


Abstract
Allopregnanolone (ALLO) is a neurosteroid that modulates synaptic and extrasynaptic GABAA receptors. We hypothesize that ALLO may be useful as first-line treatment of status epilepticus (SE). Our objectives were to (1) characterize ALLO pharmacokinetics-pharmacodynamics PK-PD after intravenous (IV) and intramuscular (IM) administration and (2) compare IV and IM ALLO safety and tolerability. Three healthy dogs and two with a history of epilepsy were used. Single ALLO IV doses ranging from 1–6 mg/kg were infused over 5 minutes or injected IM. Blood samples, vital signs, and sedation assessment were collected up to 8 hours postdose. Intracranial EEG (iEEG) was continuously recorded in one dog. IV ALLO exhibited dose-proportional increases in exposure, which were associated with an increase in absolute power spectral density in all iEEG frequency bands. This relationship was best described by an indirect link PK-PD model where concentration-response was described by a sigmoidal maximum response (Emax) equation. Adverse events included site injection pain with higher IM volumes and ataxia and sedation associated with higher doses. IM administration exhibited incomplete absorption and volume-dependent bioavailability. Robust iEEG changes after IM administration were not observed. Based on PK-PD simulations, a 2 mg/kg dose infused over 5 minutes is predicted to achieve plasma concentrations above the EC50, but below those associated with heavy sedation. This study demonstrates that ALLO is safe and well tolerated when administered at 1–4 mg/kg IV and up to 2 mg/kg IM. The rapid onset of effect after IV infusion suggests that ALLO may be useful in the early treatment of SE.
SIGNIFICANCE STATEMENT The characterization of the pharmacokinetics and pharmacodynamics of allopregnanolone is essential in order to design clinical studies evaluating its effectiveness as an early treatment for status epilepticus in dogs and people. This study has proposed a target dose/therapeutic range for a clinical trial in canine status epilepticus.
Introduction
Human status epilepticus is a life-threatening neurologic emergency defined as a prolonged seizure or recurrent seizures without complete recovery of consciousness (Trinka et al., 2015). It is the result of either a failure of the mechanisms responsible for seizure termination or an initiation of mechanisms that lead to abnormally prolonged seizures (Trinka et al., 2015). A reported 152,000 cases with 42,000 deaths (28%) occur each year in the United States (DeLorenzo et al., 1995). Companion canines have naturally occurring epilepsy at a comparable prevalence to that in people (1%–6%) and are the only other species with epilepsy at this rate (Potschka et al., 2013). Prolonged or recurrent seizures also occur in dogs, a condition known as canine status epilepticus (CSE). In dogs with epilepsy, the prevalence of SE has been reported as high as 59% and is associated with a decreased mean life span compared with dogs with epilepsy without CSE (Bateman and Parent 1999; Saito et al., 2001). Between 19% and 25% of dogs brought to a veterinary referral center for treatment of CSE or recurrent seizures or have had an episode of CSE died or were euthanized for reasons directly related to their seizure disorder (Bateman and Parent 1999; Saito et al., 2001).
Current guidelines for initial SE treatment in humans and dogs call for administration of an adequate dose of a benzodiazepine (BZD) (Alldredge et al., 2001; Glauser et al., 2016; Blades Golubovic and Rossmeisl 2017). Nevertheless, one-third to one-half of human patients do not respond (Treiman et al., 1998; Silbergleit et al., 2012). BZD-refractory SE is known as established status epilepticus (Falco-Walter and Bleck 2016). Failure of BZDs in SE may be due to several factors, including inadequate dosing, delayed initiation of treatment, or seizure etiology. Prolonged seizure duration initiates cellular changes in the affected neurons, including endocytosis of synaptic BZD-sensitive GABAA receptors (containing β2/3 and/or γ2 subunits); increased expression of BZD-insensitive GABAA receptors (containing α4 subunits, commonly in extrasynaptic regions) at synapses; and increased recruitment of AMPA receptors to synapses (Kapur 2000; Naylor et al., 2005; Chen and Wasterlain 2006). These changes, taken together, promote continuation of seizures. The failure of the first treatment carries with it the increased risk of morbidity and mortality including cardiorespiratory compromise and neuronal injury. Hence, it is critical to treat SE immediately and with the most effective available agent.
Allopregnanolone (ALLO) is a neurosteroid that was FDA-approved for the treatment of severe postpartum depression (ZULRESSO, 2019). It has been shown to be highly effective in terminating SE in rodent models, including pilocarpine and perforant path stimulation rodent models (Frye 1995; Kokate et al., 1996). Moreover, ALLO terminates BZD-refractory SE in diverse models, including the kainate model (Rogawski et al., 2013) and a model of SE induced by the nerve agent tetramethylenedisulfatetramine (Zolkowska et al., 2018). In these studies, ALLO administered 40 minutes after seizure onset, a time when the seizures are insensitive to BZD, effectively terminated behavioral and electrographic seizures. The basis through which ALLO is able to terminate BZD-refractory SE is not completely understood but has been hypothesized to relate to the ability of ALLO to act as a positive allosteric modulator of both synaptic and extrasynaptic GABAA receptors (Rogawski et al., 2013). BZD acts exclusively on synaptic GABAA receptors, which as noted above are internalized as SE progresses and become progressively unavailable (Rogawski et al., 2013). It is noteworthy that ALLO also acts on BZD-insensitive GABAA receptors that increase in abundance at synapses as SE continues (Kapur 2000). The actions of ALLO on both synaptic BZD-insensitive and extrasynaptic GABAA receptors provide a novel mechanism of action to that of BZDs (Belelli et al., 2002; Carver and Reddy 2013).
In light of the suboptimal response rate of BZDs, we hypothesize that ALLO possesses the requisite pharmacologic, physicochemical, and pharmacokinetic (PK) properties to serve as an early treatment of SE. One approach to testing this hypothesis is the use of dogs with naturally occurring epilepsy, which is similar to human epilepsy in electroencephalographic presentation and response to therapy (Berendt et al., 1999; Patterson et al., 2015). Our approach is to characterize the relationship between exposure and response in dogs as a step toward designing clinical trials in both dogs and humans. In this study, we characterized the pharmacokinetics, pharmacodynamics, and safety/tolerability of intravenous (IV) and intramuscular (IM) ALLO in dogs to provide dosing data for a clinical study of CSE, which, in turn, would provide a proof of concept to conduct studies in human SE.
Materials and Methods
Study Animals and Safety Monitoring
Five dogs with (n = 2) and without (n = 3) a history of seizures were used (Supplemental Table 1). One of the dogs with a history of seizures had recurrent seizures despite being on phenobarbital (PB) maintenance regimen. Approval was obtained through the Institutional Animal Care and Use Committee of the University of Minnesota prior to the initiation of the study. The dogs were housed at the University of Minnesota College of Veterinary Medicine. The dog on PB was previously implanted with a device that wirelessly transmits continuous intracranial electroencephalograph (iEEG) recordings (Kremen et al., 2018). On study days, each dog participating in the study was removed from its kennel to have a central-line sampling catheter implanted an hour prior to study start. On days where IV ALLO was administered, a peripheral-line catheter was also implanted for the ALLO administration to be in separate line and limb from the sampling line. Drug administration took place in a procedure room away from the dog’s kennel. The dogs were fasted prior to and fed no sooner than 2 hours after drug administration.
Study Drug
At the time the study was conducted, there was no commercial formulation of allopregnanolone. Therefore, allopregnanolone was custom synthesized with a purity of 99.9%, using a high-performance liquid chromatography assay. A concentrated solution consisting of 6% allopregnanolone and 24% sulfobutyl ether β-cyclodextrin (Dexolve) dissolved in a 0.9% sodium chloride solution (normal saline) was prepared and target concentrations were confirmed by the Rogawski laboratory at the University of California, Davis, as previously reported (Zolkowska et al., 2018). As an additional step for quality control, after the completion of each experiment, a sample of the diluted allopregnanolone solution (see below) was sent to the Rogawski laboratory for confirmation of concentration. The concentrate was shipped frozen to the University of Minnesota. The frozen ALLO was thawed overnight prior to study date and diluted with normal saline (1 part ALLO: 0 to 3 parts normal saline) prior to IV and IM administration for a final allopregnanolone concentration of 1.5–6 mg/mL. IM studies were also conducted with concentrates consisting of 11% and 14% allopregnanolone and 40% Dexolve dissolved in normal saline and not diluted prior to administration.
Starting Dose Rationale
Basal levels of ALLO in healthy, pregnant women have been reported to reach as high as 50 ng/mL (Luisi et al., 2000). Further, IM doses of 3–6 mg/kg were effective in terminating status epilepticus in mice and rats (Zolkowska et al., 2018; Dhir et al., 2020a). Using preclinical and clinical data reported in the literature (Timby et al., 2006; Irwin et al., 2015) and simple allometry, scaling coefficients for clearance and volume were calculated by taking the natural log of weight of three different species (mouse, rabbit, and human) and plotting it against the natural log of the pharmacokinetic parameter to calculate a slope from the trendline. We predicted a 5-minute infusion of 1 mg/kg was estimated to maintain plasma concentrations in the 50 ng/mL range for 30 minutes. Hence, we used a 1 mg/kg dose to initiate our studies.
Study Design
Three healthy dogs and one dog with a history of seizures were given single IV doses of ALLO ranging from 1–4 mg/kg by a 5-minute infusion via a catheterized peripheral vein (Table 1). One to two dogs were studied at each dose level. Whole blood samples (∼2 to 5 mL) were collected via a catheterized central vein at predose, at 3, 5, 15, 30, and 45 minutes postinfusion; and 1, 2, 4, and 6 hours postinfusion. A washout period of at least 1 week was used.
Number of animals per study dose and route
After the completion of the IV dose escalation study, PK studies were undertaken after IM administration in two healthy dogs and two dogs with a history of seizures (one on chronic PB) in a dose-escalation manner. However, in addition to varying the doses, we also administered different injection volumes to test the hypothesis that a high formulation concentration (consequently, a lower injection volume) would increase bioavailability. After the first 2 mg/kg IM study, the bioavailability was approximately 50%. To attempt to attain concentrations comparable to the 3 mg/kg IV dose, the next two dogs were dosed at 6 mg/kg. In total, three doses (1, 2, and 6 mg/kg) across four ALLO concentrations (3, 6, 11, and 14 mg/mL) were evaluated. Whole blood samples (∼2 to 5 mL) were collected via a catheterized central vein at predose; at 1, 3, 5, 15, 30, and 45 minutes postinjection; and at 1, 2, 4, and 6hours postinjection. A washout period of at least 1 week was used.
Intracranial EEG data were collected in the dog with electrodes implanted intracranially. Data were recorded for two IV doses (1–2 mg/kg) and one IM dose (1 mg/kg). Each collection period started the day before and finished the day immediately after study drug administration. Continuous iEEG data were collected with sampling rate of 250 Hz from four iEEG channels.
Allopregnanolone Assay
Whole blood was collected in K3EDTA-containing purple-top vacutainer tubes and centrifuged for plasma separation. The red blood cells and plasma were immediately frozen (−80°C) until analysis. An ultra high-performance liquid chromatography-mass spectrometry method developed and validated at the UC Davis Bioanalysis and Pharmacokinetics Core Facility was used to measure total plasma ALLO concentrations (Zolkowska et. al, 2018). Canine plasma was obtained commercially from BioreclamationIVT LLC and used as blank plasma standard and for dilution.
The range of linear quantification was from 15 ng/mL to 1500 ng/mL. Lower limit of quantitation was set at 15 ng/mL. The quality control of the calibration curve was performed by passing the criteria set by the Bioanalytical Method Validation FDA Guidance for Industry (U.S. Department of Health and Human Services et al., 2018, https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf). Dilution integrity for up to fivefold was confirmed. No significant interfering peaks with signal above lower limit of quantitation were observed in blank or experimental predose plasma samples.
The correlation coefficient in each run was at least 0.99 for allopregnanolone, using a least-squares linear regression with a 1/x weighting. The intrabatch precision across all concentrations and analytical runs ranged from 2.96% to 14.95%, and the intrabatch accuracy ranged from 17.33% to 3.54%. The interbatch precision across all concentrations and analytical runs ranged from 6.05% to 10.57%, and the interbatch accuracy ranged from −13.52% to −0.18%.
PK Analysis and Simulations
ALLO concentration–time data were analyzed using noncompartmental and compartmental analyses, including population approaches (Phoenix 64, Build 8.0.0.3176, Certara L.P., Princeton, NJ). Concentrations below the lower limit of quantitation were treated as missing values. Pharmacokinetic parameters determined included Cmax, time at which maximum concentration is achieved, and terminal half-life. The area under the time–concentration curve (AUC)∞ was calculated using eq. 1, where Cp is the plasma ALLO concentration, tlast is the time at which the last plasma sample was drawn, Cplast is the last measured plasma ALLO concentration, and kel is the terminal rate constant) and a linear-log trapezoidal method. The points used to calculate the terminal phase half-life were determined using the Phoenix algorithm, which determines the best fit line via minimizing r2. Percent extrapolated were less than 30% in all calculations except in one dog (Dog 3) after IM administration. A test for dose-proportionality was conducted using a one-way ANOVA test across dose-normalized AUC∞ and Cmax with the assumption of homoscedasticity.
IM bioavailability (F%) was calculated using eq. 2, where the individual dog’s AUCinf were used for each route. Clearance (CL) and volume of distribution (V) were calculated using eqs. 3 and 4, respectively. All plots were created using the GraphPad Prism 7 (Version 7.0a, GraphPad Software, Inc., La Jolla, CA).
Population compartmental modeling was performed to characterize the IV concentration-time data (Phoenix Non-Linear Mixed Effects 8.0). First order conditional estimation extended least squares method was used throughout the model building process. A proportional error model for between-subject variability was used. Additive, multiplicative, and combined error models for residual unexplained variability were evaluated. The best fit model was determined using visual inspection, goodness of fit plots (e.g., conditional weighted residual plots and observed versus predicted), objective function value, Akaike’s Information Criterion, and precision of model parameters. Simulations were conducted using parameter estimates generated from the population PK model in Phoenix NLME.
In addition to calculating bioavailability via the standard method of dividing drug exposure (AUC, determined by noncompartmental analysis) after two routes of administration, bioavailability can be calculated as the cumulative fraction absorbed using deconvolution. Like the AUC/noncompartmental analysis method, deconvolution is based on the assumptions of the linear superposition principle (Rowland and Tozer 2011). Phoenix WinNonlin Deconvolution function was performed to determine the input rate after IM administration using individual PK parameters after IV administration as the exponential terms of the unit impulse function. Concentration-time data from the IM administration were used as the response function.


Intracranial Electroencephalography Analysis and Pharmacokinetic-Pharmacodynamic Modeling
iEEG data were analyzed using custom algorithms (Matlab version 2018b) for characterizing the temporal dynamics of iEEG power in spectral bands of interest [delta (1–3 Hz), theta (>3–7 Hz), alpha (>7–12 Hz), beta (>12–25 Hz), and high beta (>20–5 Hz)] as described previously (Kremen et al., 2017). Absolute and relative power spectral densities across frequency bands were used as the pharmacodynamic response. Absolute power density refers to the absolute power (Vrms2/Hz, where Vrms is the root-mean-square voltage of the patient’s iEEG), whereas relative power density refers to the percentage of power a frequency band (such as beta) compared with the sum of all frequency bands (delta, theta, alpha, and beta). Individualized PK parameters were estimated using a population approach as described in the previous section, fixed, then indirect-link maximum response (Emax)/Imax pharmacodynamic (PD) models were explored using eqs. 5 and 6. An indirect link was used due to the hysteresis that was observed. The best fit model was determined using visual inspection and precision of model parameters.
Safety and Behavioral Response Evaluations
A modified Glasgow coma scale was used to quantitate degree of sedation predose and at scheduled blood sampling times (Supplemental Table 2). Cardiorespiratory activity (blood pressure, heart rate, and respiratory rate) was assessed at blood sampling times up to 30 minutes. In addition to sedation and vitals, the dogs were monitored for vomiting, diarrhea, and lethargy prior to and for 60 minutes after drug administration and at each blood sampling time. Behavioral and iEEG activity were monitored by veterinary staff for seizure activity. As doses were escalated, the maximal tolerated toxicity, as determined by the supervising veterinarian, was defined as 20 minutes of sedation with stable cardiorespiratory activity (respiratory rate greater than 6 or less than 60 bpm, systolic blood pressure greater than 60 mmHg, and heart rate greater than 50 or less than 160 bpm). The IM injection sites were examined for swelling, tenderness, and redness after drug administration.
Results
The demographics of the dogs are listed in Supplemental Table 1.
Pharmacokinetics after Intravenous Administration
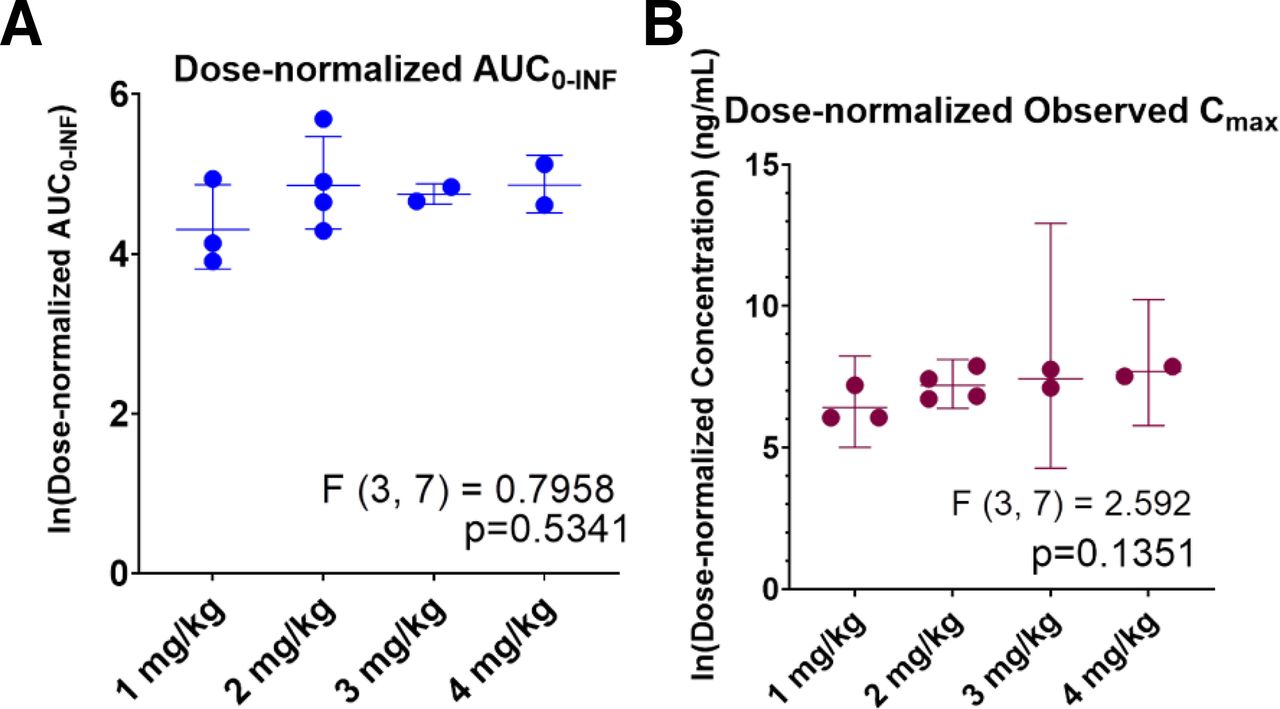
The concentration-time profiles after IV administration are shown in Fig. 1. Pharmacokinetic parameter estimates from noncompartment analysis are presented in Table 2. On average, IV ALLO exhibits a relatively short terminal phase half-life of 10–30 minutes and a large volume of distribution. One-way ANOVA tests across dose-normalized AUC∞ and Cmax suggest IV ALLO exhibits proportionality with respect to dose within the dose range studied (Fig. 2).

Plasma ALLO concentration-time profiles after 1–4 mg/kg ALLO infused IV over 5 minutes. There were a total of 11 IV infusions (3 @ 1 mg/kg, 4 @ 2 mg/kg, 2 @ 3 mg/kg, 2 @ 4 mg/kg). PB: temporary PB; cPB: chronic PB.

Dose-normalized (A) AUC∞ and (B) Cmax across 4 IV doses on a logarithmic scale. Mean values are shown with standard deviations. P values were generated using one-way ANOVA tests (n-1 groups = 3, degrees of freedom = 7) assuming homoschedasticity.
Noncompartmental PK parameter estimates after IV administration
Using a population approach, a two-compartment model with first-order elimination and proportional error best fit the ALLO concentration data after IV (Supplemental Table 3 and Supplemental Fig. 1). A random effect for volume of distribution was noninformative and therefore not included in the model. These parameter estimates were used to simulate plasma concentration-time profiles for dosing selection.
Pharmacodynamics after Intravenous Administration
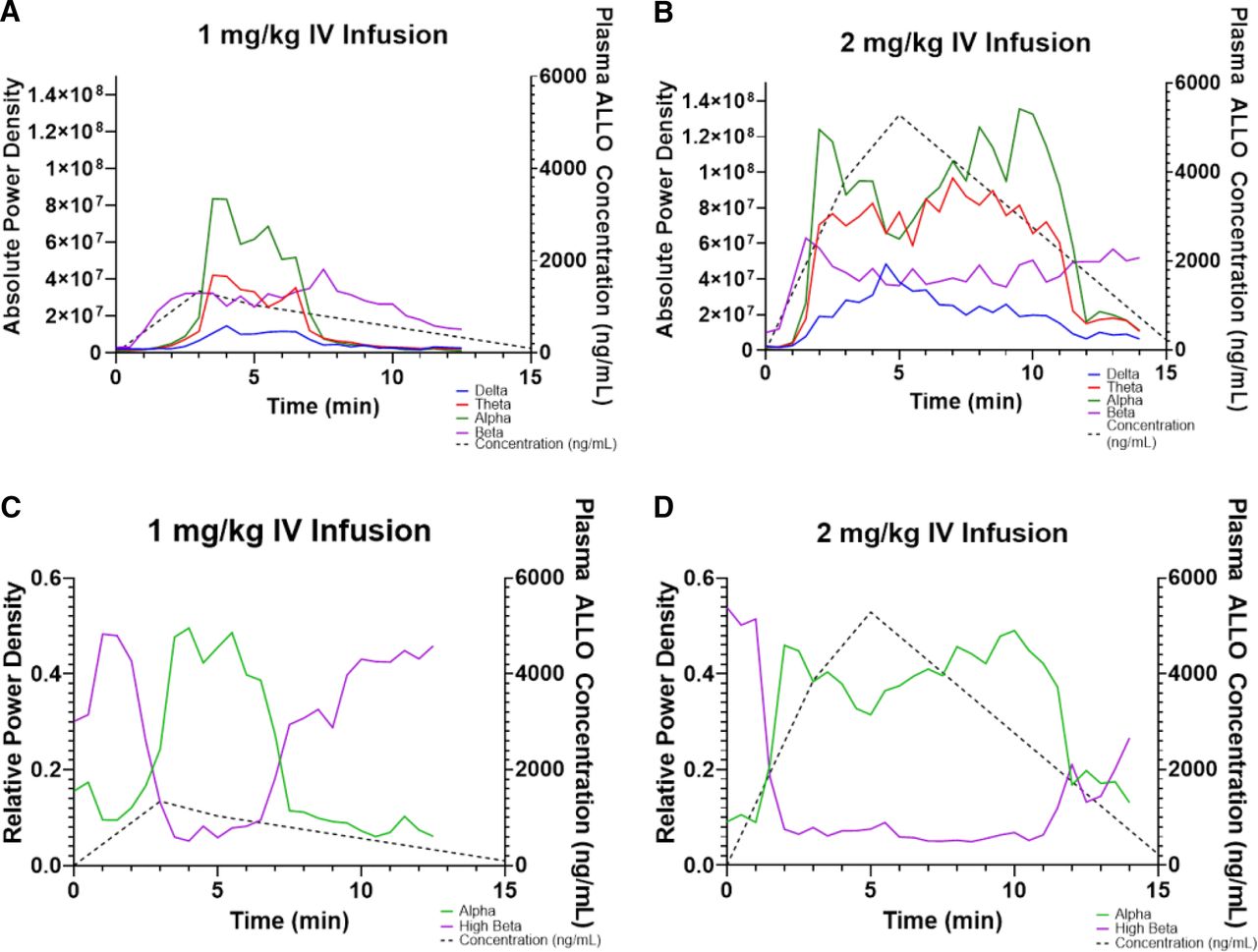
Across all iEEG frequency bands, absolute power density increased during and transiently after IV ALLO 1 mg/kg infusion (Fig. 3). The amplitude and duration of these changes increased at the higher IV dose. Concentration-EEG data were best fit by indirect-link sigmoidal Emax models (Table 3, Fig. 4). The delay equilibrium rate constants ranged between 1.3 and 5.1 minutes−1, corresponding to a delay equilibrium half-life of 8–30 seconds. The EC50 ranged between 270 and 700 ng/mL and was considerably lower for the beta frequency band, suggesting the beta frequency band is the most sensitive to ALLO. Its EC50 of 270 ng/mL in the theoretical effect compartment corresponds to ∼400 ng/mL in the plasma.

Time versus observed iEEG absolute power density (V2rms/Hz) across four frequency bands [delta (blue), theta (red), alpha (green), beta (purple)] and concentration (dashed black line) after (A) 1- and (B) 2-mg/kg IV infusions in one dog on PB. Time versus observed iEEG relative power density across alpha (green line) and high beta (purple line) frequency bands and concentration (dashed black line) after (C) 1- and D) 2-mg/kg IV infusions in one dog on PB.

(A) iEEG effect-concentration profiles after 1–2 mg/kg infused IV over 5 minutes (red and blue, respectively) and 1 mg/kg injected IM as a bolus (orange) in 1 dog on PB. Effect compartment concentrations are predicted using an indirect link between PK and sigmoidal Emax PD models. IPRED, predicted effect; DV, observed effect. (B) iEEG (relative power density) effect-concentration profiles after 1–2 mg/kg infused IV over 5 minutes (red and blue, respectively) and 1 mg/kg injected IM as a bolus (orange) in 1 dog on PB. Effect compartment concentrations are predicted using an indirect link between PK and sigmoidal Emax/Imax PD models. Colored circles are observed data; solid lines represent predicted data. IPRED, predicted effect; DV, observed effect.
PK-PD parameter estimates associated with IV infusions
High beta frequency band (20–25 Hz) relative power density decreased, whereas alpha frequency band (7–12 Hz) relative power density increased during and transiently after IV infusion. As with absolute power density, relative power density increased in amplitude and duration after the 2 mg/kg infusion. Concentration-iEEG data for change in alpha and high beta frequency band relative power density were best fit by indirect-link sigmoidal Emax and Imax models, respectively. The delay equilibrium rate constants were 0.92–2.23 minutes−1, which correspond to delay equilibrium half-lives of 19–45 seconds. The EC50 ranged between 329 and 759 ng/mL and were lower for the high beta frequency band. Its EC50 of 329 ng/mL in the theoretical effect compartment corresponded to ∼700 ng/mL in the plasma.
Pharmacokinetics after Intramuscular Administration
The concentration-time profiles after IM administration are shown in Fig. 5. After IM injection, there is an observed monoexponential decay in contrast to the biexponential decay after IV infusion, suggesting the presence of an absorption-limited terminal phase.

Plasma ALLO concentration-time profiles 1–6mg/kg injected IM as a bolus. There were a total of 5 IM injections (1 @ 1 mg/kg, 2 @ 2 mg/kg, 2 @ 6 mg/kg).
Due to the vast differences in absorption kinetics after each IM injection, individual noncompartmental PK analysis was performed (Table 4). Based on the known drug disposition after IV administration, a two-compartment model with first-order elimination, first-order absorption, and proportional error was used. There was an increasing trend in absorption rate with increasing formulation concentration, except at the 14 mg/mL formulation. The absorption rate from the 14 mg/mL formulation could not be estimated with precision, possibly because it was truly a zero-order absorption rate constant rather than first-order absorption rate constant. In contrast to IV administration, IM ALLO exhibits a terminal phase half-life ranging from 0.6–2.1 hours. There was an increasing trend in bioavailability with increasing formulation concentration and decreasing injection volume (Fig. 6A). The absorption profiles appear to follow a first-order absorption rate in most experiments (Fig. 6B). At the highest formulation concentration tested (14 mg/mL), there appears to be zero-order absorption occurring, which raises concern for the possibility of agglomeration, crystallization, and/or precipitation of drug into muscle tissue.

(A) IM bioavailability as a function of injection concentration. Each circle represents a different experiment (different dog/dose/injection concentration). Total injection volume, individual dog, and dose are labeled by the corresponding circle. (B) Cumulative fraction absorbed as a function of time. Each line represents a different experiment.
Noncompartmental PK parameter estimates after IM administration
Pharmacodynamics after Intramuscular Administration
Across all iEEG frequency bands, there were statistically significant but minimal changes to absolute power densities (Supplemental Fig. 2). As with absolute power density, decreases in high beta frequency band and increases in alpha frequency band were not as drastic as changes seen after IV infusion.
Safety and Tolerability
IV ALLO 1–3 mg/kg infused over 5 minutes and IM 1–2 mg/kg were shown to be safe and tolerable in dogs. There was a dose-dependent increase in ataxia and sedation (Table 5). At 4 mg/kg IV, dogs were immobile and briefly unarousable even with pain stimulation for 1–3 minutes with stable vital signs.
Ataxia and sedation
After IV infusion, ataxia occurred within 1.5–3 minutes after infusion start for a duration of 10–18.5 minutes. In healthy dogs, onset of sedation occurred at 3.5–5 minutes after infusion start and lasted up to 6 minutes at doses greater than 2 mg/kg. The dog on chronic PB had greater sedation at 2 mg/kg IV, which was associated with higher plasma ALLO concentrations. There were no infusion site reactions observed.
After IM injection, onset of ataxia without sedation occurred 3–5 minutes after the 6 mg/kg dose and lasted for 17–19 minutes. Of note, pain associated with injection volume was observed only at the 6 mg/kg IM dose and may have been associated with the volume infused (19.1–20.6 mL).
Discussion
Intravenous ALLO has a short terminal phase half-life and a moderately large volume of distribution in dogs (6–30 minutes, 2–6 L/kg). There was a rapid decrease in concentrations after IV administration. Given the large volume of distribution and lipophilic nature of this compound, this drop is likely due to redistribution. It is unlikely that these high concentrations immediately after dosing are due to artificially high levels at the drug administration site as blood was sampled from an extremity different from that used for drug administration. Across the doses studied, IV ALLO exhibited linear pharmacokinetics with respect to dose. This information will be useful for instances where a manipulation of a target ALLO concentration or exposure (e.g., doubling the dose to double the exposure, up to 4 mg/kg) may be necessary during clinical trials.
We found increases in absolute power spectral density across all frequency bands after IV ALLO, which is consistent to what has been reported after BZD administration. Increases in absolute and relative power density of the beta frequency band in response to BZD intervention have been widely cited (Greenblatt et al., 1989; Friedman et al., 1992; Mandema et al., 1992; Mandema and Danhof 1992; van Lier et al., 2004). The beta frequency band appears to have highest sensitivity for plasma ALLO concentrations with EC50 values twofold lower than other frequency bands.
In contrast to absolute power spectral density, IV ALLO results in a decrease in the relative power spectral density of the beta frequency band, whereas the alpha frequency band increased. Diazepam, on the other hand, elicits increases in relative power density of high beta (defined as 21–30 Hz) whereas decreasing alpha (defined as 9–10 Hz) in rats (van Lier et al., 2004). This suggests that although ALLO and BZDs potentiate GABAA activity, they induce differential effects on EEG activity. This may be due to ALLO’s activity at synaptic and extrasynaptic GABAA receptors and/or its ability to enhance the chloride channel opening duration and frequency (Carver and Reddy 2013; Greenfield 2013). BZDs only have activity at synaptic GABAA receptors and increase chloride channel opening frequency (Carver and Reddy 2013; Greenfield 2013). The pharmacodynamic effect drops rapidly after administration in relation to ALLO plasma concentrations and fairly rapid half-life. However, it is unclear whether sustained drug exposure is necessary for seizure cessation. Previous rodent studies suggest that attaining target concentrations briefly will likely be therapeutic (Zolkowska et al., 2018).
Zolkowska et al. demonstrated that IM ALLO has great potential to be useful as a first-line treatment of SE; however, given the incomplete absorption, our current formulations are not ideal for a first-line canine SE treatment (Zolkowska et al., 2018). Plasma concentrations after dosing were lower than expected compared with the same dose given IV. This observation is explained by the low absorption rates, which were dependent on formulation concentration and injection volume.
By evaluating the PK across different doses and formulation strengths, we found that a higher formulation concentration was associated with a faster absorption rate. It has been noted that injection volume does have an inverse relationship with absorption rate for IM injections independent of the water solubility of the drug (Hirano et al., 1981; Pfeffer and Van Harken 1981). Other factors that may affect the absorption rate constant include the particle size, cohesiveness of the dissolved particles, initial injection concentration, injection speed and pressure, and the physiologic state of the injection site (Hirano et al., 1981). We also observed that at the highest formulation concentration of 14 mg/mL, the estimated absorption rate constant was not as high as we expected based on the formulation-absorption trend observed with previous formulations and appeared to follow a zero-order rate. This suggests that 14 mg/mL may be too high of an initial injection concentration and may have resulted in possible agglomeration and/or precipitation of the drug. We observed that 11–14 mg/mL result in a near 100% bioavailability; however, additional studies with a larger sample size would be needed to verify this finding.
Simulations using the PK parameters from one dog (Supplemental Fig. 3) suggest an 8 mg/kg IM dose would be necessary to achieve target plasma concentration window of 500–1000 ng/mL using a 6 mg/mL formulation. However, this would result in an unacceptably large injection volume (∼27 mL for a 20 kg dog), which would limit the absorption rate and cause pain upon injection similar in severity to what was observed at the 6 mg/kg dose.
The largest limitation of the IM route of administration is the inability to attain high enough plasma concentrations to elicit iEEG changes. Strategies to increase plasma concentration using clinically relevant volumes include but are not limited to: (1) increasing its water solubility by synthesizing a prodrug or using alternative delivery systems; (2) using different administration strategies like multiple injections or changing the needle gauge and/or length; or (3) optimizing the formulation by using multiple solvents.
Based on studies done in a tetramethylenedisulfotetramine-induced SE mouse model and diisopropyl fluorophosphate-induced SE rat model, peak plasma ALLO concentrations ranging between 400–2000 ng/mL are expected to terminate BZD-refractory SE in at least 90% of animals (Zolkowska et al., 2018, 2020; Dhir et al., 2020b). Therefore, compiling all of the PK-PD and safety data from our study and what has been reported by Zolkowska et al. and Dhir et al., we propose that a 2 mg/kg dose infused over 5 minutes is an appropriate starting dose to test as a first-line agent to treat canine SE, as this dose is predicted to attain the target concentrations (500–1000 ng/mL) without causing heavy sedation (Fig. 7). Alternatively, a 1 mg/kg bolus injection may also be sufficient to achieve the targeted Cmax. In addition, a 1 mg/kg 5-minute infusion would also attain plasma concentrations that fall within the range between the EC50 and those associated with heavy sedation; however, the 2 mg/kg infusion is predicted to attain the peak concentration observed in the tetramethylenedisulfotetramine SE mouse study (900 ng/mL) as early as 3 minutes into the start of infusion.

Simulations after 5-minute IV infusions at four doses. Simulations were generated using population PK parameter values. Dashed line denotes the plasma concentration associated with heavy sedation. Dotted line denotes the plasma concentration associated to the EC50 in the theoretical effect compartment.
One major limitation of the study is the small number of animals, as is the heterogeneity among the dogs. Furthermore, although sex differences may exist in the pharmacokinetics or pharmacodynamics of ALLO, we were unable to examine the differences due to the limited number of dogs (three males and two females) in our study. Our study included only one animal on chronic PB and one with a history of witnessed seizures. Given the small number of dogs in the study, it is not possible to conclude whether concurrent use of PB induces drug-drug interaction. However, for one heathy dog, daily PB dosing was added for 2 weeks prior to the study of ALLO and the pharmacokinetics of ALLO did not appear to be affected. Our results in combination with preclinical evidence of rapid onset of antiseizure activity suggest IV ALLO shows promise as a first-line treatment of canine SE given its rapid distribution to the brain, safety, and novel mechanism of action.
Future work includes evaluating of formulations suitable for IM delivery of ALLO and conducting a pilot study of IV ALLO in treatment of canine SE. If a canine SE clinical trial proves successful, not only could the results be used to further the development of ALLO as an alternative treatment of canine SE, but it could also inform clinical trial designs for the use of IV ALLO in treating SE in people.
Acknowledgments
The authors would like to acknowledge the American Epilepsy Society Predoctoral Fellowship, American Foundation of Pharmaceutical Education Pre-Doctoral Fellowship, American Kennel Club Foundation, National Institutes of Health/National Institute of Neurologic Disorders and Stroke (NIH/NINDS), and MacMillan Epilepsy Innovation Fund who funded this research. The authors also acknowledge and thank Andrea Eckert for her care of the animals and sample collection.
Authorship Contributions
Participated in research design: Vuu, Patterson, Leppik, Rogawski, Cloyd, Coles.
Conducted experiments: Vuu, Patterson.
Contributed new reagents or analytic tools: Wu, Zolkowska.
Performed data analysis: Vuu, Worrell, Kremen, Coles.
Wrote or contributed to the writing of the manuscript: Vuu, Patterson, Wu, Zolkowska, Leppik, Cloyd, Coles.
Footnotes
- Received May 18, 2021.
- Accepted November 30, 2021.
This work was supported by the American Epilepsy Society Predoctoral Fellowship, American Foundation of Pharmaceutical Education Pre-Doctoral Fellowship, American Kennel Club Foundation [Grant 02133], National Institutes of Health/National Institute of Neurological Disorders and Stroke [Grant R21-NS072166], and MacMillan Epilepsy Innovation Fund.
No author has an actual or perceived conflict of interest with the contents of this article.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ALLO
- allopregnanolone
- AUC
- area under the concentration-time curve
- BZD
- benzodiazepine
- CL
- clearance
- CSE
- canine status epilepticus
- Emax
- maximum response
- F
- bioavailability
- iEEG
- intracranial electroencephalograph
- IM
- intramuscular
- IV
- intravenous
- PB
- phenobarbital
- PD
- pharmacodynamic
- PK
- pharmacokinetics
- PK-PD
- pharmacokinetic-pharmacodynamic
- SE
- status epilepticus
- Copyright © 2022 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}