


Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) can cause epithelial cell damage in the stomach, intestine, and colon. NSAIDs are reported to induce autophagy and apoptosis in intestinal epithelial cells; however, their role in cell damage is poorly understood. To examine the role of autophagy in cell damage, we used autophagy-related gene Atg5-conditional knockout mice, in which the Atg5 gene is only knocked out in intestinal epithelial cells. In an indomethacin (IM)–induced gastrointestinal ulcer mouse model, intestinal epithelium damage was reduced in Atg5-conditional knockout mice compared with wild-type mice. IM-induced damage in IEC6 rat intestinal epithelial cells was reduced when Atg5 was silenced (IEC6shAtg5 cells). Western blot analyses indicated that IM-induced apoptosis decreased, and the potent, oxidative stress–related extracellular signal–regulated kinase (ERK)/nuclear factor-erythroid2-like2 (Nrf2)/heme oxygenase-1 (HO-1) signaling pathway was upregulated in IEC6shAtg5 cells. An experiment using a reactive oxygen species (ROS)-sensitive fluorescent dye in IEC6shAtg5 cells revealed that the amount of ROS at the baseline and the rate of increase after IM treatment were lower than in intact IEC6 cells. The mitochondrial membrane potential at the baseline and the reduction rate in IM-treated IEC6shAtg5 cells were lower than in intact IEC6 cells, indicating that autophagy deficiency increased ROS production caused by mitochondrial disturbance. Furthermore, MnTMPyP, a manganese–superoxide dismutase mimetic, significantly inhibited IM-induced autophagy and subsequent apoptosis as well as activation of the ERK/Nrf2/HO-1 pathway. These data suggest that autophagy deficiency and subsequent activation of the ERK/Nrf2/HO-1 pathway diminished IM-induced, apoptosis-mediated intestinal epithelial cell damage, and genetic analyses of single nucleotide polymorphisms in autophagy-related genes could predict NSAID-induced intestinal injury.
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are known to cause damage such as erosion, hemorrhage, or perforation in the gastric membrane. Studies using capsule endoscopy or balloon enteroscopy recently revealed that NSAIDs cause membrane damage not only in the stomach but also in the intestine and colon (Graham et al., 2005; Sidhu et al., 2006; Maiden et al., 2007). A previous study reported that 68% of patients experience damage in the intestinal membranes at 2 weeks after the internal use of NSAIDs combined with a proton pump inhibitor (PPI) (Maiden et al., 2007). A Japanese study used a balloon enteroscopy database to analyze the frequency of NSAID-induced intestinal ulcers, and intestinal ulceration was observed in approximately 51% of patients taking NSAIDs (Matsumoto et al., 2008).
Because the pH in the stomach is very low, antacids such as H2 receptor antagonists or PPIs are efficient treatments for NSAID-induced gastric ulcers (Yeomans et al., 1998). However, antacids may not work in the intestine, because the pH is around 8. In the intestine, NSAIDs reduce ATP production in the mitochondria of intestinal epithelial cells; therefore, membrane permeability is increased by the resulting disturbance to the maintenance system in the junction between the cells (Bjarnason et al., 1993). Enteric bacteria, bile acids, and proteases thereby cross the cell membrane, resulting in the migration and activation of neutrophils. The activated neutrophils produce cytokines or nitric oxide, which induces damage in intestinal membranes (Higuchi et al., 2009).
Autophagy plays an important role in maintaining cell homeostasis during nutrient deprivation, oxidative stress, or endoplasmic reticulum stress (Kuma et al., 2004; Komatsu et al., 2005; Ogata et al., 2006). Autophagy is induced by the proteins coded by the autophagy-related genes (Atg) (Suzuki et al., 2004; Mizushima et al., 2011). This process includes 1) the formation and elongation of the isolation membrane, 2) autophagosome formation, 3) docking and fusion with lysosomes, and 4) vesicle breakdown or degradation. The Atg12/Atg5 complex, which resides in the isolation membrane, has a pivotal role in membrane elongation and thus is essential for autophagy. However, an excessive level of autophagy induces nonapoptotic cell death (Chiou et al., 2011). Sulindac sulfide, an NSAID, causes autophagy that induces the death of rat gastric mucosal cell line rat gastric epithelial cells.
Although autophagy and apoptosis are different physiologic processes, they have a complicated relationship in terms of their interactions with each other. The activation of autophagy can either promote or inhibit apoptosis, depending on the particular stimulating factors or cell types (Besirli et al., 2011; Saiki et al., 2011). Celecoxib inhibits proliferation in the SGC-7901 human gastric cancer cell line by induction of autophagy and subsequent apoptosis (Liu et al., 2014). On the other hand, when apoptosis is activated, autophagy-related proteins such as beclin-1 or Atg5 are degraded by a series of apoptosis-related proteases and caspases, which results in the gradual cessation of autophagy (Luo and Rubinsztein, 2010). Bcl-2 functions not only as an antiapoptotic protein but also as an antiautophagy protein via its inhibition of beclin-1 (Pattingre et al., 2005).
Knockout (KO) of Atg5 in mice is lethal to mouse embryos, so tissue-specific conditional Atg5 KO mice have been used to investigate autophagy in vivo (Nakai et al., 2007; Gukovsky and Gukovskaya, 2015). Here, we established intestinal epithelial cell–specific conditional Atg5 KO mice, and we examined whether autophagy may effect intestinal epithelial cell damage by an NSAID, indomethacin (IM), in vivo. Moreover, Atg5-silenced rat intestinal epithelial cells (IEC6shAtg5 cells) were used to reveal the molecular mechanisms underlying the effect of autophagy on IM-induced cell damage.
Materials and Methods
Antibodies.
An anti–poly(ADP-ribose)polymerase 1 (PARP1) antibody was purchased from Enzo Life Sciences Inc. (Farmingdale, NY). An anti–microtubule-associated protein 1 light chain 3 (LC3) antibody was obtained from MBL (Nagoya, Japan). Anti–total and phospho-extracellular signal–regulated kinase (ERK) antibodies were obtained from Cell Signaling Technology (Danvers, MA). Anti–HO-1 and anti–nuclear factor-erythroid2-like2 (Nrf2) antibodies were from Stressgen Biotech (San Diego, CA) and Santa Cruz Biotechnology Inc. (Santa Cruz, CA), respectively.
Fluorescent Dye.
MitoTracker Red CMXRos dye was purchased from Life Technologies (Carlsbad, CA).
Other Reagents.
MnTMPyP was procured from Santa Cruz Biotechnology Inc. The Total ROS/Superoxide Detection Kit was obtained from Enzo Life Sciences Inc.
Oral Administration of IM and Extraction of Intestine in Wild-Type or Atg5-Conditional Knockout Mice.
Oral administration of IM was performed as described previously (Tanaka et al., 2002). Briefly, wild-type (WT) and Atg5-conditional knockout (CKO) mice (n = 5 to 6 for each) were given IM p.o. at a dose of 10 mg/kg and euthanized 24 hours later under deep ether anesthesia. The small intestines (stomach to ileum) were excised and used for the measurement of a lesion score or Western blot analyses.
Cell Culture.
IEC6, a rat intestinal epithelial cell line, was obtained from RIKEN BioResource Center (Saitama, Japan). This cell line was maintained in Dulbecco’s modified Eagle’s medium (Life Technologies) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin.
IEC6shAtg5 cells (shAtg5 cells), in which the Atg5 gene was suppressed by short hairpin RNA, were maintained in the same medium, except that it was supplemented with 1 μg/ml puromycin (Wako Pure Chemical Industries, Osaka, Japan).
Evaluation of Intestinal Lesion Area and Protein Expression Levels.
To evaluate lesion scores, the excised tissues were treated with 2% formalin to fix the tissue walls, the tissues were opened along the antimesenteric attachment, and the area of macroscopically visible lesions was measured under a dissecting microscope with square grids (10×), summed per small intestine, and used as a lesion score. To delineate the damage, 1% Evans blue (Sigma-Aldrich, St. Louis, MO) solution was injected i.v. in a volume of 0.5 ml/animal 0.5 hours before euthanasia.
For Western blot analyses, the excised tissues were homogenized in lysis buffer (25 mM Tris-HCl, 100 mM NaCl, 1% Triton X-100, and 0.25% deoxycholate, pH 7.4) containing protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan) using a Potter-Elvehjem homogenizer.
Western Blot Analysis.
After SDS-PAGE, the gel was put into a semidry blotting system (Bio-Rad, Hercules, CA), and the proteins were transferred onto a polyvinylidene membrane (Pall, Port Washington, NY). The membrane was blocked with 5% skimmed milk in PBS containing 0.1% Tween 20 (PBST, blocking buffer), incubated in blocking buffer with the primary antibody of interest, washed three times with PBST, and then incubated with a secondary antibody conjugated with horseradish peroxidase. The membrane was visualized with Immobilon Western (Millipore, Hayward, CA), and the image was captured with Chemidoc (Bio-Rad).
Nrf2 Promoter Assay.
IEC6 cells were transfected with pGL4.37 (ARE/Hygro) and pGL4.74 (hRluc/TK) (Promega, Madison, WI) using Lipofectamine 2000 (Life Technologies).
After 48 hours, the cells were incubated with IM with or without MnTMPyP for 5 hours. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Chemiluminescence was detected using a GloMax Multi-Detection System (Promega).
Evaluation of Cell Viability.
Cell viability was measured colorimetrically using Cell Counting Kit-8 (Wako Pure Chemical Industries) following the manufacturer’s instructions. In brief, cells of interest were plated onto a 96-well plate and incubated overnight to allow the cells to attach. The cells were then incubated with or without inhibitors for various time periods (24–72 hours). To measure the daily rate of cell proliferation, the cells were incubated with a water soluble tetrazolium salts 8 reagent for 1 hour and its absorbance was measured at a wavelength of 450 nm using a microplate reader (model 680; Bio-Rad).
Evaluation of Oxidative Stress.
The Total ROS/Superoxide Detection Kit (Enzo Life Sciences Inc.) was used to evaluate whether oxidative stress increased in intact and shAtg5 cells before and after IM treatment. Briefly, the cells were plated onto a 96-well plate compatible with the fluorescence assay. Twenty-four hours later, the cells were incubated with 200 μM IM for 1 to 2 hours, which was preincubated for 30 minutes with or without 5 mM N-acetyl cysteine. Fluorescence was directly measured with a GloMax Multi-Detection System equipped with an appropriate optical kit [blue for reactive oxygen species (ROS), green for superoxide; Promega].
Measurement of Mitochondrial Membrane Potential.
Mitochondrial function was estimated using MitoTracker Red CMXRos (Life Technologies, Carlsbad, CA), a membrane potential–dependent mitochondria probe. The cells were preincubated with IM for 1 hour, after which MitoTracker Red CMXRos was added and further incubation for 1 hour was performed. Fluorescence was measured using a GloMax 96-well microplate luminometer (Promega).
Results
Evaluation of Intestinal Epithelial Injury in WT and Atg5-CKO Mice after IM Administration.
In a previous study, Narabayashi et al. (2015) reported that IM induced accumulation of cytoplasmic lipid droplets (LDs), and lipophagy (autophagic degradation of portions of LDs or whole LDs either alone or mixed with other cell contents) was promoted to remove the toxic LDs in IEC6 cells. To examine how autophagy (including lipophagy) is involved in IM-induced intestinal epithelial injury, we used Atg5-CKO mice in which autophagy did not occur only in intestinal epithelial cells. At 24 hours after the oral administration of IM to either WT or Atg5-CKO mice, the mice were euthanized and the extent of intestinal epithelial injury was evaluated. As a result, the intestinal epithelial injury was significantly decreased in Atg5-CKO mice compared with WT mice, according to macroscopic findings and ulcer index values (Fig. 1, A and B).

Role of Atg5 in injury of the small intestine after IM treatment in vitro and in vivo. (A and B) Gross appearance of intestinal lesions (A) and ulcer index values (B) after IM administration in WT and conditional Atg5KO mice. Arrows indicate epithelial erosion caused by IM-induced intestinal injuries. Ulcer index values are expressed as means ± S.E. (n = 5). (C) Cytotoxicity was estimated after treatment with various concentrations of IM in IEC6 and IEC6shAtg5 cells. Open bars, IEC6 cells; closed bars, IEC6shAtg5 cells. Statistical analysis was performed using Tukey’s method. *P < 0.05.
Evaluation of Cell Viability in IEC6 or IEC6shAtg5 Cells after IM Treatment.
To further understand the role of autophagy in intestinal epithelial injury, we established IEC6shAtg5 cells (Fig. 2A) and evaluated them in vitro. Cell damage after IM treatment in IEC6shAtg5 cells was significantly increased compared with IEC6 cells, which is consistent with the data in vivo (Fig. 1C).

Induction of autophagy and subsequent apoptosis by IM treatment in IEC6 cells, not in IEC6shAtg5 cells. The cells were treated with 200 μM IM for various durations (0, 1, 2, 4, 8, and 12 hours). (A) LC3 expression levels at each time point in IEC6 cells and IEC6shAtg5 cells were determined using Western blot analyses. The arrow and arrowhead indicate LC3-I and LC3-II, respectively. (B) The cleavage of PARP1 in IM-treated IEC6 cells and IEC6shAtg5 cells was determined. The arrow and arrowhead indicate intact and cleaved PARP1, respectively.
Evaluation of Autophagy and Apoptosis in IEC6 or IEC6shAtg5 Cells after IM Treatment.
We retested whether autophagy might occur after IM treatment by carrying out Western blot analyses using an anti-LC3 antibody. The results confirmed that autophagy increased in intact cells after IM treatment, and the increase was totally reversed in IEC6shAtg5 cells (Fig. 2A).
Next, we evaluated whether apoptosis is related to the decline of cell viability, and the cleavage of PARP1, which is the substrate of caspase-3, was evaluated by Western blot analyses. The data showed that the cleavage of PARP1 was significantly suppressed in IEC6shAtg5 cells, indicating that the augmented autophagy after IM treatment might induce apoptosis (Fig. 2B). The subsequent apoptosis is thought to deteriorate the intestinal epithelial cell damage.
Total ROS and Superoxide Production in IEC6 or IEC6shAtg5 Cells.
Autophagy plays an important role in maintaining cell homeostasis during nutrient deprivation, oxidative stress, or endoplasmic reticulum stress. To examine the mechanism by which autophagy was induced after IM treatment, we focused on oxidative stress. Total ROS and superoxide generation before and after IM treatment was estimated using a Total ROS/Superoxide Detection Kit (Enzo Life Sciences Inc.). First, we examined the level of total ROS and superoxide generation in IEC6shAtg5 cells at the baseline. As a result, both total ROS and superoxide generation were significantly increased in IEC6shAtg5 cells compared with intact cells. Second, we examined whether ROS generation was increased after IM treatment in both cells. The data showed that ROS generation was significantly increased in both cells after IM treatment (Fig. 3, A and B). The elevation of ROS generation was totally reversed by a radical scavenger, N-acetylcysteine. Interestingly, changing rates of relative ROS or superoxide production after IM treatment were significantly different from each other. The changing rate in IEC6shAtg5 cells was significantly lower than in IEC6 cells, indicating that IEC6shAtg5 cells were more tolerant of ROS than IEC6 cells (Fig. 3, C and D).

ROS generation before and after IM treatment in IEC6 and IEC6shAtg5 cells. (A and B) Both total ROS and superoxide were estimated before and after treatment with 200 μM IM in IEC6 and IEC6shAtg5 cells using a Total ROS/Superoxide Detection Kit (Enzo Life Sciences Inc.). The effect of N-acetyl cysteine (NAC) was evaluated to confirm that the assay was suitable. (C and D) The changing rates of relative ROS or superoxide production after various concentrations (0, 50, 100, or 200 μM) of IM treatment were estimated in both cells and compared with each other. *P < 0.05; **P < 0.01.
Mitochondrial Membrane Potential before and after IM Treatment in IEC6 or IEC6shAtg5 Cells.
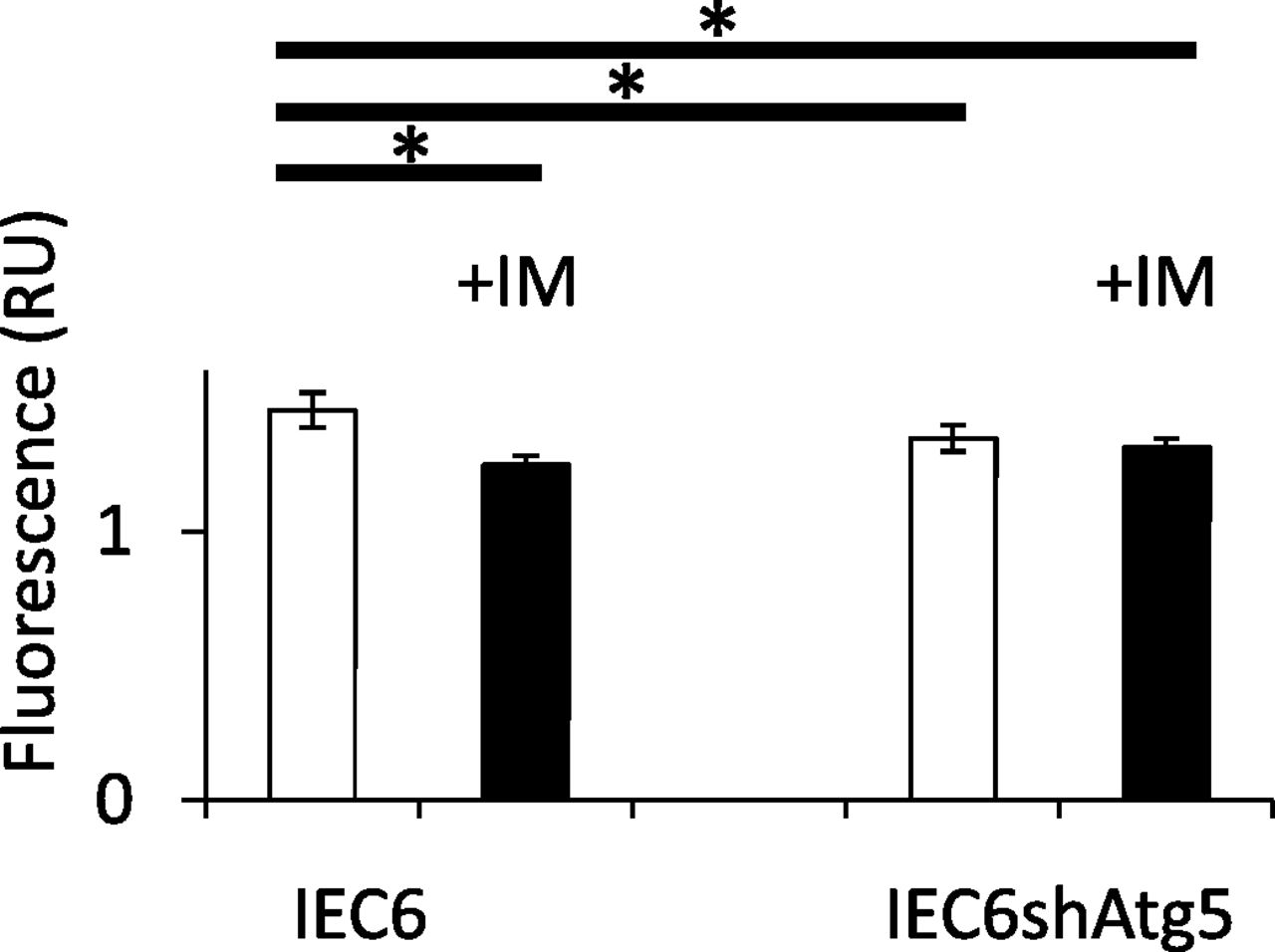
To reveal the mechanism by which ROS generation was significantly increased in IEC6shAtg5 cells compared with IEC6 cells at the baseline, and both were significantly increased in both cells after IM treatment, we measured mitochondrial membrane potentials in IEC6 cells and IEC6shAtg5 cells, which is the indicator of mitochondrial activity. If this potential is reduced, it is thought that the percentage of damaged mitochondria might be increased. As a result, the potential was significantly lower in IEC6shAtg5 cells compared with IEC6 cells at the basal line. IM treatment lowered the potentials in IEC6 cells, but not in IEC6shAtg5 cells (Fig. 4).
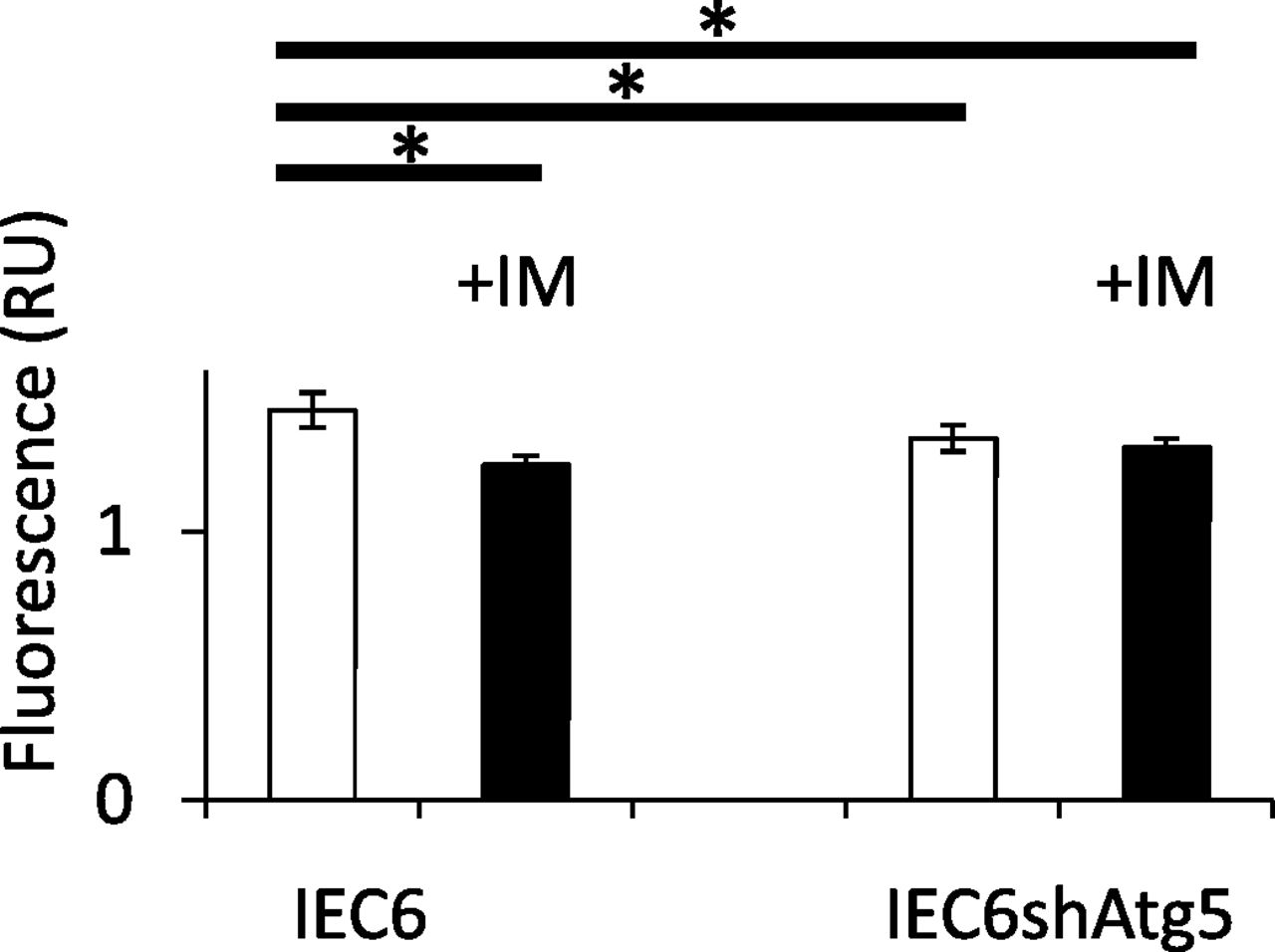
Quantitative evaluation of functionally active mitochondria before and after IM treatment in IEC6 cells and IEC6shAtg5 cells. Mitochondria staining with MitoTracker Red CMXRos, a membrane potential dependent mitochondria probe. Cells were preincubated with or without 200 μM IM for 1 h; then, MitoTracker Red CMXRos was added, and further incubated for 1 h. The fluorescence was measured using GloMax (Promega). *P < 0.05; Tukey’s test.
Activity of ERK/Nrf2/HO-1 Pathway in IEC6 or IEC6shAtg5 Cells after IM Treatment.
Autophagy is a cytoprotective pathway for degradation of cellular components within autophagic vacuoles caused by various types of cellular stress. It was previously reported that heme oxygenase-1 (HO-1) prevented intestinal epithelial cells from IM-induced ulceration in rats. To reveal whether HO-1 is involved in tolerance against ROS in IEC6shAtg5 cells, we examined whether the ERK/Nrf2/HO-1 pathway might be activated in these cells. As shown in Fig. 5, ERK phosphorylation and Nrf2/HO-1 expression were upregulated in IEC6shAtg5 cells compared with IEC6 cells. Thus, activation of the ERK/Nrf2/HO-1 pathway could be involved in the acquisition of tolerance against ROS, resulting in the reduction of cell damage in IEC6shAtg5 cells.

Changes in protein expression levels after IM treatment in IEC6 and IEC6shAtg5 cells. The cells were treated with 200 μM IM for the indicated times. (A and B) Protein expression levels of total and phosphorylated ERK (A) and Nrf2 and HO-1 (B) at each time point after IM treatment in IEC6 and IEC6shAtg5 cells were determined using Western blot analyses.
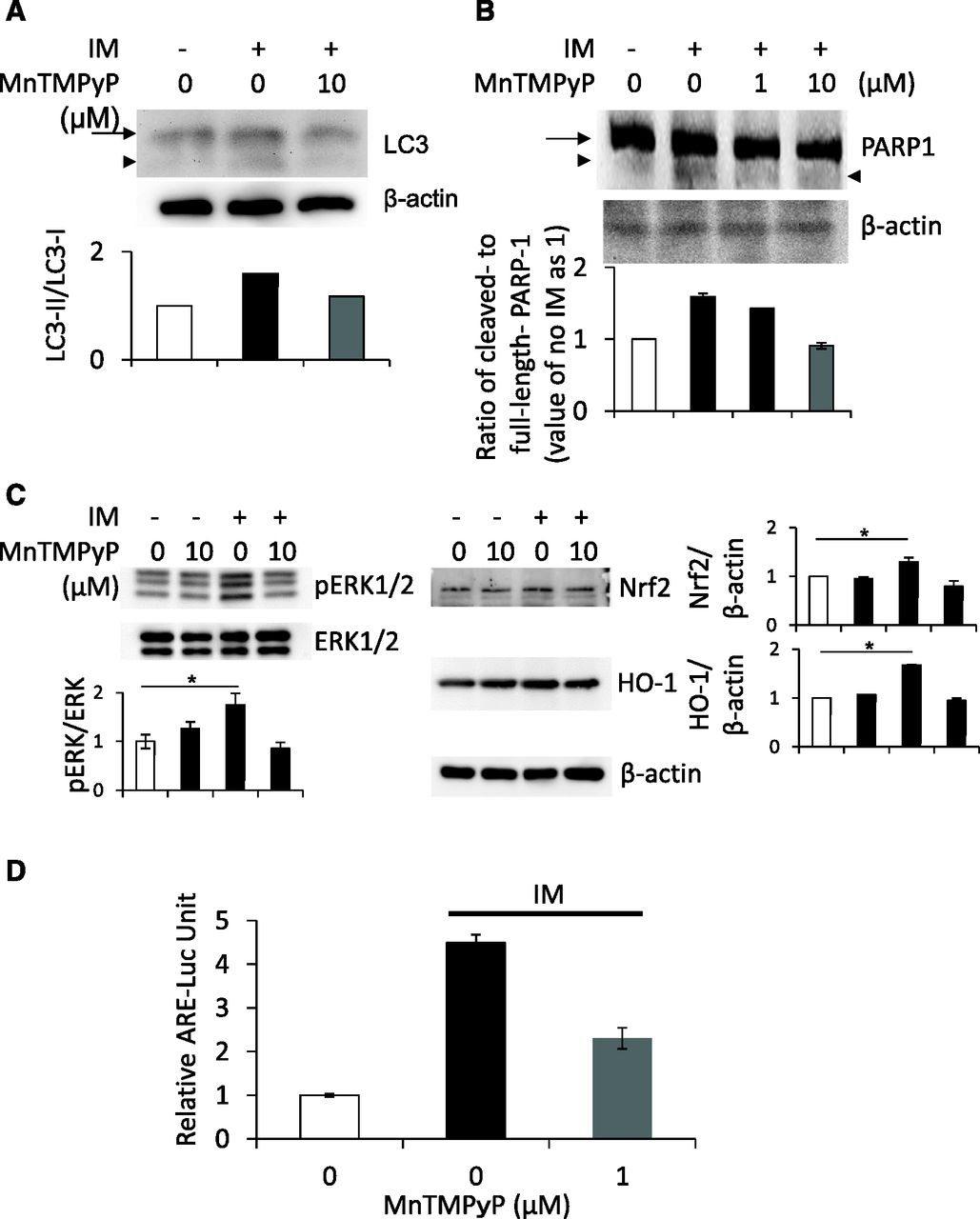
Effect of MnTMPyP on Upregulation of Autophagy, Apoptosis, and the ERK/Nrf2/HO-1 Pathway after IM Treatment in IEC6 Cells.
To prove whether ROS regulates IM-induced autophagy and subsequent apoptosis, we used a manganese–superoxide dismutase mimetic, MnTMPyP, to scavenge ROS in a series of experiments and examined the effect of MnTMPyP on the cleavage of LC3 or PARP1 after IM treatment. As a result, upregulation of LC3-II or cleaved PARP1 was totally reversed by MnTMPyP (Fig. 6, A and B). Furthermore, we examined the effect of MnTMPyP on IM-induced upregulation of the ERK/Nrf2/HO-1 pathway. Our results showed that upregulation of the pathway was also totally reversed by MnTMPyP (Fig. 6C). We also performed Nrf2 promoter analysis to examine the effect of IM with or without MnTMPyP on transcriptional activity; our results showed that Nrf2 activity was also upregulated after IM treatment, which was reversed by MnTMPyP (Fig. 6D). These results indicate that ROS promoted IM-induced autophagy and subsequent apoptosis, resulting in intestinal epithelial cell damage, and this damage was strongly blocked by upregulation of the ERK/Nrf2/HO-1 pathway.

Involvement of ROS in IM-induced autophagy, subsequent apoptosis, and the ERK/Nrf2/HO-1 pathway. After the preincubation with or without 10 μM MnTMPyP for 30 minutes, IEC6 cells were treated with 200 μM IM for another 2 hours. (A and B) Cleavage of LC3 (A) or PARP1 (B) was determined using Western blot analyses. Arrows indicate LC3-I (A) or intact PARP1 (B), and arrowheads indicates LC3-II (A) or cleaved PARP1 (B). (C) Using the same samples, the levels of ERK phosphorylation, Nrf2 stabilization, and HO-1 induction were determined using Western blot analyses. (D) After preincubation with or without 1 μM MnTMPyP for 30 minutes, IEC6 cells were treated with 200 μM IM for another 2 hours. Nrf2 promoter activity was evaluated by a luciferase assay. *P < 0.01.
Effect of Autophagy Deficiency on the ERK/Nrf2/HO-1 Pathway in Intestinal Epithelial Cells from WT and Atg5-CKO Mice.
The ERK/Nrf2/HO-1 pathway was activated to induce the acquisition of tolerance against ROS, resulting in the reduction of cell damage in IEC6shAtg5 cells (Fig. 5). To confirm whether the pathway may be activated in intestinal epithelial cells from Atg5-CKO mice as well as in IEC6shAtg5 cells, epithelial cells scraped from the intestines of WT and Atg5-CKO mice with or without IM administration were used for Western blot analyses. As shown in Fig. 7, the ERK/Nrf2/HO-1 pathway in the cells from Atg5-CKO mice was activated compared with those from WT mice without IM administration. Furthermore, the activity was significantly increased after IM treatment only in the cells derived from Atg5-CKO mice. These data suggest that the ERK/Nrf2/HO-1 pathway is activated by autophagy deficiency in vivo.

Effects of autophagy deficiency and IM administration on the ERK/Nrf2/HO-1 pathway in small intestinal mucosa. WT and Atg5 KO mice were treated with or without IM administration orally. After 24 hours, small intestines (stomach to ileum) of the euthanized animals were excised, and the mucosa was surgically scraped. The scraped tissues were collected and analyzed with Western blot analyses. *P < 0.05.
Discussion
NSAID-induced gastric membrane damage has been successfully treated with antacids such as H2 receptor antagonists or PPIs (Yeomans et al., 1998). Given that the pH of intestinal epithelium is neutral (not acidic like the stomach), it is not likely that NSAIDs will be effective, and some reports indicate that they do not ameliorate damage to the intestinal membrane (Daniell, 2012). Thus, to treat NSAID-induced intestinal membrane damage, it is very important to elucidate the mechanism.
Since autophagy is reported to take place after IM treatment as well as apoptosis in IEC6 cells (Narabayashi et al., 2015), we focused on autophagy and examined the role of autophagy in intestinal epithelial damage. Here, we showed that prevention of autophagy reduced apoptosis-mediated, IM-induced intestinal epithelial cell damage. The amelioration was attributable to activation of the ERK/Nrf2/HO-1 pathway.
In some contexts, autophagy is regarded as an adaptive response to stress, which promotes survival; in other cases, it appears to promote cell death and morbidity (Nakai et al., 2007; Kundu and Thompson, 2008). In our in vitro and in vivo experiments, prevention of autophagy reduced IM-induced intestinal epithelial cell damage, meaning that autophagy promoted cell death and morbidity. To examine the mechanism, we studied the changes in intracellular signaling pathways before and after IM treatment in IEC6 and IEC6shAtg5 cells. We previously reported that lansoprazole, a PPI, prevented IM-induced intestinal epithelial damage through upregulation of HO-1 (Yoda et al., 2010). Therefore, we examined whether the potent, oxidative stress–related ERK/Nrf2/HO-1 signaling pathway was upregulated. We found that the pathway was more activated at the baseline, and the rate of increase after IM treatment was higher in IEC6shAtg5 cells than in IEC6 cells. This indicates that autophagy deficiency makes the cells more resistant to oxidative stress through activation of the ERK/Nrf2/HO-1 pathway.
The ERK/Nrf2/HO-1 pathway is upregulated by ROS (Papaiahgari et al., 2006; Xu et al., 2013). Since the ERK/Nrf2/HO-1 pathway was activated before and after IM treatment in IEC6shAtg5 cells, it is possible that IEC6shAtg5 cells should produce more ROS than IEC6 cells. As expected, ROS production was increased in IEC6shAtg5 cells compared with IEC6 cells. It was revealed that the ROS increase activated ERK, followed by Nrf2 and HO-1. We confirmed this by the fact that activation of the pathway was inhibited by the ROS scavenger MnTMPyP. Together, upregulated ROS production might activate the ERK/Nrf2/HO-1 pathway, resulting in tolerance toward IM-induced oxidative stress. The effects of autophagy deficiency with or without IM treatment on IEC6 cells (in vitro) or mouse intestinal epithelial cells (in vivo) are summarized in Table 1.
Effect of autophagy deficiency with or without IM treatment on IEC6 cells or mouse intestinal epithelial cells
Here, we show that Nrf2 is activated through upregulation of ERK phosphorylation before and after IM treatment of IEC6shAtg5 cells. Since autophagy deficiency is reported to activate Nrf2 through direct interaction between Keap1 and p62 (Lau et al., 2010), Nrf2 might be activated in two ways: 1) upregulation of ERK phosphorylation and 2) inhibition of Keap1-mediated degradation. Nrf2 activation functions as a potent cell survival factor by the induction of not only antioxidative enzymes such as HO-1 but also detoxifying enzymes such as glutathione-S-transferase mu1 (Chowdhry et al., 2013). Therefore, Nrf2 activation might be the most important factor in terms of the protective effect after IM treatment in IEC6shAtg5 cells.
Autophagy normally recycles macromolecular aggregates produced through oxidative stress– mediated pathways and may also reduce mitochondrial production of ROS through recycling of old and damaged mitochondria (Hensley and Harris-White, 2015). Thus, autophagy is thought to be an essential cellular antioxidant pathway (Giordano et al., 2014). In our experiment, IEC6shAtg5 cells in which autophagy did not occur produced more ROS than IEC6 cells. The increase of ROS production in autophagy-deficient cells might be because autophagy mainly reduces oxidative stress by degrading dysfunctional mitochondria, which releases ROS.
We found that IM-induced intestinal epithelial cell damage might be mediated by ROS production, followed by a decrease in mitochondrial membrane potential (Chung et al., 2003). Decreased mitochondrial membrane potential is thought to induce ROS production (Hosseini et al., 2014). Since the increase in ROS (which results from decreased mitochondrial membrane potential) induced disturbance of mitochondrial membranes, it is likely that IM-induced ROS production took the form of a vicious cycle. This cycle might result in the deterioration of oxidative stress, leading to apoptosis.
Here, we demonstrated that autophagy deficiency diminishes IM-induced intestinal epithelial cell damage through activation of the ERK/Nrf2/HO-1 pathway. This does not simply mean that autophagy deteriorates IM-induced intestinal epithelial cell damage. Since autophagy is primarily a protective process for the cell, autophagy deficiency may not be protective in a different situation. If oxidative stress is increased and ROS production is harmful to the cells, autophagy deficiency will not be protective. Although oxidative stress was increased in IEC6shAtg5 cells at the baseline, the amount of ROS might not be harmful to the cells; instead, the ROS level could make the cells resistant toward oxidative stress. Furthermore, the data may also indicate that individuals with an autophagy-related disorder, such as Crohn’s disease (Scharl et al., 2012; Spalinger et al., 2014), or a functional variant of the autophagy-related gene ATG (Latiano et al., 2008; Martin et al., 2012; Kimura et al., 2014) are resistant to NSAID-induced intestinal epithelial cell damage. Genetic analyses of single nucleotide polymorphisms in ATG genes could be used to predict NSAID-induced intestinal injury.
In summary, we revealed that IM-induced intestinal epithelial cells were damaged by oxidative stress due to mitochondrial dysfunction. The cell damage was partly caused by apoptosis. In a condition in which autophagy does not function, cell damage was reduced through activation of the ERK/Nrf2/HO-1 pathway. The possible mechanism by which autophagy deficiency diminishes IM-induced intestinal epithelial cell damage is depicted in Fig. 8. Oxidative stress might be one of the most important factors inducing IM-induced intestinal epithelial cell damage. Activation of antioxidative signaling systems such as the ERK/Nrf2/HO-1 pathway could be very effective in protecting the small intestine from IM-induced injury.

Possible mechanism for the amelioration of IM-induced intestinal epithelial cell injury by autophagy deficiency. (A) In normal cells, oxidative stress is suppressed by autophagy; therefore, the ERK/Nrf2/HO-1 pathway is not activated under basal conditions. After IM treatment, the cells cannot fully prevent apoptosis-induced cell damage due to the increase of oxidative stress. (B) In autophagy-deficient cells, the ERK/Nrf2/HO-1 pathway is activated by oxidative stress even under basal conditions. The cells can minimize apoptosis-induced cell damage after IM treatment, because it is more resistant to oxidative stress due to the upregulated ERK/Nrf2/HO-1 pathway.
Authorship Contributions
Participated in research design: Nakagawa, Higuchi, Asahi.
Conducted experiments: Harada, Nakagawa, Yokoe.
Contributed new reagents or analytic tools: Nakagawa, Edogawa, Takeuchi, Inoue.
Performed data analysis: Harada, Nakagawa, Higuchi, Asahi.
Wrote or contributed to the writing of the manuscript: Asahi.
Footnotes
- Received June 2, 2015.
- Accepted September 23, 2015.
This research was supported in part by the Japan Society for the Promotion of Science [Grant-in-Aid for Scientific Research (C) 25460397 (to M.A.)] and the Ministry of Education, Science, Culture, Sports and Technology of Japan.
Abbreviations
- CKO
- conditional knockout
- ERK
- extracellular signal–regulated kinase
- HO-1
- heme oxygenase-1
- IM
- indomethacin
- KO
- knockout
- LC3
- microtubule-associated protein 1 light chain 3
- LD
- lipid droplet
- Nrf2
- nuclear factor-erythroid2-like2
- NSAID
- nonsteroidal anti-inflammatory drug
- PARP1
- poly(ADP-ribose)polymerase 1
- PPI
- proton pump inhibitor
- ROS
- reactive oxygen species
- WT
- wild type
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}