quantified against standard curves obtained after comparable extractive and chromatographic procedures (0.005–10 μM for parent anthracyclines or 0.001–10 μM for secondary alcohol metabolites; r ≥ 0.996; p < 0.0001). In the studies of myocardial strips, all the values (nanomoles per gram of tissue) were expressed as micromolar equivalents upon considering that the cardiac tissue has a density very similar to that of water (Salvatorelli et al., 2006b). The levels of anthracyclines in plasma (nanomoles per milliliter) were also expressed as micromolar equivalents normalized to the weight of the strips in the incubations. Upon consideration of factors such as the weight of myocardial strips or sample dilution, the lowest detection limit of the standard curves was many times below the level of a given analyte in myocardial strips or isolated cytosol. For epirubicinol, which always formed in lesser amounts compared with doxorubicinol or doxorubicinolone, the lowest detection limit of the HPLC assay was ∼9 or ∼360 times below its level of formation in myocardial strips or isolated cytosol, respectively. Taxanes were also quantified against proper standard curves (0.001–10 μM paclitaxel or docetaxel; r ≥ 0.995; p < 0.0001) and expressed as micromolar equivalents normalized to the weight of myocardial strips; the lowest detection limit of the HPLC assay was ∼60 times below the minimal level of either taxane in the soluble or membrane fraction of the strips.
High-Performance Liquid Chromatography Assay for DCF-Detectable Hydrogen Peroxide. In the assay for DCF-detectable H2O2, the soluble and membrane fractions of myocardial strips were extracted with 2 volumes of CH3OH/CHCl3 (1:1), and 25 μl of the upper phase was analyzed by HPLC using a Nucleosil C18 column (100 × 4.6 mm; 5 μm), operated at 25°C, and eluted at the flow rate of 1 ml/min for a total 25-min run time [15-min linear gradient from 100% 50 mM NaH2PO4, pH 4.0, to (50–50%) CH3CN-50 mM NaH2PO4, followed by a 10-min isocratic elution with (50–50%) CH3CN-50 mM NaH2PO4]. The fluorescent peak of DCF (excitation at 488 nm, emission at 525 nm) was identified by cochromatography with a DCF standard (retention time 18.3 min), and its area under the curve was corrected for that of strips extracted immediately after the loading with DCFH-DA. The differences indicated the net level of DCF formation during 4-h incubations and were quantified against a standard curve of 0.0025 to 2.5 μM DCF (r = 0.98; p < 0.0001). Basal or anthracycline-induced H2O2 formation was quantified by assuming that 1 nmol of H2O2 caused the formation of 0.43 nmol of DCF/g human myocardium; this stoichiometry was calculated by correcting total DCF formation for H2O2-independent DCFH oxidation or side reactions of DCF with cellular redox agents other than H2O2 (Rota et al., 1999; Salvatorelli et al., 2006a). The values were expressed as micromolar equivalents normalized to the weight of myocardial strips. Under such defined conditions, the lowest detection limit of the HPLC assay was ∼7 or ∼96 times below the minimal level of H2O2 formation in the soluble or membrane fraction of the myocardial strips, respectively.
Other Assays and Conditions. Proteins were measured by the bicinchoninic acid method. The myocardial release of myoglobin, troponin T, and creatine kinase isoenzyme MB was determined by an electrochemiluminescence immunoassay with an Elecsys 2010 Analyzer (Roche Diagnostics, Mannheim, Germany), according to the manufacturer's instructions (Salvatorelli et al., 2006b). Unless otherwise indicated, all the values were means ± S.E. of at least three experiments. Data were analyzed by one-way analysis of variance followed by Bonferroni's test for multiple comparisons; where indicated, unpaired Student's t test was also applied. Differences were considered significant when p was <0.05. Other details are given under Results and in the legends for figures or tables.
Results
Limited Formation of Epirubicinol in Isolated Human Heart Cytosol and Myocardial Strips. Doxorubicin and epirubicin share a tetracyclic ring system (doxorubicinone) with adjacent quinone-hydroquinone moieties and a short side chain with a carbonyl group at C13. Secondary alcohol metabolites are formed after an NADPH-dependent two-electron reduction of the side chain carbonyl group (Fig. 1A). Doxorubicin and epirubicin also contain an amino sugar (daunosamine), with epirubicin differing from doxorubicin in the axial-to-equatorial epimerization of the hydroxyl group at C4′ in this moiety. Experiments with isolated human heart cytosol, NADPH and 50 μM anthracyclines showed that the levels of formation of doxorubicinolone (the alcohol metabolite of doxorubicinone) were ∼16 or ∼43 times higher than those of doxorubicinol or epirubicinol (Fig. 1B).

Anthracycline secondary alcohol metabolite formation in human heart cytosol. A, anthracycline secondary alcohol metabolites are formed after a two-electron reduction of the side chain carbonyl group of doxorubicinone. B, in human heart cytosol, the yield of secondary alcohol metabolites was diminished by the presence of an amino sugar moiety (doxorubicin) and its epimerization at C4′ (epirubicin). The incubations (0.25-ml final volume) contained human heart cytosol (0.15 mg of protein), 0.25 mM NADPH, and 50 μM doxorubicinone, doxorubicin, or epirubicin. Secondary alcohol metabolites were measured after 4 h, as described under Materials and Methods. Values are means ± S.E. of 12 to 15 two experiments and were analyzed by one-way analysis of variance followed by Bonferroni's test for multiple comparisons. *, p < 0.0001 for the secondary alcohol metabolite of doxorubicinone (doxorubicinolone) versus doxorubicinol and epirubicinol; **, p > 0.05 for doxorubicinol versus epirubicinol (p < 0.001 by unpaired Student's t test).
We next characterized alcohol metabolite formation in human myocardial strips incubated in plasma with anthracyclines. Doxorubicin and epirubicin were used at 10 μM, similar to the plasma maximal concentrations (Cmax) produced by 5-min boluses of 60 mg of doxorubicin/m2 or 90 mg of epirubicin/m2 (Gianni et al., 1997; Grasselli et al., 2001). Under such defined conditions the strips did not release myoglobin, troponin T, or creatine kinase MB isozyme, demonstrating that the cardiac tissue did not undergo an acute damage that otherwise altered the mechanisms and levels of formation of anthracycline metabolites (Gewirtz, 1999; Salvatorelli et al., 2006a,b). An examination of 27 strips exposed to 10 μM anthracyclines demonstrated a total content of 9.1 ± 0.7 μM doxorubicin or 8.8 ± 1.1 μM epirubicin, with the individual levels ranging from 4.9 to 15 μM for doxorubicin and from 3 to 20 μM for epirubicin. That numerous strips contained anthracyclines above their formal concentration in plasma was in keeping with the known ability of myocardium to accumulate these drugs from biological fluids, but the highest accumulation factors determined in this study (1.5–2) were lower than those determined by others when studying isolated rabbit heart in laboratory buffers (factors ≥3) (Olson et al., 1988); this denoted the ability of plasma proteins to bind anthracyclines and limit their diffusion in tissues (Finlay and Baguley, 2000). Under such defined conditions doxorubicin and epirubicin distributed equally well to the soluble or whole membrane fraction of the strips, with the concentration of epirubicin in the membrane fraction being slightly higher than that of doxorubicin (Table 1). Both doxorubicinol and epirubicinol were found only in the soluble fraction of the strips, but the levels of epirubicinol were ∼40% lower than those of doxorubicinol (Table 1). The absence of doxorubicinol or epirubicinol in the membrane fraction of the strips could not be attributed to their inadequate extraction from that fraction; in fact, titrating the membrane fraction of control strips with known amounts of doxorubicin(ol) or epirubicin(ol) showed that the extraction procedure adopted in this study always recovered ≥95% of both parent anthracyclines and secondary alcohol metabolites. The extracts of epirubicin-treated strips did not contain other polar fluorescent metabolites that could be attributed to epirubicin- or epirubicinol-glucuronides (data not shown). This was consistent with the notion that epirubicin(ol) glucuronidation would be catalyzed primarily in liver by UDP glucuronosyltransferase 2B7 (Innocenti et al., 2001).
Anthracycline distribution and secondary alcohol metabolite formation in human myocardial strips
Human myocardial strips were incubated in plasma with 10 μM anthracyclines, as described under Materials and Methods. Values are means ± S.E. of six to 20 experiments and were analyzed by one-way analysis of variance followed by Bonferroni's test for multiple comparisons.
Doxorubicinone exhibited an apparently lower distribution to the soluble or membrane fractions of the strips; nevertheless, the levels of doxorubicinolone in the soluble fraction of the strips were ∼40 or ∼130% higher than those of doxorubicinol or epirubicinol, respectively. Doxorubicinolone also was found in the membrane fraction of the strips, and especially in plasma (Table 1). Control experiments demonstrated that neither plasma nor an isolated membrane fraction could reduce doxorubicinone to doxorubicinolone in the presence of NADPH (data not shown); therefore, the data suggested that doxorubicinolone equilibrated across soluble and membrane fractions and could also diffuse from the strips in plasma. To probe this latter possibility, we loaded the strips with anthracyclines and incubated them in anthracycline-free plasma that was assayed for the release of parent drugs and alcohol metabolites. As reported in Table 2, the strips released much more doxorubicinone than doxorubicin or epirubicin; they also released up to ∼50% of doxorubicinolone, but they did not release doxorubicinol or epirubicinol. In this set of experiments, the HPLC assay would have been sensitive enough to detect the efflux of at least 6 or 22% of the doxorubicinol or epirubicinol content of the strips, respectively. The lack of a measurable efflux of doxorubicinol or epirubicinol, as opposed to the sizeable efflux of doxorubicinolone, could not therefore be attributed to major limitations of the assay; instead, the data suggested that the absence of the amino sugar rendered doxorubicinolone (and doxorubicinone) more lipophilic and membrane-permeable than doxorubicin(ol) or epirubicin(ol). Thus, we calculated that, in the strips exposed to anthracyclines for 4 h, the total levels of doxorubicinolone (membrane fraction + soluble fraction + plasma) were ∼8 or ∼13 times higher than those of doxorubicinol or epirubicinol, respectively (cf. Table 1). By having considered that an extracellular diffusion of doxorubicinone probably limited doxorubicinolone formation to some extent, we concluded that the ratios of doxorubicinolone to doxorubicinol or epirubicinol were qualitatively similar to those seen with an isolated cytosol.
Myocardial efflux of anthracyclines and secondary alcohol metabolites
Human myocardial strips were incubated in plasma added with 10 μM doxorubicinone or doxorubicin or epirubicin; after 4 h, the strips were placed in anthracycline-free plasma, which was assayed for the efflux of the parent drugs or their secondary alcohol metabolites. Values were means of two experiments. The efflux was normalized to the total levels of anthracyclines or alcohol metabolites in the incubations (plasma + strips), as described under Materials and Methods. Total levels of anthracyclines were 3.1, 12.5, and 16 μM for doxorubicinone, doxorubicin, and epirubicin, respectively; total levels of alcohol metabolites were 0.52, 0.11, and 0.03 μM for doxorubicinolone, doxorubicinol, and epirubicinol, respectively.
Collectively, the experiments with isolated cytosol or whole myocardial samples showed that the presence and epimerization of an amino sugar moiety were two independent factors that limited anthracycline carbonyl reduction in the human heart, making epirubicinol formation lower than doxorubicinol formation.
Defective Taxane Modulation of Epirubicinol Formation in Isolated Human Heart Cytosol. We further exploited the cytosol model to characterize anthracycline-taxane interactions over a broad range of drug concentrations, some of which would have been too high to be safely probed in myocardial strips. In the first set of experiments, the anthracyclines were used (50 μM), whereas the taxanes were used (0.25 to 10 μM). Under such defined conditions, 0.25 to 2.5 μM paclitaxel or docetaxel stimulated doxorubicinol formation, with a peak of stimulation at 1 μM taxanes, whereas >2.5 μM paclitaxel or docetaxel gradually decreased doxorubicinol back to its basal levels (Fig. 2A). Paclitaxel and docetaxel lacked similar effects on epirubicinol formation (Fig. 2B).
We next determined the catalytic efficiency (Vmax/Km) with which the human heart cytosol formed anthracycline secondary alcohol metabolites. The Vmax/Km value of doxorubicinol formation was ∼2 orders of magnitude lower than that determined for doxorubicinolone; this was caused by both a lower Vmax value and a higher Km value. A similar pattern of lower Vmax/higher Km determined an additional ∼1 order of magnitude decrease of the Vmax/Km value of epirubicinol formation versus doxorubicinol formation (Table 3). The Vmax/Km value of doxorubicinol formation increased approximately four to five times in the presence of 1 μM taxanes but decreased to ∼70 to 80% of control values in the presence of 10 μM taxanes, similar to what was reported previously (Salvatorelli et al., 2006b; also see Table 3). Under comparable conditions, paclitaxel and docetaxel caused essentially no change of the Vmax/Km value of epirubicinol formation (Table 3). The effects of 1 or 10 μM paclitaxel or docetaxel on the Vmax/Km values therefore paralleled the absence or presence and concentration dependence of a taxane effect on the formation of epirubicinol or doxorubicinol.

Effects of paclitaxel or docetaxel on doxorubicinol or epirubicinol formation in human heart cytosol: Studies with 50 μM doxorubicin or epirubicin. The incubations (0.25-ml final volume) contained human heart cytosol (0.15 mg of protein), 0.25 mM NADPH, 0.25 to 10 μM taxanes, and 50 μM doxorubicin (A) or epirubicin (B). Secondary alcohol metabolites were measured after 4 h, as described under Materials and Methods. Values are means ± S.E. of three experiments and are expressed as percentages of control values to permit direct comparisons of incubations with different basal levels of alcohol metabolite formation (nanomoles per milligram of protein per 4 h: 1.4 ± 0.2 or 1.8 ± 0.2 for doxorubicinol in the experiments with paclitaxel or docetaxel, and 0.59 ± 0.07 or 0.46 ± 0.08 for epirubicinol in the experiments with paclitaxel or docetaxel, respectively). In A, the asterisks indicate p < 0.05 for 0.5 to 2.5 μM paclitaxel or p < 0.05 for 1 to 2.5 μM docetaxel versus taxane-free controls.
Effects of paclitaxel or docetaxel on the kinetics of doxorubicinol or epirubicinol formation in isolated human heart cytosol
All the incubations (0.25-ml final volume) contained cytosol (0.15 mg of protein) and 0.25 mM NADPH; where indicated, 1 or 10 μM paclitaxel was also included.
We previously suggested that low concentrations of taxanes stimulated doxorubicinol formation by binding with high affinity to the regulatory/allosteric site of cytoplasmic reductase(s), whereas higher concentrations inhibited doxorubicinol formation by binding with low affinity to the active site and displacing doxorubicin (Salvatorelli et al., 2006b). The apparent dissociation constants for the binding of taxanes to the high- or low-affinity sites of the reductases were approximated to 1.2 μM (1K) or 6.5 μM (2K), respectively (Salvatorelli et al., 2006b). Such a “two-site binding” mechanism explained how 1 μM taxanes stimulated doxorubicinol formation mainly by increasing the Vmax value, whereas 10 μM taxanes inhibited doxorubicinol formation mainly by increasing the Km value (Table 3). Thus, we considered that the experiments with taxanes and 50 μM doxorubicin or epirubicin could have been influenced by the different availability of anthracyclines at the active site of the reductase(s), possibly resulting in different levels of competition between taxanes and doxorubicin or epirubicin for that site; moreover, 50 μM anthracycline would be 62% of the Km value of doxorubicin but only 20% of the Km value of epirubicin. Therefore, we characterized whether the effects of taxanes on doxorubicinol or epirubicinol formation changed if doxorubicin and epirubicin were used at higher and equally saturating concentrations. In experiments with anthracyclines at three times their Km values, 0.25 to 2.5 μM paclitaxel or docetaxel caused a concentration-dependent stimulation of doxorubicinol formation, which persisted if the two taxanes were used (2.5 to 10 μM) (Fig. 3A); under comparable conditions, paclitaxel and docetaxel still caused little or no effect on epirubicinol formation (Fig. 3B). These results confirmed that paclitaxel and docetaxel lacked allosteric effects on epirubicin metabolism; they also showed that the inhibition of doxorubicinol formation induced by >2.5 μM taxanes could be relieved by increasing the concentration of doxorubicin above its Km value to compete with the 2K of taxanes at the active site of the reductase(s).
Defective Taxane Modulation of Epirubicinol Formation in Human Myocardial Strips. We next characterized alcohol metabolite formation in human myocardial strips incubated in plasma with 10 μM anthracyclines and 2 or 6 or 15 μM taxanes. Paclitaxel was formulated in 10 μl of Cremophor EL/ml plasma, similar to the Cmax of Cremophor EL during the course doxorubicin-paclitaxel infusions (Sparreboom et al., 1998), whereas docetaxel was administered in combination with the clinically relevant concentration 1 μl of polysorbate 80/ml of plasma (Loos et al., 2003). Thus, the experimental system incorporated pharmacokinetic factors such as the binding of taxanes to plasma proteins, the possible entrapment of anthracyclines or paclitaxel in Cremophor EL micelles, and the interference of polysorbate 80 with the binding of docetaxel to plasma proteins or its diffusion in tissues (Loos et al., 2003; ten Tije et al., 2003; Ng et al., 2004). Under such defined conditions, paclitaxel and doxorubicin still diffused in the strips. Paclitaxel did not affect the steady-state levels of doxorubicin or epirubicin in the soluble fraction of the strips (Fig. 4A), but it caused different effects on the levels of doxorubicinol or epirubicinol; in fact, paclitaxel did not appreciably modify the levels of epirubicinol, but it modulated those of doxorubicinol according to a bell-shaped pattern that was suggestive of the two-site binding mechanism described with doxorubicin at less than its Km value (Fig. 4B). Similar results were obtained on replacing paclitaxel with docetaxel (Fig. 4, C and D). Adding plasma with 2 to 15 μM paclitaxel or docetaxel caused a concentration-dependent distribution of either taxane also to the membrane fraction of the strips, but this was not accompanied by doxorubicinol or epirubicinol formation in that fraction; moreover, the strips exposed to anthracycline-taxane combinations did not release doxorubicinol or epirubicinol in plasma (data not shown).

Effects of paclitaxel or docetaxel on doxorubicinol (A) or epirubicinol (B) formation in human heart cytosol: studies with doxorubicin or epirubicin at three times their Km values. The experimental conditions were as described in the legend to Fig. 2, except that doxorubicin and epirubicin were used at three times their Km values (224 and 750 μM, respectively). Values were taken from representative experiments and are expressed as percentages of control values to permit direct comparisons of incubations with different basal levels of alcohol metabolite formation (4.3 and 1.3 nmol/mg protein/4 h for doxorubicinol or epirubicinol, respectively).

Effects of increasing concentrations of taxanes on the levels of doxorubicin or epirubicin and their secondary alcohol metabolites in the soluble fraction of human myocardial strips. Human myocardial strips were incubated in plasma that contained 10 μM doxorubicin or epirubicin, and 2 to 15 μM paclitaxel or docetaxel formulated in 10 μl of ethanol/ml + 10 μl of Cremophor EL/ml or 1 μl of ethanol/ml + 1 μl of polysorbate 80/ml, respectively. After 4 h, the strips were assayed for the levels of taxanes and anthracyclines or their secondary alcohol metabolites in the soluble fraction. A, relationship between the individual levels of paclitaxel and those of doxorubicin or epirubicin; values were expressed as percentages of anthracycline content in strips exposed to ethanol/Cremophor EL without paclitaxel (ranges 0.5–10 μM and 0.7–6 μM for doxorubicin or epirubicin, respectively; n = 15–21). B, relationship between the individual levels of paclitaxel and those of doxorubicinol or epirubicinol. Values are expressed as percentages of doxorubicinol content in strips exposed to ethanol/Cremophor EL without paclitaxel (ranges 0.015–0.07 μM for doxorubicin and 0.011–0.057 μM for epirubicinol, respectively; n = 15–21). C, relationship between the individual levels of docetaxel and those of doxorubicin or epirubicin; values were expressed as percentages of anthracycline content in strips exposed to ethanol/polysorbate 80 without docetaxel (ranges 0.5–4.8 μM and 0.8–3.2 μM for doxorubicin and epirubicin, respectively; n = 15–20). D, relationship between the individual levels of docetaxel and those of doxorubicinol or epirubicinol. Values are expressed as percentages of doxorubicinol content in strips exposed to ethanol/polysorbate 80 without docetaxel (ranges 0.01–0.08 μM and 0.01–0.04 μM for doxorubicinol and epirubicinol, respectively; n = 15–20).
The maximal increase of doxorubicinol levels occurred on adding plasma with 6 μM taxanes (78 ± 11% with paclitaxel or 81 ± 18% with docetaxel; n = 6–12; p < 0.05 versus taxane-free strips). It is of note that 6 μM taxane would be similar or close to the Cmax of paclitaxel or docetaxel in patients treated with doxorubicin-paclitaxel or doxorubicin-docetaxel infusions (Gianni et al., 1997; D'Incalci et al., 1998; Salvatorelli et al., 2006b). Also of note is that the taxane-induced elevation of doxorubicinol was not accompanied by the formation of paclitaxel or docetaxel metabolites nor was it blunted by a 100 μM bolus of the protein synthesis inhibitor cycloheximide (data not shown). These results support the notion that pharmacokinetically relevant concentrations of paclitaxel or docetaxel stimulated doxorubicinol formation through a direct and allosteric-like modulation of pertinent anthracycline reductases, with little or no requirement for a de novo protein synthesis (Salvatorelli et al., 2006b).
The lack of metabolic interactions between epirubicin and taxanes requires further considerations. In the soluble fraction of strips exposed to anthracyclines and 6 μM taxanes, the steady-state levels of epirubicin were very similar to those of doxorubicin (micromolar doxorubicin or epirubicin, 3.6 ± 0.7 or 3.3 ± 0.6 in the strips with paclitaxel, and 3.4 ± 0.5 or 2.8 ± 0.2 in the strips with docetaxel; n = 6–12; p > 0.05). The soluble fraction of the same strips also contained comparable amounts of taxanes (micromolar paclitaxel, 1.5 ± 0.2 with epirubicin or 1.9 ± 0.3 with doxorubicin; micromolar docetaxel, 1.7 ± 0.3 with epirubicin or 1.8 ± 0.5 with doxorubicin; n = 6–12). The steady-state levels of doxorubicin corresponded to ∼4% of its Km value, whereas those of epirubicin corresponded to ≤1.4% of its Km value; however, the previous experiments with isolated cytosol clearly showed that paclitaxel or docetaxel would not stimulate epirubicinol formation if one increased epirubicin up to three times its Km value (cf. Fig. 3). The failure of paclitaxel or docetaxel to induce an allosteric-like stimulation of epirubicinol formation in myocardial strips could not therefore be attributed to concentration-related factors such as a limited availability of epirubicin or taxanes at the active or regulatory sites of cytoplasmic reductase(s).
Inhibitor Studies. Experiments with laboratory animals or reconstituted biochemical systems showed that anthracycline secondary alcohol metabolites could be formed by an array of carbonyl- and aldehyde- or aldose-reductases, whereas experiments with human myocardium showed that doxorubicinol was formed primarily by aldehyde reductases (Slupe et al., 2005; Salvatorelli et al., 2006b). To characterize the enzymology of epirubicinol formation versus doxorubicinol formation, we treated the isolated human heart cytosol with AL1576 (inhibitor of aldehyde reductases) (Barski et al., 1995), EBPC (inhibitor of aldose reductases) (Mylari et al., 1991), and quercetin (inhibitor of human carbonyl reductases) (Holleran et al., 2004). In incubations with anthracyclines at three times their Km values, AL1576 inhibited the formation of both doxorubicinolone and doxorubicinol or epirubicinol with IC50 values <10 μM, whereas quercetin or EBPC inhibited with IC50 values that were at least 1 order of magnitude higher (Table 4). Similar results were obtained with myocardial strips incubated in plasma with 10 μM anthracyclines and 1 to 500 μM inhibitors. In these latter experiments, the partitioning of ∼5 μM AL1576 in the soluble fraction of the strips always induced 50% or greater inhibition of alcohol metabolite formation (data not shown). The anthracyclines evaluated in this study therefore shared a prevailing specificity for aldehyde reductases.
Secondary alcohol metabolite formation in isolated human heart cytosol: inhibitor studies
The incubations (0.25-ml final volume) contained human heart cytosol (0.15 mg of protein), 0.25 mM NADPH, and anthracyclines at three times their Km values (14.4, 224, and 750 μM, respectively). Secondary alcohol metabolites were measured at 4 hours in the absence or presence of inhibitors specific to aldehyde reductases (AL1576), carbonyl reductases (quercetin), and aldose reductases (EBPC). Inhibition curves were obtained with eight to 13 concentrations of each inhibitor (0.1 μM—1 mM range).
Limited Diffusion of Doxorubicinol or Epirubicinol from Plasma into Myocardial Strips. The experiments described in the preceding sections characterized the conversion of anthracyclines to secondary alcohol metabolites inside the heart. We considered that patients treated with anthracyclines exhibit measurable levels of doxorubicinol or epirubicinol also in plasma; therefore, we characterized whether doxorubicinol and epirubicinol could diffuse from plasma into the heart. To probe such a possibility, we incubated myocardial strips in plasma added with 0.1 μM doxorubicinol or epirubicinol, similar to or higher than the Cmax of either metabolite during the course of anthracycline-taxane infusions (Gianni et al., 1997; Esposito et al., 1999; Grasselli et al., 2001). Neither doxorubicinol nor epirubicinol diffused from plasma into the strips; only the lipophilic doxorubicinolone, also used 0.1 μM, diffused from plasma and reached a myocardial level of 0.09 ± 0.01 μM (mainly localized to the soluble fraction).
Taxanes and Anthracycline-Derived ROS. One-electron addition to the quinone moiety of anthracyclines generates a semiquinone free radical that reduces oxygen to ROS like superoxide anion and its dismutation product H2O2. In the light of the low antioxidant defenses of the heart compared with other tissues, such ROS might play a role in inducing cardiotoxicity (Gewirtz, 1999; Minotti et al., 2004a). We characterized whether the taxanes influenced the formation of anthracycline-derived ROS in a manner that could explain the different pattern of cardiotoxicity induced by doxorubicin-taxane or epirubicin-taxane combinations. After 4-h incubation in plasma the basal concentration of H2O2 in the strips averaged 0.53 ± 0.06 μM (0.2–1.1 μM range), mostly localized to the membrane fraction (0.48 ± 0.06 μM; 0.1–1 μM range; n = 15). Adding plasma with the ethanol-Cremophor EL cosolvent system did not alter the pattern of H2O2 formation in the strips, whereas adding plasma with ethanol-Cremophor EL + 10 μM doxorubicin caused an increased formation of H2O2 in the membrane fraction (Fig. 5).
The increased formation of H2O2 induced by doxorubicin in the membrane fraction of the strips was consistent with the prevailing role of mitochondrial reductases in the one-electron reduction of the quinone moiety of anthracyclines (Gewirtz, 1999; Minotti et al., 2004b). Doxorubicin-derived H2O2 probably diffused from membranes in the soluble fraction, but the latter never exhibited a measurable increase of H2O2 (Fig. 5). As mentioned under Materials and Methods, the strips were loaded with a concentration of DCFH-DA that was high enough to measure H2O2 in both soluble and membrane fractions of the strips; moreover, we reported that an extracellular diffusion of H2O2 would account for no more than ∼5 to 8% of the total H2O2 measured inside the strips (Salvatorelli et al., 2006a). The lack of a measurable increase of H2O2 in the soluble fraction, therefore, reflected biochemical events such as the elimination of H2O2 through the pseudoperoxidatic activity of myoglobin, which is highly abundant in that fraction and uses anthracyclines as reducing substrates (Menna et al., 2002). It is noteworthy that adding plasma with 10 μM doxorubicin + 6 μM paclitaxel did not cause more H2O2 formation compared with doxorubicin alone (Fig. 5). In other experiments, doxorubicin was held constant at 10 μM, whereas paclitaxel was used over the usual 2 to 15 μM range. Under such defined conditions, paclitaxel exhibited a concentration-dependent accumulation in both membrane and soluble fractions of the strips, but this never augmented H2O2 formation compared with doxorubicin alone (Fig. 6, A and B).

Doxorubicin-dependent ROS formation in human myocardial strips. Human myocardial strips were loaded with 50 μM DCFH-DA and then incubated in plasma added with 10 μl of ethanol/ml + 10 μl of Cremophor EL/ml. Where indicated plasma was added also with 10 μM doxorubicin, with or without 6 μM paclitaxel. After 4 h, the soluble or membrane fraction of the strips was assayed for ROS by a DCF-based assay for H2O2. Values were means ± S.E. of three experiments. The single asterisks indicate p < 0.05 for H2O2 in the membrane fraction of strips exposed to ethanol-Cremophor EL with doxorubicin, or doxorubicin + paclitaxel, versus H2O2 in the membrane fraction of strips exposed to ethanol-Cremophor EL only.

ROS and taxane levels in human myocardial strips exposed to doxorubicin and increasing concentration of paclitaxel. The experimental conditions were as described in the legend to Fig. 5, except that plasma was added with 2, 6, or 15 μM paclitaxel. After 4 h, the strips were assayed for paclitaxel or DCF-detectable H2O2. Values are means ± S.E. of three experiments. A, membrane fraction. B, soluble fraction.
Neither 10 μM epirubicin nor 10 μM epirubicin + 6 μM paclitaxel increased H2O2 formation in the membrane fraction of myocardial strips (Fig. 7A). In previous studies, the failure of epirubicin to form H2O2 was attributed to its limited localization to mitochondrial reductases, probably caused by a concomitant partitioning and pH-dependent accumulation of epirubicin in acidic organelles such as lysosomes, recycling endosomes, or vesicles of the trans-Golgi network. Bafilomycin A1, a known inhibitor of the vacuolar H+-ATPase inhibitor that acidifies such organelles, prevented an anomalous distribution of epirubicin and favored its partitioning toward the mitochondrial reductases that converted anthracyclines to ROS (Salvatorelli et al., 2006a).1 In the present study, the exposure of myocardial strips to bafilomycin A1 allowed 10 μM epirubicin to increase H2O2 formation in the membrane fraction of the strips; however, the simultaneous addition of 6 μM paclitaxel caused no further stimulation of H2O2 formation (Fig. 7B). The effect of bafilomycin A1 was highly specific to H2O2 formation induced by epirubicin in the membrane fraction; bafilomycin A1 did not increase H2O2 levels in the soluble fraction (Fig. 7B) nor did it alter epirubicinol formation or H2O2 formation induced by doxorubicin (data not shown). Also of note is that 6 μM paclitaxel distributed equally well in the absence or presence of bafilomycin A1 (micromolar paclitaxel in the soluble fraction: 2.5 ± 0.6 with epirubicin and 2.7 ± 0.4 with epirubicin + bafilomycin A1; micromolar paclitaxel in the membrane fraction: 2.2 ± 0.5 with epirubicin and 2.3 ± 0.4 with epirubicin + bafilomycin A1; n = 3–6).
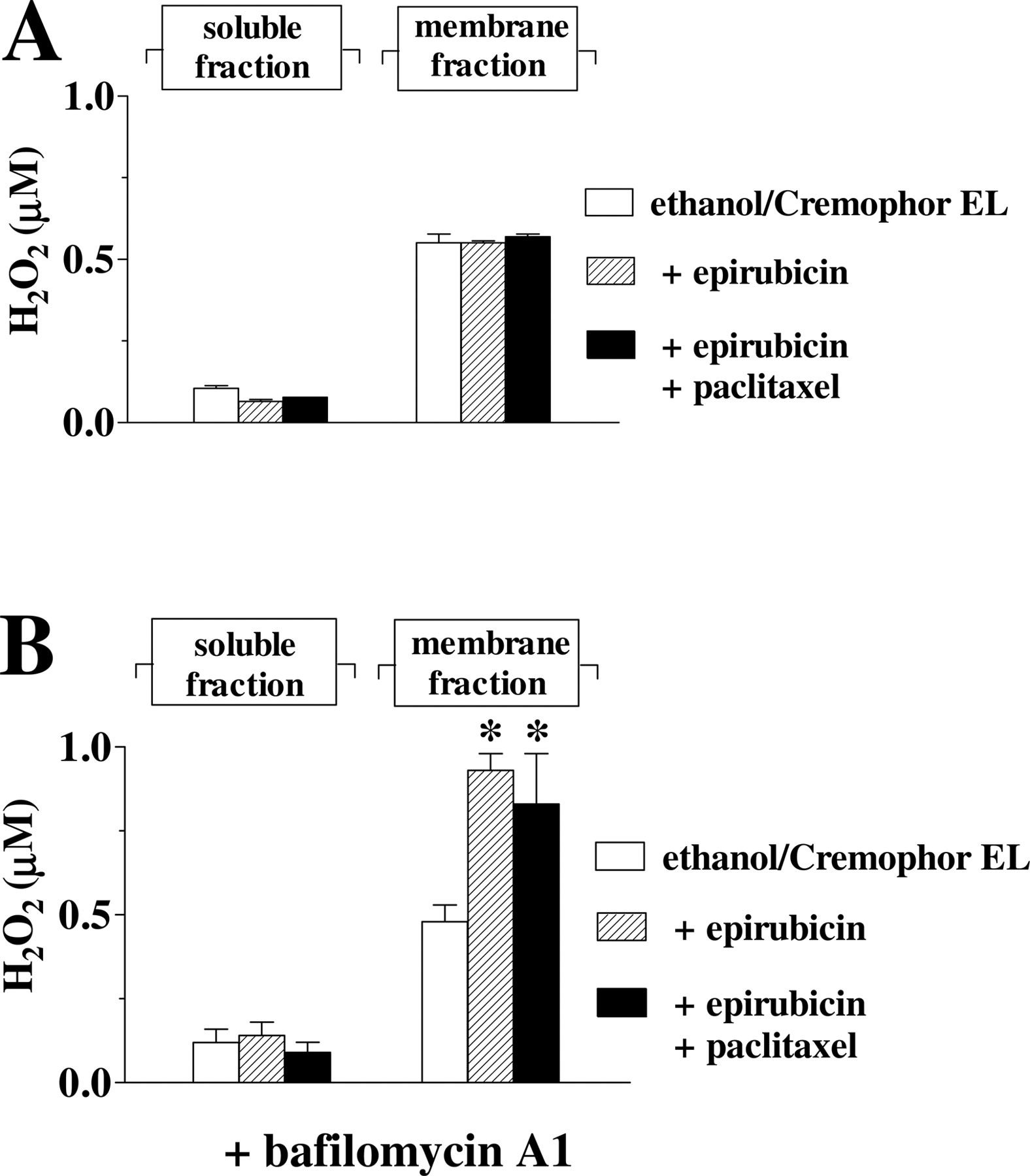
Epirubicin-dependent ROS formation in human myocardial strips. A, human myocardial strips were loaded with 50 μM DCFH-DA and then incubated in plasma added with 10 μl of ethanol/ml + 10 μl of Cremophor EL/ml; where indicated plasma was added also with 10 μM epirubicin, with or without 6 μM paclitaxel. After 4 h, the soluble or membrane fraction of the strips was assayed for ROS by a DCF-based assay for H2O2. Values are means ± S.E. of three to six experiments. B, strips were also exposed to bafilomycin A1 (300 nM during the loading with DCFH-DA and 90 nM during the subsequent 4-h incubation in plasma). *, p < 0.05 for H2O2 in the membrane fraction of strips exposed to ethanol-Cremophor EL with epirubicin, or epirubicin + paclitaxel, versus H2O2 in the membrane fraction of strips exposed to ethanol-Cremophor EL only.
These experiments confirmed that only doxorubicin could form ROS in human myocardial strips, and they showed that such a metabolic pattern would not be influenced by a concomitant administration of taxanes.
Discussion
In laboratory animals, the development of anthracycline-induced CHF correlated with a myocardial accumulation of secondary alcohol metabolites, which often proved to be more potent than their parent drugs at diminishing the expression and/or activity of many important cell constituents (Forrest et al., 2000; Gambliel et al., 2002; Minotti et al., 2004a; Charlier et al., 2005). Therefore, pharmacokinetic studies of cancer patients sought to establish whether the higher than expected cardiotoxicity of some anthracycline-taxane combinations could be caused by anomalous increases of secondary alcohol metabolites in plasma. Paclitaxel reduced the systemic elimination of doxorubicinol and increased its plasma levels during the course of doxorubicin-paclitaxel infusions (Gianni et al., 1997); however, paclitaxel increased also the plasma levels of epirubicinol and epirubicinol glucuronide during infusion of the less cardiotoxic paclitaxel-epirubicin combination (Esposito et al., 1999; Grasselli et al., 2001). Docetaxel did not alter doxorubicin(ol) elimination (D'Incalci et al., 1998), whereas it increased the plasma levels of epirubicinol in some studies (Rischin et al., 2002) but not in others (Esposito et al., 1999). In examining such inconsistencies, one should also consider that doxorubicinol and epirubicinol would be too polar to diffuse from plasma in the heart and to induce cardiotoxicity by this route. Accordingly, our present work reports that pharmacokinetically relevant concentrations of doxorubicinol and epirubicinol did not diffuse from plasma into human myocardial strips; only doxorubicinolone, secondary alcohol metabolite of the hydrophobic/lipophilic aglycone doxorubicinone, diffused from plasma in myocardial strips during the course of the experiments. These observations support the concept that the cardiotoxicity of anthracycline-taxane combinations would be determined by the levels of doxorubicinol or epirubicinol only if paclitaxel or docetaxel stimulated the formation of such metabolites inside the heart (Salvatorelli et al., 2006b).
In the present study, we showed that the presence and epimerization of daunosamine were two independent factors that limited the catalytic efficiency with which cytoplasmic reductases converted anthracyclines to secondary alcohol metabolites, making epirubicinol formation lower than doxorubicinol formation. We also demonstrated that paclitaxel and docetaxel caused allosteric effects that stimulated the formation of doxorubicinol, but not of epirubicinol, in isolated human heart cytosol or in myocardial strips exposed to pharmacokinetically relevant concentrations of anthracyclines and taxane-cosolvent formulations. Comparisons between doxorubicin and epirubicin therefore show that epirubicin would have favorable characteristics such as a limited conversion to epirubicinol and a defective taxane-stimulation of epirubicinol formation.
The different patterns of basal or taxane-modulated alcohol metabolite formation could not be attributed to a metabolization of doxorubicin or epirubicin by different reductases, because inhibitor studies showed that doxorubicin and epirubicin and their aglycone doxorubicinone shared a prevailing specificity for aldehyde reductases. In this regard, it may be worth noting that the catalytic activity of aldehyde reductases increases with the hydrophobicity of substrates (Barski et al., 1995), which may explain how these reductases metabolized doxorubicinone much better than the more polar doxorubicin and epirubicin. Equally important is that the binding of NADPH to aldehyde reductases and the subsequent release of alcohol products and NADP+ are accompanied by such complex conformational changes that also substrate-related steric factors such as daunosamine epimerization could come into play and determine the orientation and turnover of anthracyclines at the active site of the reductases. As indirect evidence of such a possibility, we would note that epirubicin is slightly more hydrophobic than doxorubicin (Wielinga et al., 2000), but it always formed less alcohol metabolite than doxorubicin. Future studies will explore whether substrate-related steric factors influenced also the binding of taxanes to the regulatory site of the reductases and its coupling with an improved electron transfer from the reductases to doxorubicin but not epirubicin.
The basal levels of doxorubicinol or epirubicinol in the soluble fraction of human myocardial strips always averaged ≤1% of the amount of doxorubicin or epirubicin available to the reductases. This figure should be opposed to the much higher levels of doxorubicinol in the heart of patients deceased after anthracycline administration; in these latter studies, the concentration of doxorubicinol often equaled that of doxorubicin (Stewart et al., 1993). Such a discrepancy may be explained by considering that the post mortem samples were derived from patients treated with several doses of doxorubicin, whereas the myocardial strip model adopted in our study mimicked doxorubicinol formation that occurred on a single administration of doxorubicin. Also of note is that the myocardial strips released detectable amounts of doxorubicin or epirubicin but not of doxorubicinol or epirubicinol; that the strips released much higher amounts of the hydrophobic/lipophilic doxorubicinone or doxorubicinolone confirmed that the cardiac clearance of anthracyclines was inversely related to their polarity. These factors anticipate that the chronic administration of doxorubicin or epirubicin would be accompanied by a gradual cardiac accumulation of doxorubicinol or epirubicinol over doxorubicin or epirubicin. Moreover, the ability of taxanes to stimulate doxorubicinol formation while not inducing its diffusion in plasma would translate into an increased cardiac accumulation of doxorubicinol and a consequent development of CHF at lower than expected cumulative doses of doxorubicin. This may not occur if the taxanes failed to stimulate alcohol metabolite formation, as was the case for epirubicin-paclitaxel or epirubicin-docetaxel combinations.
There is controversy about whether ROS caused less or more cardiotoxicity than secondary alcohol metabolites (Gewirtz, 1999; Minotti et al., 2004a). We previously reported that epirubicin formed essentially no ROS compared with doxorubicin in human myocardial strips (Salvatorelli et al., 2006a), an observation that was confirmed in this present work. Inasmuch as epirubicin also formed less alcohol metabolite than doxorubicin, we proposed that both a defective conversion to ROS and a limited formation of epirubicinol might render epirubicin less cardiotoxic than doxorubicin if the two anthracyclines were used as single agents (Salvatorelli et al., 2006a). Here, we have shown that paclitaxel never increased ROS formation in myocardial strips exposed to doxorubicin or epirubicin. Paclitaxel failed to promote the conversion of epirubicin to ROS also when bafilomycin A1 was used to prevent its accumulation in membrane compartments other than mitochondria.2 In highlighting the lack of a taxane effect on anthracycline-induced ROS formation, these results suggest that the higher than expected cardiotoxicity of doxorubicin-taxane combinations would be caused quite specifically by the stimulation of doxorubicinol formation, whereas the better tolerability of epirubicin-taxane combinations would be determined primarily by a defective stimulation of epirubicinol formation.
Epirubicin is receiving growing attention as a valuable alternative to doxorubicin, especially if anthracyclines were considered for combination with drugs that could aggravate cardiotoxicity (Glück, 2005); however, the choice of replacing doxorubicin with epirubicin was based on observational rather than mechanistic considerations. We have shown that a defective taxane stimulation of epirubicinol formation offers mechanism-based insight into the better cardiac tolerability of epirubicin-taxane combinations as opposed to equivalent doxorubicin-taxane regimens. This information may help to improve the clinical use of anthracyclines and taxanes in many clinical settings and serve as experimental guidelines for exploring the safety or toxicity of anthracyclines in combination with other anticancer agents.
Acknowledgments
We thank Dr. Raimondo De Cristofaro (Catholic University School of Medicine, Rome, Italy) for helpful discussions and Dr. Ruggero De Paulis and Mirella De Cotiis (University of Tor Vergata and European Hospital, Rome, Italy) for providing myocardial samples.
Footnotes
-
↵1 The small size of human myocardial strips did not allow for an accurate subcellular fractionation and direct measurements of epirubicin in one organelle or another. The distribution of epirubicin to acidic vesicles rather than mitochondria was demonstrated by monitoring ROS formation in the absence or presence of bafilomycin A1 and by confocal microscopy visualization of epirubicin, mitochondria, and acidic vesicles in H9c2 cardiomyocytes (Salvatorelli et al., 2006a).
-
↵2 The anomalous distribution of epirubicin to acidic organelles probably was favored by its slightly higher lipophilicity and consequent facilitated diffusion in the vacuoles, followed by protonation of the amino group of daunosamine and vacuolar retention of the drug (Wielinga et al., 2000; Salvatorelli et al., 2006a).
-
This work was supported by Associazione Italiana Ricerca sul Cancro and Ministero dell' Universita' e Ricerca Scientifica e Tecnologica (Cofin 2004, FIRB RBNE 014HJ3–002, and Center of Excellence on Aging at the University of Chieti) (to G.M.).
-
E.S. and P.M. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.116160.
-
ABBREVIATIONS: CHF, congestive heart failure; ROS, reactive oxygen species; EBPC, ethyl-1-benzyl-3-hydroxy-2(5H)-oxopyrrole-4-carboxylate; AL1576, 2,7-difluorospirofluorene-9,5′-imidazolidine-2′,4′-dione; DCFH-(DA), dichlorofluorescin-(diacetate); DCF, dichlorofluorescein; HPLC, high-performance liquid chromatography.
- Received October 25, 2006.
- Accepted November 21, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}