


Abstract
Sympathetic hyperinnervation is the leading cause of fatal ventricular arrhythmia (VA) after myocardial infarction (MI). Cardiac mast cells cause arrhythmias directly through degranulation. However, the role and mechanism of mast cell degranulation in sympathetic remodeling remain unknown. We investigated the role of oxytocin (OT) in stabilizing cardiac mast cells and improving sympathetic innervation in rats. MI was induced by coronary artery ligation. Western blotting, immunofluorescence, and toluidine staining of mast cells were performed to determine the expression and location of target protein. Mast cells accumulated significantly in peri-infarcted tissues and were present in a degranulated state. They expressed OT receptor (OTR), and OT infusion reduced the number of degranulated cardiac mast cells post-MI. Sympathetic hyperinnervation was attenuated as assessed by immunofluorescence for tyrosine hydroxylase (TH). Seven days post-MI, the arrhythmia score of programmed electrical stimulation was higher in vehicle-treated rats with MI than in rats treated with OT. An in vitro study showed that OT stabilized mast cells via the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) signaling pathway. Further in vivo studies on OTR-deficient mice showed worsening mast cell degranulation and worsening sympathetic innervation. OT pretreatment inhibited cardiac mast cell degranulation post-MI and prevented sympathetic hyperinnervation, along with mast cell stabilization via the PI3K/Akt pathway.
SIGNIFICANCE STATEMENT This is the first study to elucidate the role and mechanism of oxytocin (OT) in inflammatory-sympathetic communication mediated sympathetic hyperinnervation after myocardial infarction (MI), providing new approaches to prevent fatal arrhythmias.
Introduction
Acute myocardial infarction (AMI) is a serious clinical disease known to have very high mortality due to the serious complications of ventricular arrhythmia (VA), including ventricular tachycardia (VT) and ventricular fibrillation (VF) (Wu and Vaseghi, 2020; Yang et al., 2021). Previous studies have shown that sympathetic hyperinnervation is one of the primary causes of ventricular arrhythmogenesis and sudden cardiac death. The role of inflammatory cells such as macrophages, lymphocytes, and neutrophils in the pathogenesis of myocardial infarction (MI) is well defined (Yin et al., 2016). However, mast cells, multifunctional immune cells best known for their role in immediate hypersensitivity and chronic allergic reactions, have not been adequately studied in MI. Recently, accumulating evidence suggests that mast cell degranulation increases and cardiac remodeling worsens after myocardial infarction (Engels et al., 1995; Patella et al., 1997, 1998; Jaggi et al., 2007). However, the role and mechanism of mast cell degranulation in sympathetic remodeling remain unknown.
Cardiac mast cells are present in low numbers and in an immature state around vasculature and mammalian hearts. However, this value increases 2- to 6-fold in cardiac diseases such as MI, hypertension, and ischemia-reperfusion models (Varricchi et al., 2020). As cardiac mast cells mature, they degranulate particles and release mediators, including histamine, tumor necrosis factor alpha (TNF-α), transforming growth factor beta (TGF-β), tryptase, and chymase, which can either stimulate collagen synthesis leading to fibrosis (e.g., in ischemia-reperfusion) or induce matrix metalloproteinase activation, resulting in collagen degradation and ultimately ventricular dilatation. Furthermore, mast cell–derived renin promotes local angiotensin formation and norepinephrine release, causing arrhythmias in an ischemia-reperfusion model (Mackins et al., 2006). Therefore, stabilizing mast cells may be an effective strategy to protect MI hearts from inflammation and neural remodeling.
Oxytocin (OT), a cardiovascular hormone, a neuroendocrine hormone, with its receptor (OTR), was first reported to play an important role in cardiovascular homeostasis by regulating blood volume through the release of atrial natriuretic peptide (ANP) release from the cardiac atria (Japundžić-Žigon et al., 2020).The role of OT in modulating immune and anti-inflammatory responses is mediated by neutrophils, macrophages, and T lymphocytes in MI animal models (Jankowski et al., 2010). Accumulating evidence suggests that sympathetic remodeling after MI is closely linked to inflammation and occurs primarily at the periphery of the infarction, where inflammatory cells accumulate and regulate nerve remodeling through nerve growth factor (NGF) secretion. However, the role of OT in mast cell function and inflammation-induced sympathetic hyperinnervation in rat hearts remained unclear.
Considering the possible effects of OT on cardiac mast cells, we hypothesized that treatment with the hormone would attenuate inflammation in a rat model of MI and improve sympathetic hyperinnervation, thereby providing new targets for the treatment of life-threatening arrhythmias after MI. Specifically, we examined the effects and mechanisms of OT on cardiac mast cell function and sympathetic remodeling post-MI and in in vitro mast cells. Furthermore, we confirmed the deleterious effects of OTR deficiency on mast cell destabilization and sympathetic innervation in OT knockout mice.
Materials and Methods
Myocardial Infarction Animal Model
The study was approved in accordance with the protocols and guidelines of the Laboratory Animal Ethics Committee of Shandong First Medical University Affiliated Qianfoshan Hospital (Protocol number: S 030).
Protocol 1.
Adult male Sprague Dawley rats (250–300 g) were used as research subjects. Myocardial infarction (MI) surgery was performed after a 7-day acclimatization period. Briefly, rats were anesthetized with 1% pentobarbital sodium (45 mg/kg body weight). Endotracheal intubation was performed carefully. Mechanical ventilation was achieved by connecting the endotracheal tube to a ventilator (HX-300S; TME, Chengdu, China), cycling at 80 breaths per minute with a tidal volume of 1.5 ml per 100 g body weight. The left coronary artery (LAD) was routinely ligated approximately 2 to 3 mm from its origin, between the pulmonary artery conus and the left atrium. In the control group, the suture was placed around the LAD without tying it. To confirm infarction, ST elevation, regional cyanosis, and wall motion abnormalities were assessed (Supplemental Fig. 1; Supplemental Video).
Rats were randomly assigned to ensure that there were approximately equal numbers of survivors in each group. The groups were designated as follows: Control, MI, and MI + OT groups (MI rats received OT therapy). OT was delivered subcutaneously using the Alzet mini-osmotic pump (model 2001) at [125 ng/(kg.h)] for 7 days from chest-opening procedure until approximately 24 hours before the end of the experiments to eliminate pharmacological actions. Seven days post-MI, the animals were killed by cervical dislocation after anesthesia, and their hearts were collected for further studies.
Protocol 2.
OTR knockout mice were developed by Professor Chuanyong Liu of Shandong University (Tang et al., 2019) and kindly donated by the National Institutes of Biomedical Innovation. This strain was backcrossed with C57BL/6 J mice in our laboratory. Homozygous knockout (KO) and wild-type (WT) mice were used in this study. We created an MI model in 12- to 13-week-old male KO mice (KO + MI) and WT littermates (WT + MI) by ligation of the left coronary artery, as described in protocol 1. All animals were randomly assigned to the following groups: Control, OTR+/− MI, OTR−/− MI groups.
In Vivo Electrophysiological Experiments.
Before the cardiac LAD artery was ligated, an implantable transmitter (Telemetry Research) was embedded in the rat abdomen to record ECG signals. ECG data were collected using PowerLab software. All animals were monitored with the transmitter for 7 days, and the data were saved for further analysis. Before sacrifice, a programmed stimulation protocol was applied using electrodes implanted on the epicardial surface of the left ventricle (LV). Arrhythmias were triggered using an electrical Bloom stimulator (Chengdu Electronic Machine Company) and then induced by ventricular stimulation at a basic cycle length of 150 milliseconds (S0), with single (S1), double (S2), and triple (S3) additional stimuli delivered after eight paced beats. Pacing protocols were halted if they induced sustained ventricular tachycardia (VT). Ventricular tachyarrhythmias, including VT and fibrillation, were considered nonsustained if their duration lasted <15 beats and sustained if the duration lasted >15 beats. The experimental protocols were completed within 10 minutes. All procedures were performed and recorded using an animal biological function experimental system (LEAD-7000; JJET, China). The ventricular arrhythmia (VA) scores were calculated as described in our previous study (Kim et al., 2019).
Historical Preparation.
The rat hearts were frozen in liquid nitrogen immediately after euthanasia. The tissues were then embedded in an optimal cutting temperature compound (OCT) for sectioning. All slices were 7 μm thick. Sections were fixed in acetone for 10 minutes at room temperature. Slices were washed with tap water and tissues were stained with 0.5% toluidine blue for 10 minutes. Ammonia (1%) was used to reduce background staining. The slices were then dehydrated with an alcohol gradient. Neutral balsam was used to cover the tissues for permanent storage. Mast cells were visualized using an optical microscope.
Cell Culture.
The murine mast cell line (P815) was a gift from Professor Chuanyong Liu of Shandong University. P815 was used to characterize mast cell degranulation because other mast cell lines such as HMC-1 do not degranulate (Hughes and McNagny, 2015). P815 was planted on poly-D-lysine (PDL)-coated coverslips separately and cultured for 48 hours in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS in 5% CO2 and 95% air. Stimulation reagents included mast cell degranulation trigger C48/80 (6 mg/ml) (Akerlund et al., 1985), OT (0.01 ng/ml), phosphoinositide 3-kinase (PI3K) inhibitor, LY294002 (6.4 μM), and a selective OTR antagonist, atosiban (1 μM) (Zhang et al., 2022).
Immunohistochemistry.
The infarct size was evaluated using heart tissue sections stained with Masson’s trichrome (Jiancheng, China) according to standard protocols. The digitized images were analyzed using planimetry, and the infarcted area was expressed as the percentage of stained fibrotic area over the entire LV. Only hearts with large infarctions (>30%) were analyzed according to clinical relevance. For cardiac mast cell staining, cardiac tissues were prepared using a cryostat and then stained with 0.05% toluidine blue. Briefly, a 1% stock solution of toluidine blue in 70% ethanol was dissolved in 0.5% NaCl (pH 2.2–2.3). The slides were immersed in the staining solution for 30 minutes, washed twice with distilled water, dehydrated using a series of increasing concentrations of ethanol, and finally immersed in butyl acetate ester. Cover slips were applied using Eukitt mounting medium, and the slides were allowed to dry overnight. The number of intact mast cells (IMCs) and degranulated mast cells (DMCs) in the sections were observed and counted, and the degranulation rate of mast cells [MCD = DMCs/(IMCs + DMCs) × 100%] was calculated.
For tissue immunofluorescence, hearts were harvested after the hemodynamic study and immersed overnight in 30% sucrose in phosphate-buffered saline (PBS), embedded in Tissue-Tek OCT compound (Sakura Finetek), and frozen in an isopentane bath on dry ice. For cell immunofluorescence, cells were washed three times with PBS for 5 minutes each, followed by fixation with 4% paraformaldehyde (PFA) for 20 minutes at room temperature. The coverslips were washed three times with PBS. Triton X-100 (0.5%) was used to penetrate cell membranes and facilitate antibody entry into cells. Coverslips were blocked with 10% bovine serum albumin (BSA) for 30 minutes. The samples were incubated with anti-TH Ab (ab112, 1:500; Abcam), anti-OTR Ab (ab87312, 1:500; Abcam), and anti-Tryptase Ab (ab2378, 1:200; Abcam) overnight at 4°C, followed by a 2-hour incubation with FITC-conjugated rabbit anti-sheep (1:200; Bethyl), Alexa 545-conjugated goat anti-rabbit (1:100; PeproTech) or FITC-conjugated rabbit anti-mouse (1:200; BioLegend) secondary antibodies. The sections were counterstained with DAPI (Life Technologies) to identify nuclei. Quantification of the fraction of sympathetic nerve fibers was expressed as the ratio of the labeled nerve fiber area to the total area, whereas papillary muscles were excluded from the study because variable sympathetic innervation has been reported.
Ten microscopic fields in a 20× field (0.13 mm2) were randomly selected by a blinded investigator. All images were captured using an Olympus LCX100 Imaging System. All images were analyzed using ImageJ software (version 1.38×; National Institutes of Health).
Free Calcium (Ca2+) Measurement.
P815 cells were activated with or without C48/80 and then with or without OT and LY294002 for 10 minutes. The medium was discarded, and the suspension was incubated in Tyrode’s buffer supplemented with Fluo-3 acetoxymethyl ester (Fluo-3-AM) (Invitrogen) at 37°C for 20 minutes. After discarding the Fluo-3-AM, the cells were resuspended in Tyrode’s buffer and stimulated with 100 ng/ml DNP-HSA. Baseline calcium levels were recorded at 488 nm excitation/520 nm emission using the AccuriTM C6 flow cytometer (BD Biosciences, USA) (10,000 events). Cells were then exposed to stimulations for 10 minutes, and the mean fluorescence intensity was recorded. The FCS Express 4 software was used for data presentation (De Novo Software, Glendale, CA).
Flow Cytometry.
A FACSTAR equipped with a Consort 30 Data system (Becton Dickinson, Mountain View, CA) was used. The excitation of the argon-ion laser was 488 nm at a power of 150 mW. A nozzle with a diameter of 70PM was used. Mast cells were identified by size and shape using forward scatter (FSC) and by granularity using side scatter (SSC). A minimum of 1 × 104 cells from each group (n = 6 per group) were used for each analysis. After identification by fluorescence-activated cell sorter (FACS), mast cells (1 × 105) were incubated with various treatment concentrations for 3 minutes at room temperature. The percentage of mast cell degranulation was calculated as previously described (Perretti et al., 1990).
Western Blot.
P815 cells were treated with C48/80, C48/80 + OT, and C48/80 + OT + atosiban for 15 minutes and harvested for western blot analysis. The normal medium Protein Extraction Kit (Beyotime Institute of Biotechnology, Jiangsu, China) was used, and protein concentration was measured with the BCA Protein Assay Reagent Kit (Pierce). Equal amounts of total protein (80 μg of protein per lane) were resolved on 5%–10% SDS-PAGE gels and transferred onto polyvinylidene difluoride (PVDF) membranes using the semidry transfer method. The membranes were blocked with 5% nonfat dry milk in PBST (PBS containing 0.05% Tween 20) and incubated overnight at 4°C with an anti–p-Akt (phosphorylated protein kinase B) rabbit mAb (CST, 1:2000) and anti-GAPDH rabbit pAb (1:2000; Abcam). Incubated membranes were immersed in solution with HRP-conjugated secondary antibodies (1:5000; GenScript, Piscataway, NJ), kept at 37°C for 1 hour and subsequently detected by the ECL substrate (Millipore, Billerica, MA) and visualized using a FluorChem E Imager (Protein-Simple, Santa Clara, CA). The densities relative to GAPDH were analyzed using ImageJ software (NIH).
Statistical Analysis.
Statistical analysis was performed with SPSS 17.0. Quantitative data are presented as the mean ± S.E. Differences among groups were analyzed by t test, analysis of variance (ANOVA) with Turkey’s post hoc tests. A value of P < 0.05 was considered statistically significant.
Results
Mast Cells Degranulate in Infarcted Rat Heart.
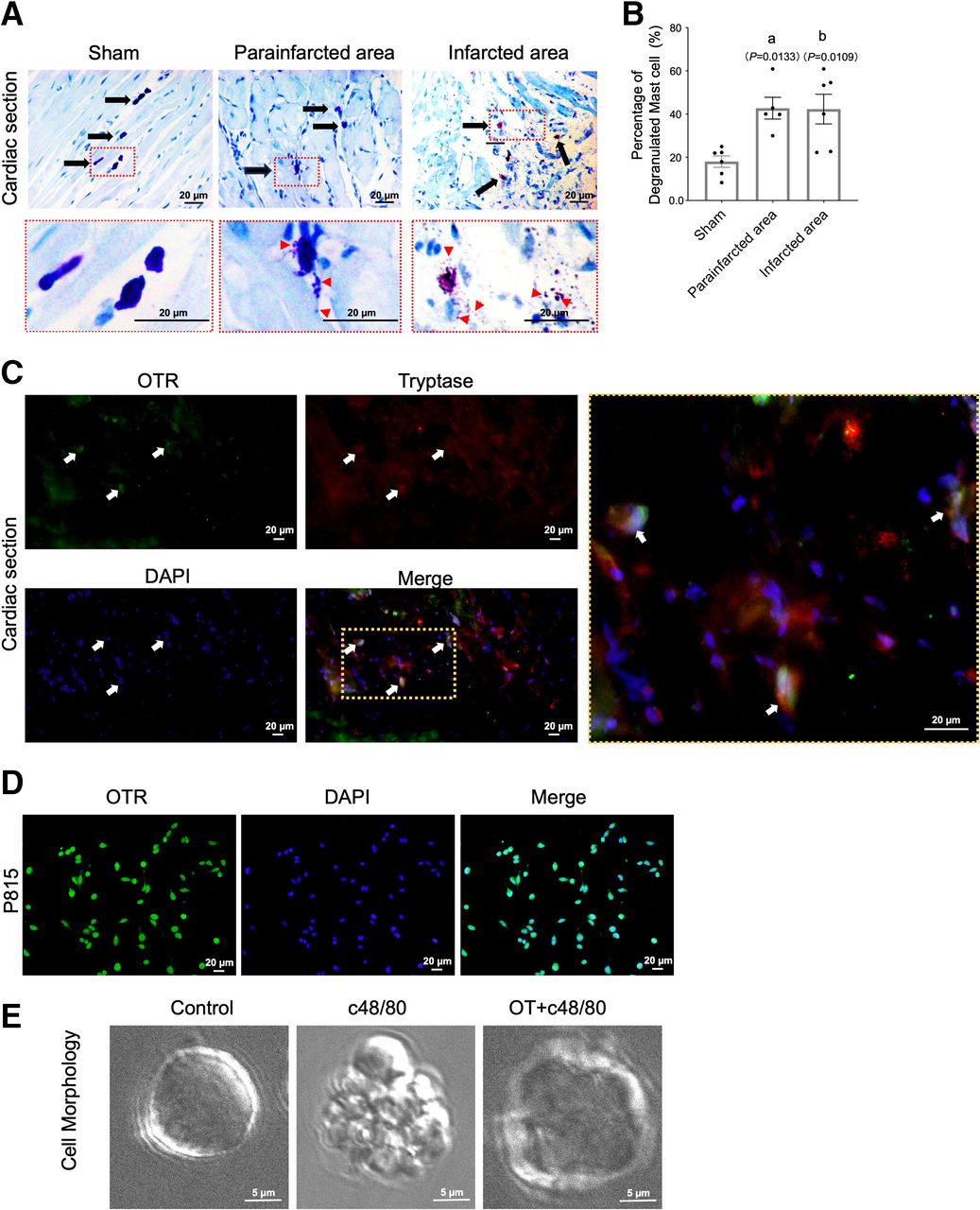
The degranulation of mast cells is a prerequisite for their effect in the environment. To evaluate the degree of cardiac mast cell degranulation, we used toluidine blue staining as an indicator to index mast cell distribution and morphology in cardiac tissue. These results showed that mast cells were small and continuous with dense cell membranes in mock cardiac tissue. However, in the infarcted tissue, mast cells release granules in both the infarcted and peri-infarcted areas, and the mast cell granules travel a long distance in the heart, which is almost three times the mast cell size or more. The percentage of degranulated mast cells increased significantly after MI (Fig. 1, A and B). Costaining with tryptase and OTR showed that OTR was expressed in mast cells from both infarcted border tissues and P815 cells (Fig. 1, C and D). Furthermore, exposure to C48/80 resulted in a significant increase in the number of degranulated cells compared with the control group (Fig. 1E), whereas preconditioning with OT significantly reversed the degranulation process.

Number of degranulated cardiac mast cells increases after MI and is inhibited by the OT-OTR system. (A) Representative cardiac Toluidine blue staining of mature mast cells in granulated and degranulated states found in healthy myocardium, peri-infarcted areas, and infarcted areas. Arrows indicate representative cardiac mast cells. (B) Quantitation of mast cells degranulation (n = 8). (C) Double immunostaining for OTR (green) and mast cell tryptase (red) in the vehicle‐MI group shows a limited distribution of OTR on mast cells in peri-infarcted tissue; (D) P815 cells show positive staining of OTRs on cell membranes. (E) Cell morphology, as observed using a scanning electron microscope, showed that OT administration reduces mast cell degranulation in cells stimulated with C48/80. (A and B) P <0.05 compared with the sham group.
OT Administration Suppressed Mast Cell Degranulation via the PI3K/Akt Pathway.
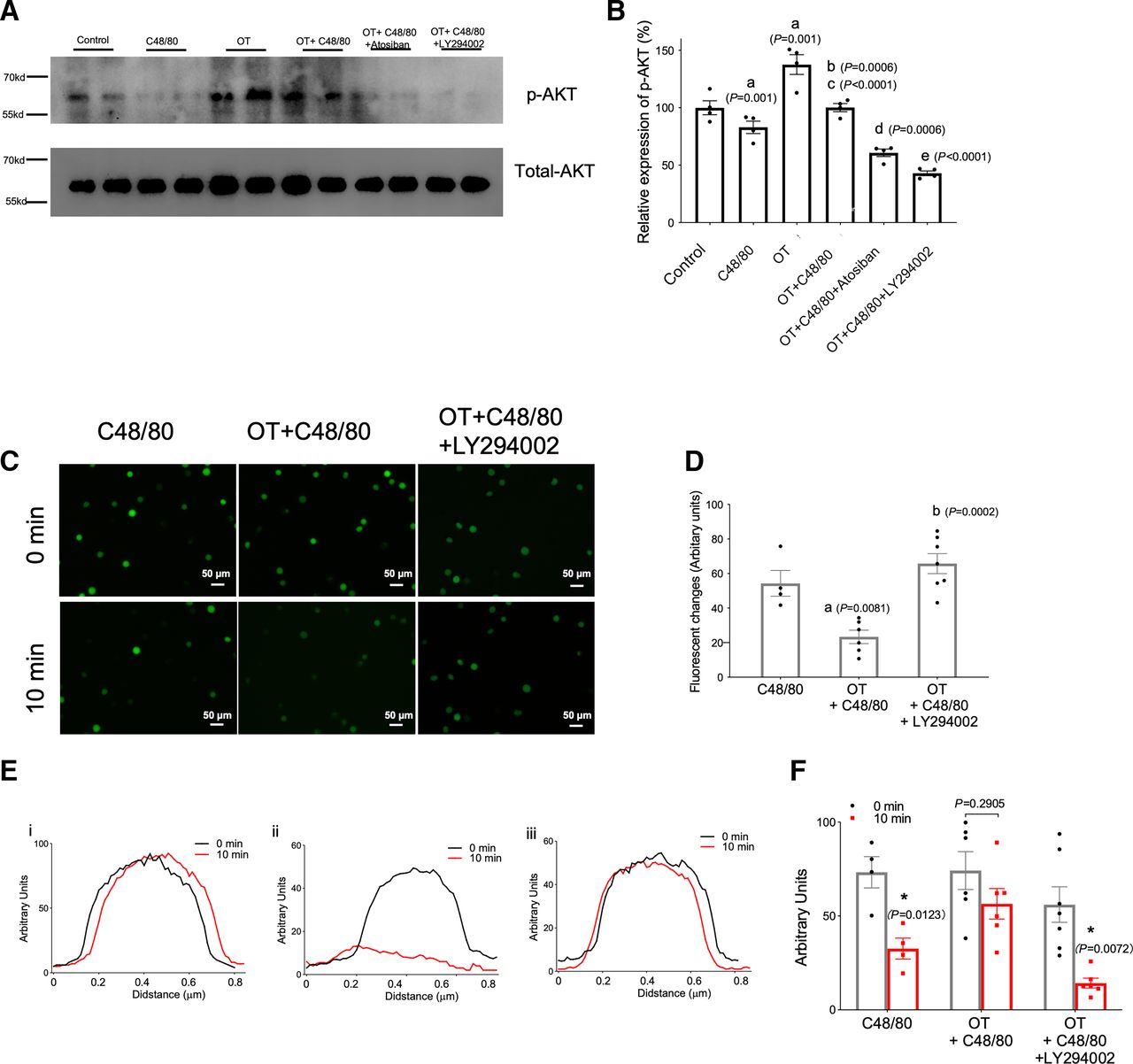
To further investigate the role of the PI3K/Akt signaling pathway in mast cell degranulation, a specific PI3K-inhibitor (LY294002) was used. C48/80 resulted in an apparent increase in both mast cells and degranulation, as detected using flow cytometry, which was attenuated by OT in P815 cells (P < 0.05) (Fig. 2, A–C). In addition, the protective role of OT was completely blocked by OTR, ruling out the off-target effects of OT. Further intervention studies examining the upstream and downstream relationship between OT and PI3K/Akt showed that OT treatment significantly increased p-Akt expression compared with the control group, whereas C48/80 exposure p-Akt expression was downregulated, which was reversed by OT treatment. Furthermore, atosiban blocked the OT-induced upregulation of p-Akt to a lesser extent than LY294002 (Fig. 3A; Supplemental Fig. 2). C48/80 increased fluorescent intensity of Ca2+ staining increased significantly, which was reversed by OT treatment. LY294002 further abolished the effects of OT pretreatment (Fig. 3, B and C). Similarly, OT administration reversed the Ca2+ release caused by C48/80, whereas in the presence of LY294002, OT failed to induce an increase in intracellular Ca2+ concentration (Fig. 3, D and E). These data suggest that PI3K/Akt is the downstream pathway of OT.

OT-OTR system significantly reduced mast cell degranulation induced by C48/80. (A and B) Flow cytometry analysis of P815 cells by forward scatter (upper panel) and side scatter (lower panel). (C) P1 cell percentage relative to control group. (A) P <0.05 compared with control group; (B) P <0.05 compared with C48/80 group; (C) P <0.05 compared with OT group.

OT stabilized mast cells through the PI3K/Akt pathway. (A) Representative western blot illustrating p-Akt protein levels (upper panel) in mast cells under various stimuli. p-Akt expression was quantified relative to total Akt levels and showed that C48/80 decreased p-Akt levels significantly in mast cells whereas the OT-OTR system inhibited p-Akt levels (lower panel). (B and C) Fluorescent images of Ca2+ staining and quantification of fluorescent changes. (D and E) OT reduced Ca2+ release from mast cells stimulated with C48/80. Ca2+ release was measured using flow cytometry in activated cells. Data are represented as the mean ± S.D. for three experiments (n = 3) and were analyzed using analysis of variance (ANOVA). (A) P <0.05 compared with control group; (B) P <0.05 compared with C48/80 group; (C) P <0.05 compared with OT group; (D) P <0.05 compared with OT + C48/80 group; (E) P <0.05 compared with OT + C48/80 + atosiban group. *P <0.05.
OT Administration Ameliorated Sympathetic Hyperinnervation and Improved Cardiac Function Post-MI.
We found that continuous administration of OT significantly reduced the number of mast cells as well as degranulated cardiac mast cells on the seventh day post-MI (Fig. 4, A–C). Notably, the mortality rate within 7 days was 2/20 (10%) in the OT-treated group and 8/35 (23%) in the MI group (P < 0.01). This represents a 2.3-fold reduction in the acute mortality rate. Masson’s trichrome staining shows formation of infarcted fibrosis in the infarcted hearts (blue color) with the ventricles becoming thinner in the hearts of the MI group. After the treatment of OT, less collagen fibrosis and significantly reduced infarct areas were detected (Fig. 4D), indicating the protective effects of OT in MI.

OT stabilized cardiac mast cells, ameliorated neural remodeling, and attenuated arrhythmias in MI rats. (A) OT administration reduces mast cell degranulation in the infarcted heart. (B and C) Quantitation of mast cell degranulation and mast cell number (n = 8). (D) Representative original color images of Masson’s trichrome staining of the infarcted area and quantitation of infarct size. (E) Immunofluorescent staining for tyrosine hydroxylase (magnification 200×) among three groups. (F) Fraction of the nerve density area (%) at the infarcted border. (G and H) Typical inducible ventricular arrhythmias (VAs) and comparisons of the arrhythmia scores across three groups. Each column with a bar represents the mean ± S.D. (n = 8). (A) P < 0.05 compared with the sham group; (B) P < 0.05 compared with the MI-vehicle group.
To investigate the mechanisms by which OT alleviates VA, we measured sympathetic nerve proliferation by staining the tyrosine hydroxylase (TH) nerve in the MI heart. TH-immunostained nerve fibers appeared to be aligned along the long axis of neighboring myofibers. Significant sprouting of sympathetic nerve fibers was observed in MI rats. In contrast, administration of OT significantly reduced nerve density in the infarcted border zone (Fig. 4, E and F). Ventricular pacing was performed to elucidate the physiological effects of attenuated sympathetic hyperinnervation. The arrhythmia score in the Sham group was 0 (Fig. 4, G and H). In infarcted rats, ventricular tachyarrhythmia was induced by programmed stimulation. OT treatment significantly reduced the inducibility of ventricular tachyarrhythmias as assessed by the inducibility quotient (both P < 0.05; see Materials and Methods), suggesting that the improved arrhythmias after MI may be due to stabilized cardiac mast cells.
OTR Deficiency Exacerbated Sympathetic Hyperinnervation after MI.
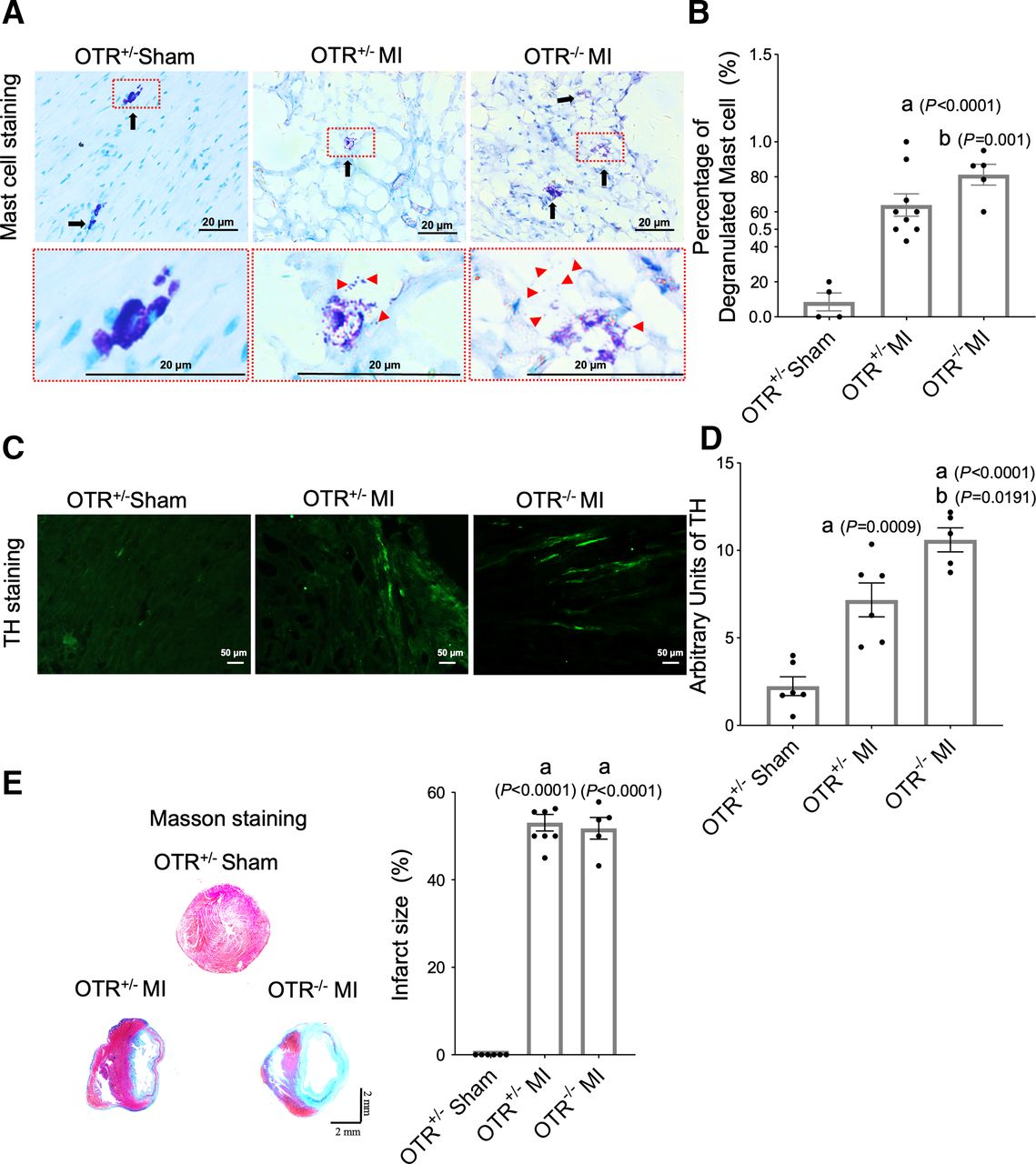
We further confirmed the exacerbated effects of the OT-OTR system in OTR−/− mice. OTR knockout mice exhibited increased mast cell degranulation (Fig. 5, A and B). Moreover, sympathetic hyperinnervation was aggravated (Fig. 5, C and D). There were no significant differences in infarction size between the two groups (Fig. 5, E and F). This result explains the cardioprotective role of OT-OTR system in improving neural remodeling.

OTR deficiency resulted in increased mast cell degranulation and neural remodeling in MI rats. (A) OT administration reduces mast cell degranulation in the infarcted heart. (B and C) Quantitation of mast cell degranulation and mast cell number (n = 8). (D) Representative original color images of Masson’s trichrome staining of the infarcted area and quantitation of infarct size. (E) Immunofluorescent staining for tyrosine hydroxylase (magnification 200×) among three groups. Each column with a bar represents the mean ± S.D. (n = 8). (A) P < 0.05 compared with the sham group; (B) P < 0.05 compared with the MI-vehicle group.
Discussion
This study provides crucial insights into the cardioprotective effects of OT on sympathetic hyperinnervation and post-MI cardiac remodeling. These mechanisms are mediated in part by the stabilization of cardiac mast cells via the OTR-PI3K/Akt axis.
OT is a cardiovascular hormone with cardioprotective effects (Gutkowska and Jankowski, 2012). Under several positive stressors such as serotonin or 5-hydroxytryptamine (5-HT) 1A, a sudden cardiovascular protective response can be elicited, which subsequently promotes the secretion of endogenous OT through neuroendocrine responses (Osei-Owusu et al., 2005). However, the endogenous OT system is inefficient and cannot secrete sufficient OT for cardiac protection. Therefore, exogenously supplemented OT as drug has been used to treat ischemic cardiac diseases (Gutkowska and Jankowski, 2012; Alizadeh and Mirzabeglo, 2013). Most of the cardioprotective effects of OT are demonstrated through direct effects on cardiomyocytes (CMs) (Kung et al., 2011; Gonzalez-Reyes et al., 2015) or indirect mechanisms involving arginine-vasopressin (AVP) and nitric oxide (NO) (Danalache et al., 2007). OT exerts its effects through specific receptor OTR, which has also been identified in tissues including the heart, colon, kidney, pancreas, and neurons (Gutkowska et al., 1997; Gimpl and Fahrenholz, 2001; Jankowski et al., 2004). OTR was also reported to participate in the process of sepsis-mediated oxidative stress, inflammation, and cardiac dysfunction (Merz et al., 2020). Interestingly, in the present study, we confirmed the expression of OTR mainly on cardiac mast cells rather than in the myocardium. Rudolph et al. (1998) firstly identified OT as endogenous mediator that may inhibit the activation of uterine mast cells. Qian et al. innovatively found the preventive and therapeutic effect of OT on mast cell degranulation and inflammation inhibition as well as cardiac function improvement in ischemia/reperfusion (I/R) model (Xiong et al., 2020). Given the above findings, we investigated the role and mechanism of OT/OTR pathway in mast cell stabilization and evaluated neuronal and cardiac remodeling in an infarcted rat as well as OTR−/− mice model.
Macrophages are the most commonly studied link between cardiac remodeling and postinfarction inflammation. However, the role of mast cells is still poorly understood. We found obvious degranulation of cardiac mast cells with markedly increased tryptase release in the infarcted border tissue post-MI. Degranulation of tissue mast cells is involved in inflammation and tissue remodeling through the release of cytotoxic mediators, thereby contributing deleterious effects on the cardiovascular system (Merz et al., 2020). Gain- and loss-of-function of OT/OTR system by continuous administration of OT and genetic OTR knockout further confirmed the effect of OT on the stabilization of cardiac mast cells. We further investigated the role and mechanism of OT in mast cell degranulation stimulated by C48/80 exposure at the cellular level. Previous studies with PI3K/Akt have typically focused on the release of proinflammatory factors and cytoskeletal regulation. The PI3K inhibitor LY294002 abolished the prevention of degranulation by OT in mast cells, whereas OT reversed the downregulation of p-Akt by C48/80 and was completely blocked by OTR. The data showed that PI3K/Akt acted downstream of OT in regulating mast cell degranulation. However, we cannot exclude other pathways through which OT regulates mast cell degranulation. For example, mast cell degranulation is regulated by intracellular calcium release from the endoplasmic reticulum and activation of protein kinase C (PKC), which depends on the activation of G-protein–coupled receptors (GPCRs) on the mast cell surface (Kimura et al., 1998; Nguyen et al., 2002; Kuehn and Gilfillan, 2007). Eastmond et al. (1997) reported that NO directly inhibited the IgE-mediated secretory function of mast cells. OT promotes NO synthesis by cardiac myocytes and dorsal root ganglion (Jankowski et al., 2010; Gutkowska et al., 2014, Gong et al., 2015). Gong et al. further explored the NO pathway and pointed out that OT evoking a concentration-dependent increase of intracellular Ca2+ was responsible for the activation of neuronal nitric oxide synthase (NOS1) and endothelial NOS (NOS3), thus producing inhibition of mast cell in Ca2+-NOS–dependent pathway (Gong et al., 2015). Therefore, we cannot exclude the notion that OT induces NO synthesis and release from cardiac myocytes, which may result in NO preventing degranulation of cardiac mast cells.
A recent study reported that mast cell stabilizers were effective in improving cardiac VAs during I/R (Krystel-Whittemore et al., 2015). Germination of sympathetic nerves caused by inflammation is the main cause of ventricular arrhythmias. We focused on the effect of OT on sympathetic innervation. Surprisingly, we found that OT has been proven to be an effective therapeutic molecule for inhibiting sympathetic hyperinnervation and ultimately resolving cardiac arrhythmias in the early stages of MI. In contrast, OTR results in impaired sympathetic innervation and deterioration of cardiac function. The direct roles of neural remodeling and mast cell degranulation were not defined in the present study. Mast cells synthesize and release NGF (Leon et al., 1994), a neurotrophic factor critical for promoting aggressive growth of sympathetic nerves, leading to undesirable arrhythmias. In future studies, we plan to investigate mast cell degranulation in relation to NGF secretion and sympathetic innervation using a mast cell depletion model. For cardiac remodeling protection, we also found reduced infarct size and mortality with continuous OT treatment. In addition to stabilizing mast cells, several benefits may lead to cardiac protection, including ANP release from CMs, protection against oxidative stress in vascular cells, and promotion of endothelial cell migration (Gutkowska et al., 2014). Interestingly, infarcted fraction showed no significant difference between OTR−/− and OTR+/− mice, as the endogenous OT system is conversely inefficient for cardiac protection.
Limitations
In the present study, we confirmed successful infarction via accessing the blanching of the myocardium in the affected area, ST elevation, regional cyanosis, and ventricular motion abnormalities. Still, we couldn’t totally exclude technique bias of incomparable infarct size among groups. The troponin I from plasma was another excellent biomarker that can be used to evaluate myocardial injury (Santulli et al., 2015, Zhang et al., 2017). In future studies, plasma troponin I measurement 24 hours after surgery should be performed as a standardized protocol in minimizing the bias caused by model. Besides, because OT can target pathogenic events in multiple cell types, these undefined cells may be inflammatory cells such as macrophages (Barouch et al., 2001), lymphocytes (Ralainirina et al., 2012), or cardiomyocytes. Further studies are needed to clarify how the OT-OTR system may contribute to cardiac remodeling in other cell types. Despite these limitations, our study clearly shows that the OT-OTR system can stabilize mast cells and inhibit neural and cardiac remodeling.
Conclusions
This study demonstrated a novel role of OT-OTR signaling in sympathetic hyperinnervation post-MI. Its mechanism of action is mediated in part by the stabilization of cardiac mast cells through the PI3K/Akt pathway.
Data Availability
The authors declare that all of the data supporting the findings of this study are available within the paper and its Supplemental Material.
Authorship Contributions
Participated in research design: Y. Li, Hu.
Conducted experiments: Yin, Wang, Ge, Yu, Jing.
Contributed new reagents or analytic tools: Yin, W. Yan, Liu.
Performed data analysis: Gong, S. Yan, Wang, X. Li.
Wrote or contributed to the writing of the manuscript: Yin, Han, Y. Li, Hu.
Footnotes
- Received January 2, 2024.
- Accepted May 31, 2024.
This work was supported by grants from the National Natural Science Foundation of China [Grant 81570305] (to S.Y.); Natural Science Foundation of Shandong Province [Grant ZR2020MH024] (to H.H.), [Grant ZR2023MH182] (to W.H.), and [Grant ZR2020MH023] (to Y.L.); Shandong First Medical University Talent Introduction Funds (to S.W.); and Qianfoshan Hospital Foundation of National Natural Science Foundation of China [Grant QYPY2019NSFC0801(A000951)] (to Y.L.).
No author has an actual or perceived conflict of interest with the contents of this article.
This article is posted as a preprint in Research Square [https://doi.org/10.21203/rs.3.rs-3565452/v1].
↵1 J.Y. and Y.W. contributed equally to this work.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- Akt
- protein kinase B
- KO
- knockout
- LAD
- left coronary artery
- MI
- myocardial infarction
- NGF
- nerve growth factor
- NO
- nitric oxide
- OT
- oxytocin
- OTR
- oxytocin receptor
- p-Akt
- phosphorylated Akt
- PI3K
- phosphoinositide 3-kinase
- TH
- tyrosine hydroxylase
- VA
- ventricular arrhythmia
- VT
- ventricular tachycardia
- WT
- wild-type
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}