


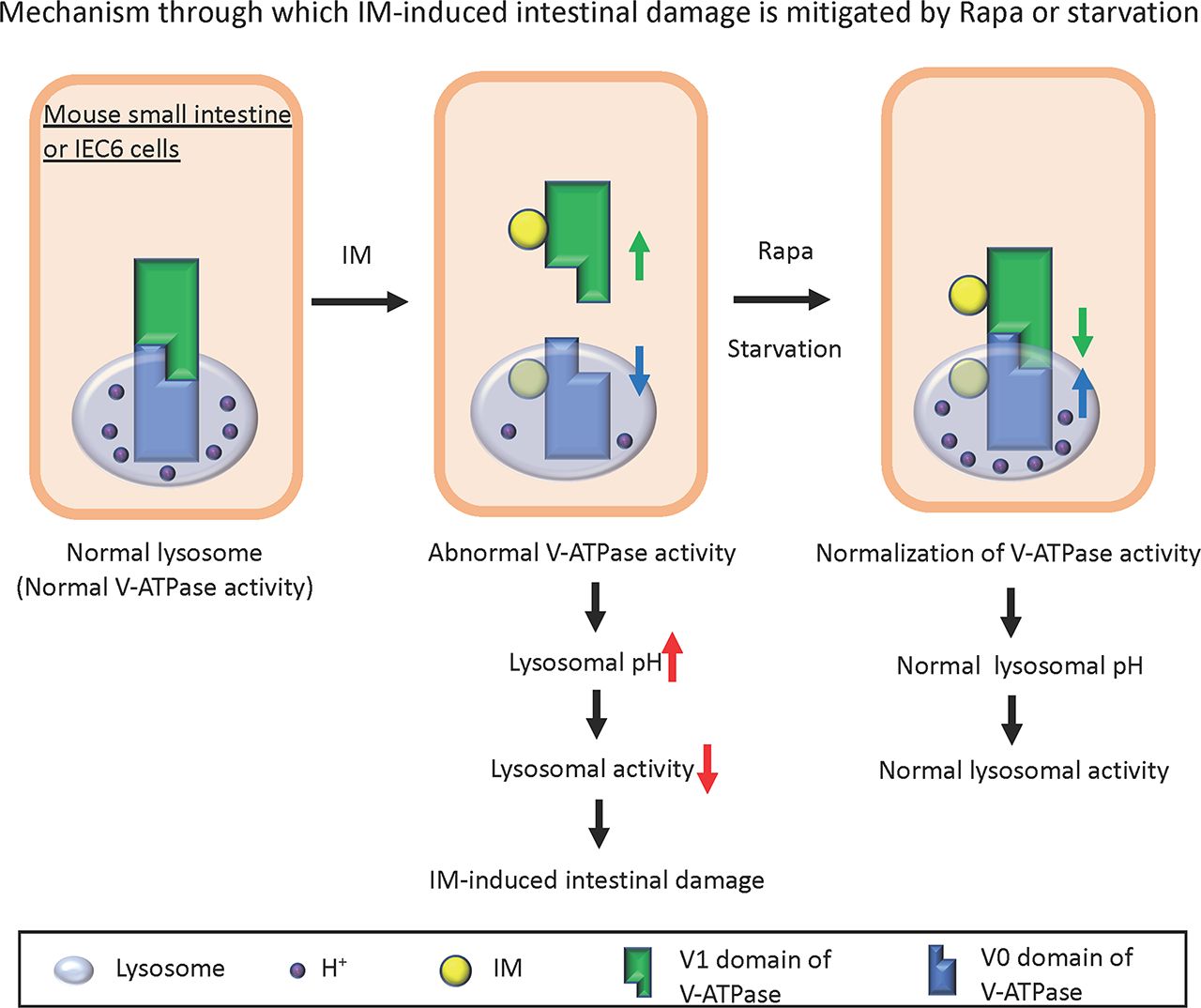
Visual Overview

Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) possess anti-inflammatory, antipyretic, and analgesic properties and are among the most commonly used drugs. Although the cause of NSAID-induced gastric ulcers is well understood, the mechanism behind small intestinal ulcers remains elusive. In this study, we examined the mechanism through which indomethacin (IM), a prominent NSAID, induces small intestinal ulcers, both in vitro and in vivo. In IEC6 cells, a small intestinal epithelial cell line, IM treatment elevated levels of LC3-II and p62. These expression levels remained unaltered after treatment with chloroquine or bafilomycin, which are vacuolar ATPase (V-ATPase) inhibitors. IM treatment reduced the activity of cathepsin B, a lysosomal protein hydrolytic enzyme, and increased the lysosomal pH. There was a notable increase in subcellular colocalization of LC3 with Lamp2, a lysosome marker, post IM treatment. The increased lysosomal pH and decreased cathepsin B activity were reversed by pretreatment with rapamycin (Rapa) or glucose starvation, both of which stabilize V-ATPase assembly. To validate the in vitro findings in vivo, we established an IM-induced small intestine ulcer mouse model. In this model, we observed multiple ulcerations and heightened inflammation following IM administration. However, pretreatment with Rapa or fasting, which stabilize V-ATPase assembly, mitigated the IM-induced small intestinal ulcers in mice. Coimmunoprecipitation studies demonstrated that IM binds to V-ATPase in vitro and in vivo. These findings suggest that IM induces small intestinal injury through lysosomal dysfunction, likely due to the disassembly of lysosomal V-ATPase caused by direct binding. Moreover, Rapa or starvation can prevent this injury by stabilizing the assembly.
SIGNIFICANCE STATEMENT This study elucidates the largely unknown mechanisms behind small intestinal ulceration induced by indomethacin and reveals the involvement of lysosomal dysfunction via vacuolar ATPase disassembly. The significance lies in identifying potential preventative interventions, such as rapamycin treatment or glucose starvation, offering pivotal insights that extend beyond nonsteroidal anti-inflammatory drugs–induced ulcers to broader gastrointestinal pathologies and treatments, thereby providing a foundation for novel therapeutic strategies aimed at a wide array of gastrointestinal disorders.
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are frequently used for chronic conditions such as pain, fever, and rheumatoid arthritis (Laine, 2001; Thakur et al., 2018), with their demand steadily increasing. Although NSAIDs are known to have side effects such as mucosal injury, methods have been established to address gastric ulcers, including the concurrent use of proton pump inhibitors (PPIs) with NSAIDs (Kavitt et al., 2019; Hnepa et al., 2021). Recent investigations employing capsule and balloon endoscopes have disclosed that NSAIDs not only damage the gastric membrane but also those of the intestines and colon (Graham et al., 2005). The frequent occurrence of small intestinal ulcers poses a clinical challenge (Watanabe et al., 2020; Domper Arnal et al., 2022). Consequently, there is a pressing need to develop novel preventive and therapeutic agents against NSAID-induced small intestinal ulcers. Indomethacin (IM) is a widely used NSAID and an antirheumatic drug (Gliszczynska and Nowaczyk, 2021). Experimentally, IM is often chosen to induce gastric ulcers due to its higher ulcerogenic potential compared with other NSAIDs (Suleyman et al., 2010).
Autophagy is an intracellular degradation mechanism that transports cytoplasmic constituents to lysosomes. It plays a pivotal role in cell survival, differentiation, development, and homeostasis (Mizushima et al., 2002). Central autophagy pathways, such as the mammalian target of rapamycin (Rapa) complex 1 (mTORC1) pathway, are vital for cellular maintenance (Taneike et al., 2016; Ranek et al., 2019; Oeing et al., 2020). During autophagy, cells generate double-membraned vesicles known as autophagosomes, which merge with lysosomes to degrade their enclosed substances (Levine and Kroemer, 2008). This entire procedure is also termed autophagy flux. The transformation of LC3I to LC3II serves as an autophagy indicator. The p62 protein is another autophagy marker, and its expression levels frequently gauge the degradation capabilities of autophagic lysosomes. An interruption in autophagy flux can result in abnormal accumulation of LC3II and p62 proteins (Kabeya et al., 2000; Mizushima and Komatsu, 2011).
In prior research, we demonstrated that autophagy plays a role in IM-induced small intestinal damage using Atg5-conditional knockout mice and small intestinal cell lines where Atg5 was knocked down (Harada et al., 2015). Given recent reports suggesting that IM hampers autophagy flux by interfering with lysosomal functionality (Vallecillo-Hernandez et al., 2018), we investigated the potential disruption of autophagy flux in IM-induced small intestinal ulcers. After confirming the interrupted autophagy flux post IM treatment both in vitro and in vivo, we delved into the causes behind this disruption. In this study, we discuss a novel mechanism behind IM-induced small intestinal ulcers and put forth a potential therapeutic approach for alleviating the intestinal injury.
Materials and Methods
Antibodies.
The anti-ATP6V1A (V1A) antibody was purchased from Gene Tex (Irvine, CA). Anti-ATP6V1B1 plus ATP6V1B2 (V1B), anti-ATPV0d1 (V0d), and anti-p62 antibodies were purchased from Abcam (Cambridge, UK). Additionally, anti-LC3 and anti–α-tubulin antibodies were purchased from MBL (Nagoya, Japan). Secondary horseradish peroxidase–conjugated antibodies were purchased from SouthernBiotech (Birmingham, AL).
Chemicals.
IM was sourced from Sigma-Aldrich (St. Louis, MO). Chloroquine (CQ) and bafilomycin A1 (Baf A1) were obtained from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). LysoSensor Green DND-189 (#L7535) was purchased from Thermo Fisher Scientific (Waltham, MA). Rapa was procured from Merck (La Jolla, CA), and Magic Red Cathepsin B assay kit (#6134) was acquired from Immunochemistry Technologies (Bloomington, MN).
Mouse Model of Small Intestine Inflammatory Disease.
The mouse model for acute intestinal inflammatory disease was established using IM treatment as previously described (Yoriki et al., 2013; Horibe et al., 2016). Briefly, WT mice were administered IM (10 mg/kg, i.p.) for 24 hours. Subsequently, mice were euthanized, and the tissues were harvested for experiments. All animal procedures were performed according to the guidelines of the Osaka Medical and Pharmaceutical University Animal Care and Use Committee (approval protocol number: AM23-081).
Histopathology.
Small intestines were immediately excised from euthanized mice 24 hours after IM administration. The excised portions of the small intestines were rinsed with PBS, fixed in 10% formalin, embedded in paraffin, and then sectioned transversely at 5 μm for H&E staining and Evans blue staining.
Real-Time Polymerase Chain Reaction Analysis.
Total RNA was extracted from isolated small intestine tissues using RNAzol (RN190; Molecular Research Center, Cincinnati, OH). Total RNA was reverse transcribed with the PrimeScript RT reagent Kit (Takata Bio Inc., Kusatsu, Japan). Quantitative real-time polymerase chain reaction was performed using QuantStudio 5 Real-Time PCR System (Applied Biosystems, Waltham, MA) using PowerTrack SYBR Green Master Mix (Thermo Fisher Scientific, Langenselbold, Germany) and the following primers: 1) IL-6: 5′-ACAACCACGGCCTTCCTTCCCTACTT-3′ and 5′-CACGATTTCCCAGAGAACATGTG-3′ and 2) GAPDH: 5′-ATGACAACTTTGTCAAGCTCATTTGC-3′ and 5′-GGTCCACCACCCTGTTGCTGC-3′ for 40 cycles (95°C for 5 seconds, 60°C for 30 seconds).
Cell Culture.
IEC6 cells were purchased from the Riken Bioresource Center (Riken BRC, Tsukuba, Japan). The cells were maintained in Dulbecco’s modified Eagle’s medium growth medium (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. IEC6 cells were exposed to 200 μM indomethacin for 4 hours, either alone or in combination with 10 μM chloroquine or 10 μM bafilomycin A1.
Pull-Down Assay Using IM-Immobilized FG Beads.
A pull-down assay was conducted using IM-immobilized NH2-FG beads (TAS8848N1130; Tamagawa Seiki, Nagano, Japan) in line with the manufacturer’s protocol (https://fgb.tamagawa-seiki.com/en/data/pdf/protocolE005.pdf) (Liu et al., 2012; Aono et al., 2018). Briefly, 20 mM IM (resulting in a final concentration of 10 mM) was activated by incubating with an equivalent molarity of succinimide and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) in 1000 μL of N, N′-dimethylformamide (DMF) for 2 hours at room temperature (22–28°C) using a microtube mixer. This activated IM was then immobilized onto NH2-FG beads by incubating with 2.5 mg of the beads for 16 hours at room temperature, again using the microtube mixer. Following this, IM-immobilized NH2-FG beads (0.5 mg) were incubated with 500 μg of mouse heart homogenates and IEC6 cell lysates at 4°C for 4 hours. The pull-down eluates were subsequently subjected to western blot analysis employing the relevant antibodies.
Immunofluorescence.
IEC6 cells were seeded onto a glass-bottom dish and incubated for 24 hours to facilitate cell attachment. Then, cells were incubated with 200 μM IM for 4 hours, either alone or in combination with low-glucose/0.5% serum or Rapa. Subsequently, the cells were fixed with 4% paraformaldehyde followed by permeabilization using 0.1% Triton X-100 and stained with anti-LC3 and anti-Lamp2 antibodies before being subjected to secondary labeling with Alexa Fluor–conjugated secondary antibodies. The fluorescence staining was imaged by confocal microscopy, SP8 (Leica Microsystems, Wetzlar, Germany).
Western Blotting.
IEC6 cells were lysed using a lysis buffer composed of 1% Nonidet P-40, 20 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 10% glycerol, 5 mM sodium pyrophosphate decahydrate, 10 mM sodium fluoride, 1 mM sodium orthovanadate, 10 mM β-glycerophosphate, and 1 mM phenylmethylsulfonyl fluoride (pH 7.4). This buffer also contained a protease inhibitor cocktail set V (FUJIFILM Wako Pure Chemical Corporation). Protein concentrations in the lysates were measured using a BCA protein assay kit (Thermo Fisher Scientific). These lysates underwent western blot analysis. After separation with SDS-PAGE, the gel was placed into a semidry blotting system (Bio-Rad, Hercules, CA). Proteins were then transferred to a polyvinylidene fluoride membrane (Merck Millipore, Billerica, MA). This membrane was blocked using 5% skimmed milk in Tris-buffered saline with 0.1% Tween 20 (blocking buffer). It was then incubated with the indicated antibodies in the blocking buffer, rinsed three times in Tris-buffered saline with 0.1% Tween 20, and subsequently incubated with secondary antibodies conjugated with horseradish peroxidase. The membrane was visualized using either Luminata Crescendo or Forte Western HRP Substrate (Merck Millipore), and the resulting image was captured using the Fusion FX7 system (Peqlab, Erlangen, Germany).
Immunoprecipitation.
Immunoprecipitation was performed by using SureBeads Protein G Magnetic Beads (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Total protein extracts (250 μg) were dissolved in homogenization buffer [50 mM Hepes (pH7.4), 5 mM sodium pyrophosphate, 10 mM sodium fluoride, 1 mM sodium orthovanadate, 10 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (#162-26031; FUJIFILM Wako Pure Chemical Corporation)] and incubated with the corresponding primary antibodies (overnight, 4°C). Normal rabbit IgG (#2729; CST, Danvers, MA) was used as a negative control. The pull-down eluates were used for western blot analysis with the antibodies of interest.
Magic Red and LysoSensor Staining.
IEC6 cells were seeded onto a glass-bottom dish and incubated for 24 hours to facilitate cell attachment. Then, cells were incubated with 200 μM IM for 4 hours, either alone or in combination with low-glucose/0.5% serum or Rapa. Subsequently, the cells were stained with either Magic Red (1:250 dilution) or LysoSensor (1:500 dilution) for 15 minutes. Samples were then directly examined using a confocal microscope, SP8 (Leica Microsystems).
Statistical Analysis.
The intensities of western blot bands were quantified using Fiji/ImageJ software (National Institutes of Health, Bethesda, MD). Values are presented as mean ± S.D. based on n = 3 or more determinations. An F test was employed to assess the equality of variances, which was then followed by an unpaired Student’s t test to compare two mean values. For multiple comparisons, a one-way analysis of variance combined with Tukey’s test was applied as appropriate. The Mander’s coefficient (tM1) was calculated with Coloc2 via the Fiji/ImageJ software (National Institutes of Health). P < 0.05 was considered significant.
Results
IM Disrupts Autophagy Flux in IEC6 Cells.
We have previously demonstrated that autophagy plays a role in IM-induced small intestinal damage using Atg5 conditional knockout mice and IEC6 cells (Harada et al., 2015). Given that IM was reported to disrupt autophagic flux by inducing lysosomal dysfunction in gastric cancer cells (Vallecillo-Hernández, J. et al., 2018), we assessed autophagy-related protein levels, such as LC3 and p62, 4 hours post-treatment with 200 μM IM in IEC6 cells. Both the LC3-II:LC3-I ratio and p62 protein levels in IEC6 cells saw a significant rise following IM treatment (Fig. 1). This escalation remained unchanged with CQ or Baf A1 pretreatments (Fig. 1), indicating that IM does not amplify autophagy but rather disrupts autophagy flux.

Indomethacin (IM) disrupts autophagy flux in IEC6 cells. (A) IEC6 cells were exposed to 200 μM IM for 4 hours, either alone or in combination with 10 μM CQ or 10 μM Baf A1. Subsequently, LC3 and p62 expression levels were assessed using western blotting. (B) Quantitative analysis of the LC3-II/LC3-I ratio as shown in (A). Values are presented as the mean ± S.D. (n = 3) (*P < 0.05). (C) Quantitative assessment of p62 protein expression from (A). Values are expressed as the mean ± S.D. (n = 3) (*P < 0.05).
IM Does Not Inhibit Autophagosome-Lysosome Fusion in IEC6 Cells.
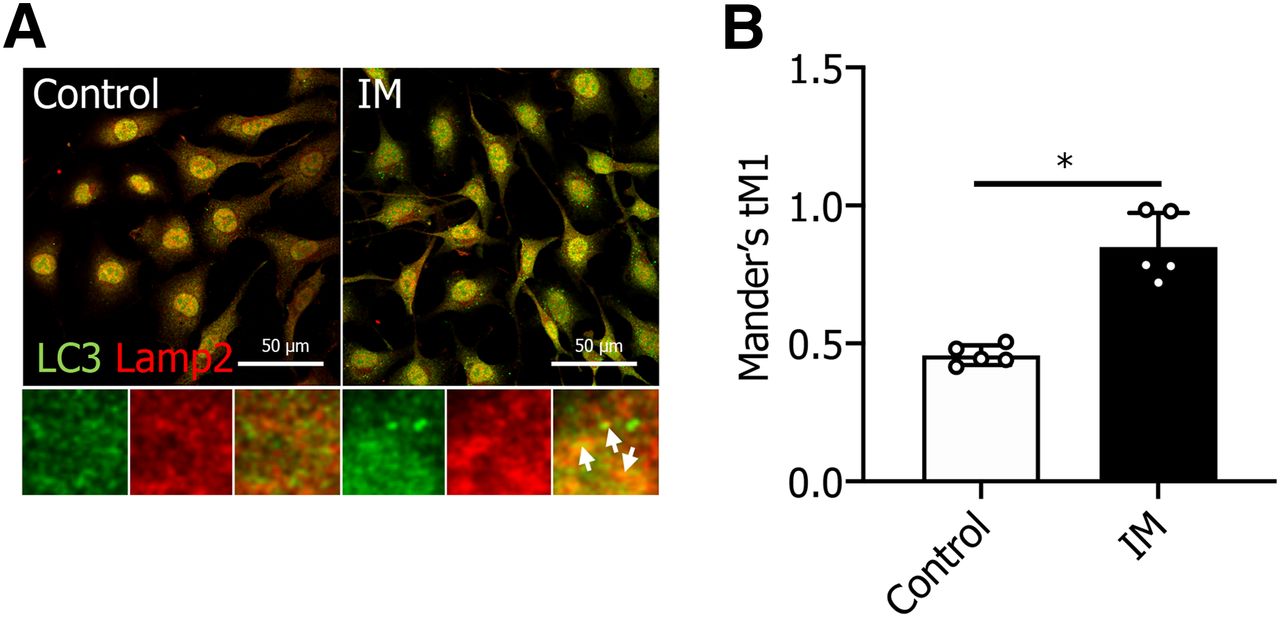
To discern the mechanism through which IM disrupts autophagic flux, we investigated the effect of IM on autophagosome-lysosome fusion. Staining for LC3 (an autophagosome marker, depicted in green) and Lamp2 (a lysosome marker, depicted in red) was carried out to assess the impact of IM on this fusion. We noted a marked increase in overlapping regions (illustrated in yellow) between LC3 and Lamp2 following IM treatment (Fig. 2). This indicates that the autophagosome successfully fused with the lysosome, but the LC3 within the lysosome was not degraded post IM treatment.

IM does not inhibit autophagosome-lysosome fusion in IEC6 cells. (A) IEC6 cells were stained for Lamp2 (red) and LC3 (green) to examine the effects of IM. White arrows highlight the fusion of autophagosomes with lysosomes. Scale bar, 50 μm. (B) Quantification of confocal images (Mander’s colocalization) between Lamp2 and LC3 as depicted in (A). Values are expressed as the mean ± S.D. (n = 5) (*P < 0.05).
Rapa or Glucose Starvation Reverses Lysosomal Dysfunction and pH Neutralization after IM Treatment in IEC6 Cells.
The disruption of autophagy flux can be attributed not only to the anomalies in autophagosome-lysosome fusion but also to lysosomal dysfunction. To determine whether lysosomal dysfunction is a contributing factor to the disrupted autophagy flux in IEC6 cells, we treated these cells with IM and then stained them with a Magic Red Cathepsin B assay kit or LysoSensor Green DND-189 to assess lysosomal activity and pH, respectively. In both staining methods, the fluorescent signals were notably diminished in IM-treated IEC6 cells compared with the control cells (Fig. 3, A–D). This suggests that cathepsin B activity decreases and lysosomal pH tends toward neutral following IM treatment. To counteract the diminished lysosomal activity post IM treatment, we endeavored to activate autophagy flux in IEC6 cells either by administering Rapa (Gupta et al., 2017; Mu et al., 2022) or through glucose starvation using low-glucose/0.5% serum (Chen et al., 2014; Karabiyik et al., 2021). As depicted in Fig. 3, A–D, both Rapa treatment and glucose starvation successfully restored the decreased lysosomal activity and pH acidification. Furthermore, we examined whether there was a synergistic effect between the two treatments. No significant increase in MagicRed and Lysosensor fluorescence intensity was observed with both treatments (Supplemental Fig. 3), indicating a possible lack of synergy.

IM reduces lysosomal activity and acidification, but these effects are mitigated by Rapa treatment or low-glucose (glu) conditions in IEC6 cells. (A) IEC6 cells were exposed to 200 μM IM and subsequently stained with Magic Red dye to assess lysosomal activity. Scale bar, 50 μm. (B) Quantitative analysis of the Magic Red fluorescence intensity per cell shown in (A). (C) IEC6 cells were treated with 200 μM IM and then stained with LysoSensor dye to measure lysosomal pH. Scale bar, 50 μm. (D) Quantitative evaluation of LysoSensor fluorescence intensity per cell as depicted in (C). (E) Lysates from IEC6 cells treated with IM, either alone or in combination with low-glucose/0.5% serum or Rapa, were subjected to coimmunoprecipitation using anti-V0D antibody. This was followed by western blotting using the anti-V1A antibody. (F) Lysates from IM-treated IEC6 cells, either alone or combined with low-glucose/0.5% serum or Rapa, were subjected to coimmunoprecipitation using the anti-V1A antibody. This was followed by western blotting using the anti-V0D antibody. FCS, fetal calf serum; IP, immunoprecipitation; n.s., not significant.
IM Disrupts the Assembly of Vacuolar ATPase, which Is Reversed by Rapa or Glucose Starvation in IEC6 Cells.
Given that lysosomal dysfunction can result from reduced pH acidification, we sought to understand the reasons behind this decrease in pH. We turned our attention to vacuolar ATPase (V-ATPase), a principal protein integral to lysosomal pH acidification. V-ATPase modulates ATP hydrolysis and proton transport based on intracellular conditions. The binding and separation of the V1 and V0 domains are pivotal for its function, with the assembly of the V1 and V0 domains being essential for the activity of V-ATPase (Forgac, 2007; Vasanthakumar and Rubinstein, 2020). With this in mind, we assessed the binding of V1A, a subunit of the V1 domain, to V0d, a subunit of the V0 domain, in the presence and absence of IM treatment. Our findings indicated that IM disrupted the assembly of V-ATPase. Remarkably, this disassembly was counteracted by either Rapa or glucose deprivation (Fig. 3, E and F). These observations suggest that treatments with Rapa or glucose reduction can mitigate IM-triggered lysosomal dysfunction, likely through the restoration of V-ATPase activity by stabilizing its assembly.
IM Induces Multiple Ulcers and Inflammation in the Small Intestines of Mice.
To elucidate the mechanism behind NSAID-induced small intestinal ulcer formation, we used an IM-induced small intestine ulcer mouse model. Following IM administration in this model, multiple ulcers and increased inflammation were observed. Evans blue staining showcased the multiple ulcers brought about by IM (Fig. 4A). H&E staining revealed evident tissue damage and accompanying inflammation after IM exposure (Fig. 4C). The ulcer ratio (ulcer area:total area) in the small intestines of IM-treated mice was significantly higher than that of the control mice (Fig. 4B). Additionally, the concentrations of IL-6, a proinflammatory cytokine, spiked noticeably post IM treatment (Fig. 4D).

IM induces ulcers and inflammation in the mouse small intestine. (A) Histologic evaluation of small intestinal lesions caused by IM as visualized using Evans blue staining. The red circle highlights an ulcerated area. Scale bar, 5 mm. (B) Quantification of the ulcer ratio (ulcer area:total area) as depicted in (A). Values are presented as the mean ± S.D. (n = 4) (*P < 0.05). (C) Histologic examination of small intestinal lesions due to IM using H&E staining. Scale bar, 5 mm. (D) Relative mRNA expression levels of interleukin 6 were determined in mouse hearts following IM treatment. Values are expressed as mean ± S.D. (n = 3) (*P < 0.05).
Rapa Treatment or Fasting Mitigates Ulcer Formation and Inflammation after IM Administration in Mouse Small Intestine.
We subsequently investigated if Rapa treatment or fasting could mitigate the IM-induced small intestinal ulcers in mice. Prior to IM administration, mice were either treated with Rapa or subjected to fasting. Evans blue staining revealed a significant reduction in IM-induced small intestinal ulcer formation in both of these groups compared with the control groups (Fig. 5, A–D). A pathologic evaluation H&E staining further confirmed that the damage to the IM-induced small intestinal mucosal tissue was notably reduced in both groups compared with the control groups (Fig. 5, E and F). To verify the effect of IM administration and the pretreatment with Rapa or fasting on the V-ATPase assembly observed in IEC6 cells, we assessed the binding of V1A to V0d, both with and without IM administration. The findings indicated that IM disrupted the V-ATPase assembly, but this disruption was alleviated by either Rapa treatment or fasting (Fig. 5, G and H). These data suggest that Rapa treatment or fasting can counteract IM-induced lysosomal dysfunction, likely due to an increase in V-ATPase activity by stabilizing the assembly in both the mouse small intestine and IEC6 cells.

Fasting or Rapa mitigates IM-induced small intestinal ulceration in mice. (A) Histologic examination of small intestinal lesions caused by IM administration, with or without Rapa, using Evans blue staining. Scale bar, 5 mm. (B) Histologic examination of small intestinal lesions caused by IM administration, with or without fasting, using Evans blue staining. Scale bar, 5 mm. (C) Quantification of the ulcer ratio (ulcer area:total area) as depicted in (A). Values are expressed as the mean ± S.D. (n = 4) (*P < 0.05). (D) Quantification of the ulcer ratio (ulcer area:total area) as depicted in (B). Values are expressed as the mean ± S.D. (n = 4) (*P < 0.05). (E) Histologic examination of small intestinal lesions caused by IM administration, with or without Rapa, using H&E staining. Scale bar, 100 μm. (F) Histologic examination of small intestinal lesions caused by IM administration, with or without fasting, using H&E staining. Scale bar, 100 μm. (G) Homogenates of the small intestine from IM-treated mice, with or without fasting or Rapa, were subjected to coimmunoprecipitation using the anti-V0d antibody. This was followed by western blotting with the anti-V1A antibody. (H) Homogenates of the small intestine from IM-treated mice, with or without fasting or Rapa, were subjected to coimmunoprecipitation using the anti-V1A antibody, followed by western blot analysis with the anti-V0d antibody. IP, immunoprecipitation.
IM Binds to V-ATPase In Vitro and In Vivo.
Following the observation of increased lysosomal pH post IM treatment, we speculated that IM might associate with a protein essential for maintaining lysosomal pH homeostasis, such as V-ATPase. To determine this association, we used IM-immobilized beads in IEC6 cells. Postincubation of these beads with the cytoplasmic extracts of IEC6 cells, we assessed the binding of IM to V-ATPase using antibodies targeting V-ATPase subunits (V1A, V1B, and V0d). Our findings revealed that IM did bind to V-ATPase (Fig. 6A), with this binding capacity increasing in a dose-dependent manner (Fig. 6B).

IM binds to V-ATPase in a concentration-dependent manner. (A) Beads immobilized with IM (+) or empty (-) were incubated with cytoplasmic extracts from IEC6 cells. Bound proteins were eluted using Laemmli dye, then subjected to SDS-PAGE and subsequent western blotting using anti-V1A, anti-V1B2, and anti-V0d antibodies. (B) Beads immobilized with 2, 5, or 10 mM of IM (+) or empty (-) were incubated with cytoplasmic extracts from IEC6 cells. After eluting bound proteins with Laemmli dye, they were analyzed using SDS-PAGE and western blotting using anti-V1A, anti-V1B2, and anti-V0d antibodies. (C) Beads immobilized with IM (+) or empty (-) were incubated with homogenates from mouse small intestine. Bound proteins were eluted using Laemmli dye and subsequently analyzed using SDS-PAGE and western blotting using anti-V1A, anti-V1B, and anti-V0d antibodies.
To ascertain if this binding also took place in the mouse small intestine, similar to the IEC6 cells, we investigated the association of IM with V-ATPase in mouse small intestinal tissue. Using the same methodology of incubating IM-immobilized beads with the cytoplasmic extracts of mouse small intestinal homogenates, we again employed antibodies against V-ATPase subunits (V1A, V1B, and V0d) to evaluate this potential binding. The results affirmed that IM binds to V-ATPase in mouse small intestine similarly to its binding in IEC6 cells (Fig. 6C). To validate that the IM-immobilized beads retained the same bioactivity as that of IM, we analyzed the expression levels of LC3 and p62 (Supplemental Fig. 1) and assessed lysosomal activity and pH (Supplemental Fig. 2) after treatment with either IM or IM-NH2 beads in IEC6 cells. This allowed us to contrast the effects on autophagy flux, lysosomal activity, and pH acidification. As anticipated, no significant alterations in bioactivities were observed.
Discussion
Recently, NSAIDs have been discovered to induce not only gastric or duodenal ulcers but also small intestinal ulcers at a high frequency. In the case of gastric or duodenal ulcers, gastric acid is known to exacerbate mucosal injury, making PPIs, which suppress gastric acid secretion, a targeted treatment. However, PPIs not only fail to counteract NSAID-induced small intestinal injury but may even pose a risk factor for it (Washio et al., 2016). This suggests that small intestinal ulceration occurs independently of gastric acid. The infiltration of enteric bacteria into the small intestinal epithelium might be the most significant virulence factor (Takeuchi and Satoh, 2015). When the intestinal mucosal barrier function is compromised due to prostaglandin deficiency or mitochondrial dysfunction, enteric bacteria invade the small intestinal epithelium. Subsequently, pattern-recognition receptors such as TLRs and nod-like receptors activate the innate immune system (Watanabe et al., 2011; Wen et al., 2013; Lamkanfi and Dixit, 2014). Though research is ongoing to elucidate the mechanism behind NSAID-induced small intestinal ulceration, the etiology following NSAID consumption remains unclear. We have identified a novel mechanism through which IM causes small intestinal injury and a potential remedy for the damage. A previous study indicated that IM disrupts autophagic flux by inducing lysosomal dysfunction in gastric cancer cells, potentially affecting the sensitivity of these cells to cytotoxic agents (Vallecillo-Hernandez et al., 2018). Our findings mirror this in IEC6 cells and mouse small intestines, leading us to conclude that this phenomenon plays a role in the mechanism through which IM induces small intestinal ulcers. Our evidence from IEC6 cells and mice demonstrates that IM interrupts autophagy flux by causing lysosomal dysfunction. This mechanism is possibly due to IM binding to V-ATPase, leading to decreased lysosomal pH acidification as a result of V-ATPase disassembly. The dysfunction of the lysosome is thought to cause problems in the degradation of many proteins, glycolipids, etc., but what specific molecules are affected and how they are damaged in the small intestine should be investigated in the future.
In our study, IM treatment of IEC6 cells elevated the LC3-II/I ratio and p62 level, and the elevation remained unchanged with CQ or Baf A1 pretreatment (Fig. 1). This indicates that IM does not amplify autophagy but rather disrupts autophagy flux. Predominantly, two main mechanisms cause disrupted autophagy flux: irregular autophagosome-lysosome fusion and lysosomal dysfunction. As displayed in Figs. 2 and 3, A–D, the disruption was attributed to lysosomal dysfunction rather than fusion abnormalities.
The assembly of the V1 and V0 domains of V-ATPase was found to be compromised by IM treatment in both IEC6 cells (Fig. 3, E and F) and mouse small intestinal homogenates (Fig. 5, G and H). This lysosomal dysfunction may be due to the disassembly of V-ATPase. Further examination revealed that IM-bound beads coimmunoprecipitated with V-ATPase subunits in both IEC6 cells (Fig. 6, A and B) and mouse small intestinal homogenates (Fig. 6C). Given this evidence, we theorize that IM's binding to V-ATPase may disrupt the assembly of the V1 and V0 domains of V-ATPase, leading to reduced lysosomal pH acidification and function. An acidic environment (pH 4.5–5.0) is vital for most hydrolytic enzymes in lysosomes (Mindell, 2012). Maintaining lysosomal pH is crucial because any deviation in the proton gradient disrupts the lysosomal degradation machinery, resulting in diseases such as Fabry disease, a lysosomal storage disorder (Desnick et al., 2003; Zarate and Hopkin, 2008; Xu et al., 2014). Thus, lysosomal dysfunction could be a factor in IM-induced small intestinal damage. Noting that autophagy flux was interrupted post IM treatment, we postulated that interventions activating autophagy, such as starvation (Stipanuk, 2009) or Rapa treatment (Neufeld, 2010), might mitigate the damage. Disassembly of the V1 and V0 domains was reversed by either Rapa treatment or starvation in both mouse small intestinal homogenates and IEC6 cells. Notably, ulcers that appeared in the mouse small intestines post IM administration were ameliorated by either Rapa treatment or fasting.
We established a mouse model to study IM-induced small intestinal injury. Consistent ulceration and significant inflammation were observed following IM administration (Fig. 4). Given the pronounced changes after IM administration, demonstrating the efficacy of Rapa treatment or starvation became straightforward. This mouse model could prove useful for testing other therapeutic agents against IM-induced small intestinal damage.
In summary, we suggest that IM induces small intestinal injury through lysosomal dysfunction resulting from V-ATPase disassembly. This injury can be prevented by either Rapa treatment or fasting.
Acknowledgments
The authors thank Takehiro Nagatsuka, Yumiko Okumura, and Natsuko Hayashi (Osaka Medical and Pharmaceutical University) for their technical assistance.
Data Availability
The authors declare that all the data supporting the findings of this study are contained within the paper.
Authorship Contributions
Participated in research design: Shirakawa, Yokoe, Nakagawa, Takeuchi, Asahi.
Conducted experiments: Shirakawa, Yokoe, Nakagawa.
Performed data analysis: Shirakawa, Yokoe, Nakagawa, Moriwaki, Takeuchi, Asahi.
Wrote or contributed to the writing of the manuscript: Yokoe, Asahi.
Footnotes
- Received October 24, 2023.
- Accepted April 19, 2024.
This research was supported by the Grant-in-Aid for Scientific Research [Grant 22K07972] from the Japan Society for the Promotion of Science (to T.T.).
The authors have no conflicts of interest to declare.
↵1 M.S. and S.Y. contributed equally to this work.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- Baf A1
- bafilomycin A1
- CQ
- chloroquine
- IM
- indomethacin
- NSAID
- nonsteroidal anti-inflammatory drug
- PPI
- proton pump inhibitor
- Rapa
- rapamycin
- V0d
- ATPV0d1
- V1A
- ATP6V1A
- V1B
- ATP6V1B2
- V-ATPase
- vacuolar ATPase
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}