


Abstract
The effect of dotinurad [(3,5-dichloro-4-hydroxyphenyl)(1,1-dioxo-1,2-dihydro-3H-1λ6-1,3-benzothiazol-3-yl)methanone] was compared with that of commercially available uricosuric agents—namely, benzbromarone, lesinurad, and probenecid. Its effect on urate secretion transporters was evaluated using probe substrates for respective transporters. Dotinurad, benzbromarone, lesinurad, and probenecid inhibited urate transporter 1 (URAT1) with IC50 values of 0.0372, 0.190, 30.0, and 165 μM, respectively. Dotinurad weakly inhibited ATP-binding cassette subfamily G member 2 (ABCG2), organic anion transporter 1 (OAT1), and OAT3, with IC50 values of 4.16, 4.08, and 1.32 μM, respectively, indicating higher selectivity for URAT1. The hypouricemic effects of dotinurad and benzbromarone were evaluated in Cebus monkeys. Dotinurad, at doses of 1–30 mg/kg, concomitantly decreased plasma urate levels and increased fractional excretion of urate (FEUA) in a dose-dependent manner. On the contrary, benzbromarone, at a dose of 30 mg/kg, showed a modest effect on plasma urate levels. The inhibitory effect of dotinurad on urate secretion transporters was evaluated in Sprague-Dawley rats, with sulfasalazine and adefovir as probe substrates of ABCG2 and OAT1, respectively. Drugs, including febuxostat as a reference ABCG2 inhibitor, were administered orally before sulfasalazine or adefovir administration. Dotinurad had no effect on urate secretion transporters in vivo, whereas benzbromarone, lesinurad, probenecid, and febuxostat increased the plasma concentrations of probe substrates. These results suggested dotinurad is characterized as a selective urate reabsorption inhibitor (SURI), which is defined as a potent URAT1 inhibitor with minimal effect on urate secretion transporters, including ABCG2 and OAT1/3, because of its high efficacy in decreasing plasma urate levels compared with that of other uricosuric agents.
SIGNIFICANCE STATEMENT Our study on the inhibitory effects on urate transport showed that dotinurad had higher selectivity for urate transporter 1 (URAT1) versus ATP-binding cassette subfamily G member 2 (ABCG2) and organic anion transporter (OAT) 1/3 compared to other uricosuric agents. In Cebus monkeys, dotinurad decreased plasma urate levels and increased fractional excretion of urate in a dose-dependent manner. To determine the inhibitory effect of dotinurad on urate secretion transporters, we studied the movement of substrates of ABCG2 and OAT1 in rats. Dotinurad had no effect on these transporters, whereas the other uricosuric agents increased the plasma concentrations of the substrates. These results suggested dotinurad as a potent and selective urate reabsorption inhibitor is characterized by increased efficacy with decreasing plasma urate levels.
Introduction
Gout is a form of acute arthritis induced by deposition of monosodium urate crystals in the joints. Environmental (food and drink) and genetic (Lesch-Nyhan syndrome and familial juvenile hyperuricemic nephropathy) factors have been reported to play a central role in the etiology of gout (Kuo et al., 2015). Hyperuricemia, a common pathogenetic factor in the development of gout, is typically defined by a serum urate concentration of >6.8 or 7.0 mg/dl (Terkeltaub, 2010; Neogi, 2011). Hyperuricemia has been reported as a risk factor for the onset and development of chronic renal diseases and a predictive factor for metabolic syndrome (Iseki et al., 2004; Obermayr et al., 2008; Yu et al., 2016).
Urate-lowering therapies with either type of urate control drug—urate production inhibitors or uricosuric agents—are indicated for patients with hyperuricemia. Benzbromarone, a commercially available uricosuric agent, effectively lowers serum urate levels. However, because of its rare but severe idiosyncratic hepatotoxic adverse effects, it is not approved in several European Union countries and the United States (Lee et al., 2008). In 2015, lesinurad, another uricosuric agent, was approved for use in the United States. However, it has been approved as 200-mg tablets in combination with urate production inhibitors because of its modest effect and increased serum creatinine at higher doses. There is a need for a novel uricosuric agent because 85% of patients develop hyperuricemia due to insufficient urate excretion (Nakamura, 2003); thus, the use of a uricosuric agent is recommended by the Japanese Guideline for the Management of Hyperuricemia and Gout (Japanese Society of Gout and Nucleic Acid Metabolism 2019).
Uricosuric agents are known to exert their pharmacological function by inhibiting the function of urate transporter 1 [URAT1; solute carrier family 22 member 12 (SLC22A12)], which is localized in the brush-border membrane of the renal proximal tubules and functions as a urate reabsorption transporter (Enomoto et al., 2002). Other urate reabsorption transporters are also present in the renal tubules: GLUT9 (URATv1 and SLC2A9) in the basolateral membrane and organic anion transporter (OAT) 4/10 (SLC22A11/13) in the brush-border membrane (Nigam and Bhatnagar, 2018). Urate secretion transporters are also present in the renal tubules: OAT1/2/3 (SLC22A6/7/8) in the basolateral membrane and ATP-binding cassette subfamily G member 2 (ABCG2; breast cancer resistance protein), MRP2/4 (ABCC2/4), and NPT1/4 (SLC17A1/3) in the brush-border membrane (Sato et al., 2010; Nigam and Bhatnagar, 2018). The presence of ABCG2 has also been reported in the brush-border membrane of the intestines, where it functions as an extrarenal urate excretion transporter (Ichida et al., 2012). Inhibition of urate secretion transporters has been reported to cause hyperuricemia. For example, probenecid, a nonselective OAT inhibitor, has been reported to elevate blood urate levels in potassium oxonate–treated rats (Yonetani et al., 1980). In ABCG2-knockout mice, a decrease in urate excretion into the intestines has been reported with a concomitant increase in blood urate levels (Ichida et al., 2012). Furthermore, several urate production inhibitors and benzbromarone were reported to inhibit urate transport by ABCG2, and their interaction probably occurs in the clinical setting (Miyata et al., 2016). These data suggest that selective urate reabsorption inhibitors (SURIs), which are defined as potent URAT1 inhibitors that do not affect urate secretion transporters including ABCG2 and OAT1/3, exhibit more potent hypouricemic effects than nonselective urate reabsorption inhibitor.
In the present study, we first compared the inhibitory effect of dotinurad [(3,5-dichloro-4-hydroxyphenyl)(1,1-dioxo-1,2-dihydro-3H-1λ6-1,3-benzothiazol-3-yl)methanone], a novel agent that exhibits uricosuric effect in rodents, on urate transport in cells overexpressing URAT1, ABCG2, and OAT1/3 with that of benzbromarone, lesinurad, and probenecid. We then investigated the hypouricemic effects of dotinurad and benzbromarone in Cebus monkeys. Finally, the inhibitory effects of these drugs on urate secretion transporters were evaluated in Sprague-Dawley rats, using sulfasalazine and adefovir as probe substrates of ABCG2 and OAT1, respectively. Considering our findings, we discuss the potency of dotinurad as a uricosuric agent and its mechanism of action.
Materials and Methods
Drugs and Materials.
Dotinurad (also called FYU-981; Fig. 1) was synthesized by Fuji Yakuhin Co., Ltd. (Saitama, Japan). Benzbromarone was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Lesinurad was purchased from Selleck Chemicals, LLC (Houston, TX). Probenecid and febuxostat were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Adefovir was purchased from LKT Laboratories, Inc. (St. Paul, MN). Sulfasalazine was purchased from Sigma-Aldrich Co., LLC. (St. Louis, MO). [14C]-Urate was synthesized by Moravek, Inc. (Brea, CA). The other reagents that were used in the present study are commercially available.

Chemical structures of dotinurad and commercially available uricosuric agents. Dotinurad (A), benzbromarone (B), lesinurad (C), and probenecid (D).
Determination of Xanthine Oxidase Activity.
Xanthine oxidase activity was measured with a spectrophotometer U-3000 (Hitachi High-Technologies Corporation, Tokyo, Japan) at 295 nm by following the absorbance change for an initial 1 minute. Assay mixture with 100 μM xanthine dissolved in 0.1 M NaOH (1% final) and drugs dissolved in dimethylsulfoxide (DMSO; 1% final) in 100 mM pyrophosphate buffer, pH 8.5, containing 0.2 mM EDTA was preincubated for 5 minutes, and reactions were started by adding xanthine oxidase (from bovine milk) to the mixture at a final concentration of 3.2 mU/ml under the aerobic conditions at 25°C. Assay was performed in triplicate.
Study of [14C]-Urate Uptake in URAT1-Overexpressing MDCKII Cells or OAT1- and OAT3-Overexpressing HEK293 Cells.
Cells were seeded into 96-well tissue culture plates at a density of 1 × 105 cells/well and cultured at 37 ± 1°C and a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium with 4.5 g/l glucose for 24 hours. Before initiating the experiment, culture medium was removed and cells were washed twice in 100 µl of Hanks’ balanced salt solution (HBSS). Uptake experiments were performed at 37 ± 1°C in 50 µl of HBSS containing 20 μM [14C]-urate and test articles dissolved in DMSO. Vehicle control contained 1% DMSO instead of test articles. URAT1-overexpressing MDCKII cells and OAT1-overexpressing HEK293 cells were washed after 10 minutes of incubation, whereas OAT3-overexpressing HEK293 cells were washed after 5 minutes of incubation in 100 µl of ice-cold HBSS, followed by cell lysis with 50 µl of 0.1 M NaOH. Transporter activity was determined by liquid scintillation counting of an aliquot from each well. Assay was performed in triplicate. The amount of translocated [14C]-urate was determined for each well in counts per minute using the MicroBeta2 liquid scintillation counter (PerkinElmer, Inc., Waltham, MA). Relative urate transport activity was reported as a percentage of the control and was calculated using the following equation: relative transport of urate (% of control) = (A − B)/(C − D) × 100, where A represents the amount of translocated [14C]-urate in the presence of test article in transfected cells, B represents the amount of translocated [14C]-urate in the presence of test article in mock cells, C represents the amount of translocated [14C]-urate in the presence of 1% DMSO in transfected cells, and D represents the amount of translocated [14C]-urate in the presence of 1% DMSO in mock cells.
Study of [14C]-Urate Uptake by Membrane Vesicles Obtained from ABCG2-Overexpressing HEK293 Cells.
Membrane vesicles were prepared from ABCG2-overexpressing cells (SOLVO Biotechnology, Hungary), and assay was performed in the presence of 70 μM urate containing 20 μM [14C]-urate and 4 mM ATP or AMP to distinguish between transporter-mediated uptake and passive diffusion into the vesicles. In 96-well plates, 50 μl of membrane vesicle suspended in 75 μM urate and 30 μM [14C]-urate containing ice-cold transport buffer (250 mM sucrose, 10 mM MgCl2, 10 mM Tris-HCl, pH 7.4) were added. Test articles dissolved in DMSO, 0.75 μl, were added to the membrane vesicle mixture. Vehicle control contained 1% DMSO instead of test article. The mixtures were preincubated for 15 minutes at 37 ± 1°C. Reactions were initiated by adding 25 μl of prewarmed 12 mM MgATP (or 12 mM AMP as a background control) in transport buffer. After 3 minutes, reactions were quenched by adding 200 μl of ice-cold washing buffer (250 mM sucrose, 100 mM NaCl, 10 mM Tris-HCl, pH 7.4) and immediately filtered using glass fiber filters. The filters were washed five times with 200 μl of ice-cold washing buffer and air dried. The amount of substrate inside the filtrated vesicles was determined by liquid scintillation counting. Assay was performed in triplicate. Relative urate transport activity was reported as a percentage of the control and was calculated using the following equation: relative transport of urate (% of control) = (A − B)/(C − D) × 100, where A represents the amount of translocated [14C]-urate in the presence of test article and ATP, B represents the amount of translocated [14C]-urate in the presence of test article and AMP, C represents the amount of translocated [14C]-urate in the presence of 1% DMSO and ATP, and D represents the amount of translocated [14C]-urate in the presence of 1% DMSO and AMP.
Animals and Housing.
For the evaluation of hypouricemic effects, 5- to 10-year-old male Cebus monkeys (Cebus paella, weight range 2.85–3.70 kg) bred in Shin Nippon Biomedical Laboratories, Ltd. (Tokyo, Japan) were used for pharmacological studies. The monkeys were housed individually in stainless steel cages in an air-conditioned animal room under a 12-/12-hour light/dark cycle at 26 ± 3°C and a relative humidity of 55% ± 20%. Animals were fed a pellet diet (New World Primate Diet 5040; Purina Mills, LCC) and fresh apples daily, except 1 day before drug administration. Water was provided ad libitum throughout the study. The experimental procedures were performed in accordance with the Animal Care and Utilization Guidelines of Shin Nippon Biomedical Laboratories, Ltd.
For the evaluation of inhibitory effects on urate secretion transporters, 7-week-old male Sprague-Dawley rats (weight range 180–220 g) bred in Japan SLC, Inc. (Shizuoka, Japan) were used. The rats were housed in wire-mesh cages in an air-conditioned animal room with a 12-/12-hour light/dark cycle at a temperature of 22 ± 4°C and a relative humidity of 60% ± 20%. Animals that did not develop abnormalities after a 1-week acclimatization period were selected for the study. Rats were fed a CE-2 pellet diet (Crea Japan Inc., Tokyo, Japan) and tap water via automatic stainless steel nozzles ad libitum throughout the study. Study protocols were designed and refined, taking animal reduction into consideration, and were approved by the Animal Care and Utilization Committee of Fuji Yakuhin Research Laboratories.
Study of the Hypouricemic Effects of Dotinurad and Benzbromarone in Cebus Monkeys.
Five Cebus monkeys that were fasted for 18 hours before drug administration orally received 1, 5, and 30 mg/kg dotinurad; 30 mg/kg benzbromarone; and 0.5% methylcellulose (MC) as control, respectively. Blood samples (about 1 ml) obtained from the saphenous vein at; before; and 2, 4, 8, and 24 hours after drug administration using a heparinized needle were kept on ice. Plasma was obtained from the blood samples by centrifugation at 3000 rpm for 10 minutes at 4°C. Urine samples were collected 0–4, 4–8, and 8–24 hours after drug administration. Urate and creatinine levels in the samples were measured by a U-3000 spectrophotometer using an Iatro LQ UAII (Mitsubishi Chemical Medience, Corp., Tokyo, Japan) and L-type Wako Creatinine F (Wako Pure Chemical Industries, Ltd.). Each treatment was administered in 13-day intervals to wash out drugs, and treatment and sample collection were performed as crossover experiments. Fractional excretion of urate (FEUA) was calculated as the ratio of urate clearance to creatinine clearance. Urinary urate excretion was calculated as urinary urate concentration × urine volume.
Determination of Drug Pharmacokinetics in Cebus Monkeys.
Plasma drug concentration was measured using the same samples described in the previous section. Plasma samples were deproteinated with thrice volume of methanol and centrifuged at 3000 rpm for 10 minutes at 4°C. Drug concentrations were measured using high-performance liquid chromatography (HPLC) using the Alliance 2695 HPLC system (Waters Corporation, Milford, MA). Area under the plasma concentration-time curve at 0–24h (AUC0–24h) were calculated using the trapezoidal rule. The concentration of 6-hydroxybenzbromarone, a major benzbromarone metabolite, was also measured in the plasma obtained from benzbromarone-treated animals.
Study of Concomitant Sulfasalazine and Hypouricemic Agent Treatment in Sprague-Dawley Rats.
Sprague-Dawley rats that were fasted for 18 hours before drug administration orally received 20 mg/kg febuxostat, 50 mg/kg benzbromarone, 1.3 mg/kg dotinurad, and 0.5% MC as control, respectively (n = 4, a total of 16 rats were used). Drug dosages used in the present study were calculated based on their clinically maximal doses detailed in Supplemental Table 1. Thirty minutes after the administration of these drugs, sulfasalazine suspended in 0.5% MC was orally administered at a dose of 20 mg/kg. For drug bioavailability (F) calculations, 5 mg/kg sulfasalazine dissolved in 50 mM Tris-saline was intravenously administered via the tail vein. Blood samples (about 200 μl) were obtained from the jugular vein at 0.083 (intravenous only), 0.25, 0.5, 1, 2, 4, 8, and 12 hours after sulfasalazine administration using a heparinized needle and kept on ice. Plasma was obtained from the blood samples by centrifugation at 3000 rpm for 10 minutes at 4°C. Plasma samples were deproteinated, and sulfasalazine concentration was measured as previously described by using liquid chromatography–tandem mass spectrometry (LC-MS/MS) on an Agilent 1100 Series HPLC Value System (Agilent Technologies, Inc., Santa Clara, CA) and API3000 (AB SCIEX, LCC., Framingham, MA) (Miyata et al., 2016). F values were calculated using the following equation: F (%) = (AUC0–inf value of oral administration/20)/(AUC0–inf value of intravenous administration/5) × 100.
Study of Concomitant Adefovir and Uricosuric Agent Treatment in Sprague-Dawley Rats.
Sprague-Dawley rats that were fasted for 18 hours before drug administration orally received 100 mg/kg probenecid, 67 mg/kg lesinurad, 50 mg/kg benzbromarone, 1.3 mg/kg dotinurad, and 0.5% MC as control (n = 6, a total of 30 rats were used). Thirty minutes after drug administration, 3 mg/kg adefovir dissolved in saline was intravenously administered via the tail vein to all animals. Blood samples (about 200 μl) were obtained from the jugular vein at 0.083, 0.25, 0.5, 1, 2, and 4 hours after adefovir administration using a heparinized needle and kept on ice. Plasma was obtained from blood samples by centrifugation at 3000 rpm for 10 minutes at 4°C. Adefovir was detected in these samples as previously described (Jullien et al., 2003). In brief, plasma was derivatized by incubation at 80°C in 0.34% chloroacetaldehyde solution for 50 minutes and detected via the excitation and emission spectra at 236 and 420 nm wavelengths, respectively, using the Alliance 2695 HPLC system (Waters Corporation).
Statistical Analysis.
In the study of xanthine oxidase activity, IC50 values were calculated using probit method. In cell-based urate uptake studies, the concentration-response curves were analyzed using the GraphPad Prism 7.03 software (GraphPad Software, La Jolla, CA), and a four-parameter logistic equation was applied to calculate the IC50 values. In the studies of Cebus monkeys and Sprague-Dawley rats, the mean and S.D. were calculated, and differences from the control group were analyzed using Dunnett’s multiple comparison test (at significance levels of 5% and 1%). In the study of Cebus monkeys, AUC0–24h was calculated using the trapezoidal rule in Microsoft Excel 2010 (Microsoft Corporation, Redmond, WA), and unbound fraction of maximum plasma concentration was calculated using Fu in Supplemental Table 3. In the study of Sprague-Dawley rats, pharmacokinetic parameters were analyzed using the Phoenix WinNonlin 6.4 software (Pharsight Corp. Sunnyvale, CA).
Results
Effects of Uricosuric Agents on Xanthine Oxidase Activity.
Benzbromarone inhibited xanthine oxidase activity with an IC50 value of 15.4 μM. Dotinurad, lesinurad, and probenecid did not have any effect up to 100, 300, and 1000 μM, respectively.
Inhibitory Effect of Uricosuric Agents on Urate Uptake by URAT1-Overexpressing MDCKII Cells.
Dotinurad, benzbromarone, lesinurad, and probenecid inhibited urate transport by URAT1-overexpressing MDCKII cells in a concentration-dependent manner at concentrations of 0.003–3, 0.003–3, 0.3–300, and 10–1000 μM (Fig. 2) and IC50 values of 0.0372, 0.190, 30.0, and 165 μM, respectively (Table 1).

Concentration-response curve of dotinurad and commercially available uricosuric agents on urate transport by URAT1-overexpressing MDCKII cells. Each value is the mean ± S.D. of urate transport relative to the control (n = 3) and described by GraphPad Prism 7.03 software (GraphPad Software).
IC50 values of dotinurad and commercially available uricosuric agents against urate transport by URAT1 and urate secretion transporters
For measurement of transport activities, MDCKII-URAT1 (passage numbers 3–11), HEK293-OAT1 (passage number 8), HEK293-OAT3 (passage number 5), and HEK293-ABCG2 membrane vesicles (SOLVO Biotechnology) were used. IC50 was calculated using a four-parameter logistic equation. Each value is the mean + S.E.M. (n = 2 to 3).
Inhibitory Effect of Uricosuric Agents on Urate Uptake by ABCG2-, OAT1-, and OAT3-Overexpressing HEK293 Cells.
Dotinurad inhibited urate transport in a concentration-dependent manner in ABCG2-, OAT1-, and OAT3-overexpressing HEK293 cells at concentrations of 0.3–300, 0.1–100, and 0.03–30 μM and IC50 values of 4.16, 4.08, and 1.32 μM, respectively. The ratios of IC50 values of ABCG2-, OAT1-, and OAT3-overexpressing cells to that of URAT1-overexpressing cells were 112, 110, and 35.5, respectively (Tables 1 and 2). Benzbromarone inhibited urate transport in ABCG2-, OAT1-, and OAT3-overexpressing cells with IC50 values of 0.289, 3.14, and 0.967 μM, respectively, and IC50 value ratios to that of URAT1-overexpressing cells were 1.52, 16.5, and 5.09, respectively. Lesinurad inhibited urate transport with IC50 values of 26.4, 6.99, and 1.07 μM, respectively, and IC50 value ratios to that of URAT1-overexpressing cells were 0.880, 0.233, and 0.0357, respectively. Probenecid inhibited urate transport with IC50 values of 433, 10.9, and 2.37 μM, respectively, and IC50 value ratios to that of URAT1-overexpressing cells were 2.62, 0.0661, and 0.0144, respectively.
IC50 ratios for ABCG2 and OAT1/3 to URAT1 of dotinurad and commercially available uricosuric agents
Each value was calculated as the IC50 of each compound for ABCG2, OAT1, and OAT3/IC50 for URAT1.
Hypouricemic Effects of Dotinurad and Benzbromarone in Cebus Monkeys.
Effects of dotinurad and benzbromarone on plasma urate levels, FEUA, and urinary urate excretion were assessed in Cebus monkeys. Dotinurad dose-dependently lowered the plasma urate levels, with its maximum effect at 8 hours (Fig. 3A). Changes in plasma urate level between 0 and 8 hours (ΔPUA) were lower than that of control by 0.28, 0.97 (P < 0.05), and 1.79 mg/dl (P < 0.01) at doses of 1, 5, and 30 mg/kg, respectively. Furthermore, dotinurad dose-dependently increased FEUA (Fig. 3B). The 0- to 4-hour FEUA increased by 180% at a dose of 30 mg/kg compared with the control (P < 0.01). On the contrary, the effect of benzbromarone at a dose of 30 mg/kg was modest; the ΔPUA was lower than that of control by 0.46 mg/dl, and the 0- to 4-hour FEUA increased by 30% compared with the control. Figure 3C details the urinary excretion amount of urate in Cebus monkeys 0–8 hours after dotinurad and benzbromarone administration. Dotinurad dose-dependently increased the urinary urate excretion to 16.2, 22.8, and 25.3 mg at doses of 1, 5, and 30 mg/kg, respectively, compared with the control, which was 13.7 mg. On the contrary, benzbromarone increased the urinary urate excretion to 19.4 mg at a dose of 30 mg/kg. Results of creatinine clearance and urate clearance are shown in Supplemental Table 2.
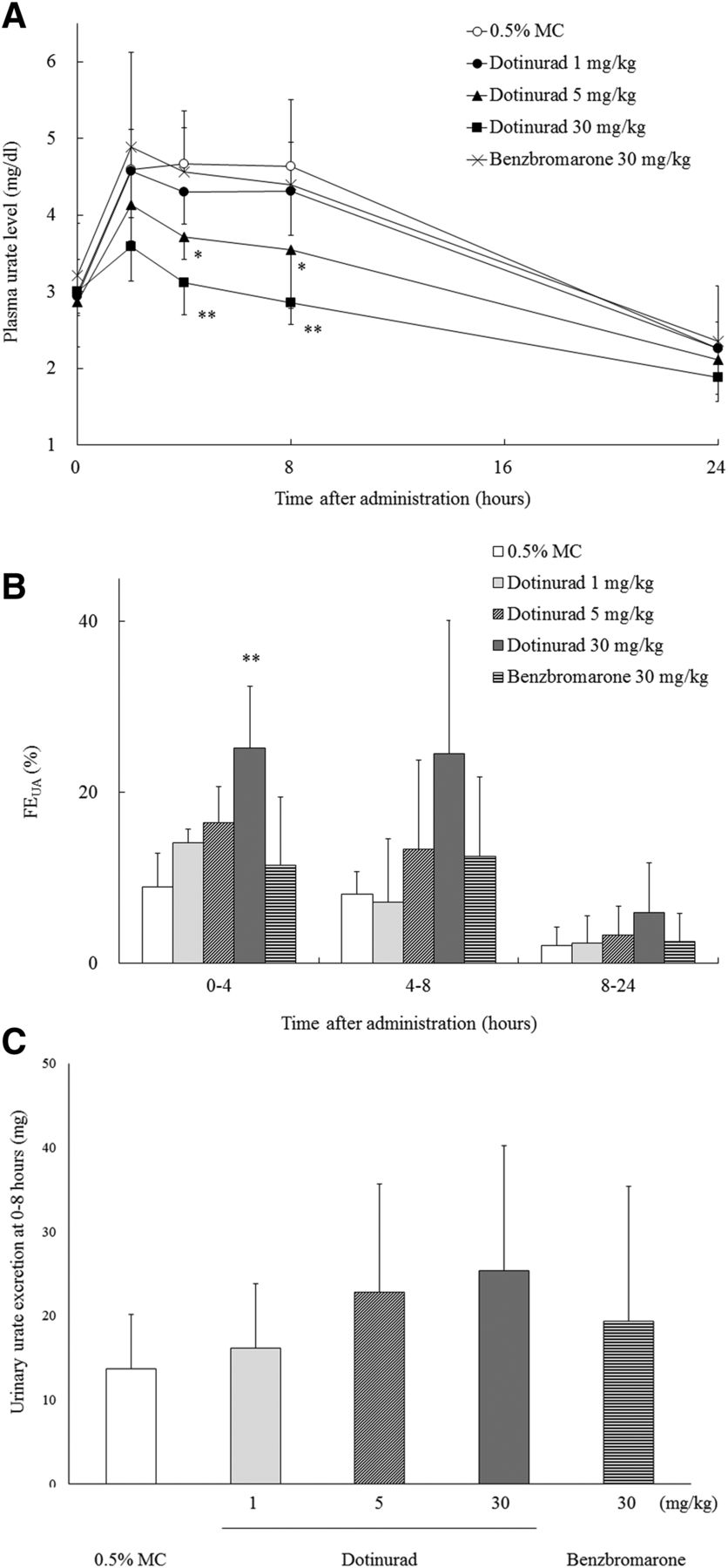
Effects of dotinurad and benzbromarone on plasma urate level, FEUA, and urinary urate excretion in Cebus monkeys. Data are represented as change in plasma urate level (A); FEUA at 0–4, 4–8, and 8–24 hours after dosing (B); and the amount of urinary urate excretion for 8 hours after dosing (C). Each value is the mean ± S.D. of four to five animals. *P < 0.05; **P < 0.01 for significant differences from the control at each time point using the Dunnett’s multiple comparison test.
Pharmacokinetic Parameters of Dotinurad and Benzbromarone in Cebus Monkeys.
The plasma concentration of dotinurad increased in a dose-dependent manner with Cmax and AUC0–24h values of 107 μg/ml and 780 μg⋅hours/ml, respectively, at a dose of 30 mg/kg (Table 3). On the contrary, Cmax and AUC0–24h values of benzbromarone were 20.9 μg/ml and 95.2 μg⋅hours/ml, respectively, at a dose of 30 mg/kg. In the plasma obtained from benzbromarone-treated animals, Cmax and AUC0–24h values of 6-hydroxybenzbromarone were 12.7 μg/ml and 80.9 μg⋅hours/ml, respectively. Unbound fraction of maximum plasma concentration of dotinurad at doses of 1, 5, and 30 mg/kg and benzbromarone at a dose of 30 mg/kg were 0.0144, 0.0800, 0.750, and 0.564 μg/ml, respectively.
Pharmacokinetic parameters of dotinurad and benzbromarone in Cebus monkeys
Plasma samples were deproteinated and drug concentrations were measured using HPLC. AUC0–24h was calculated using the trapezoidal rule. Each value is the mean ± S.D. of five animals.
Effect of Hypouricemic Agents on the Plasma Concentration of Sulfasalazine in Sprague-Dawley Rats.
Effects of febuxostat, benzbromarone, and dotinurad on the plasma sulfasalazine concentration were assessed in Sprague-Dawley rats. Febuxostat and benzbromarone significantly increased the plasma sulfasalazine concentration (Fig. 4). Febuxostat increased the AUC0–inf value to 5012 ng⋅hours/ml (P < 0.01) from a control value of 713 ng⋅hours/ml and F to 14.1% from a control value of 2.0%. Benzbromarone also increased the AUC0–inf value to 2888 ng⋅hours/ml (P < 0.01) and F to 8.1%. Dotinurad did not have any effect on the pharmacokinetic parameters of sulfasalazine (Table 4).

Effect of dotinurad and other hypouricemic agents on the plasma concentration of sulfasalazine in Sprague-Dawley rats. Hypouricemic agents or their vehicle as control (0.5% MC) was orally administered to Sprague-Dawley rats fasted for 18 hours. After 30 minutes, sulfasalazine was orally administered. Blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, and 12 hours after sulfasalazine administration. Plasma samples were deproteinated, and sulfasalazine concentrations were measured using liquid chromatography–tandem mass spectrometry (LC-MS/MS). Each value is the mean ± S.D. of three to four animals. **P < 0.01, significant differences from the 0.5% MC control group after Dunnett’s multiple comparison test.
Pharmacokinetic parameters of sulfasalazine and effects of concomitant treatment with hypouricemic agents in Sprague-Dawley rats
Pharmacokinetic parameters were calculated using Phoenix WinNonlin. F values were calculated using the following equation: F = (AUC0–inf value of oral administration/20)/(AUC0–inf value of intravenous administration/5) × 100. Values are reported as the mean ± S.D. of three to four animals.
Effect of Uricosuric Agents on the Plasma Concentration of Adefovir in Sprague-Dawley Rats.
Effects of probenecid, benzbromarone, lesinurad, and dotinurad on the plasma concentration of adefovir were assessed in Sprague-Dawley rats. Probenecid and lesinurad significantly increased the plasma adefovir concentration (Fig. 5). Probenecid increased the plasma concentration of adefovir at 0.083–0.5 hour (P < 0.01). Lesinurad also increased the plasma concentration of adefovir at at 0.083–0.5 (P < 0.01) and 1 hour (P < 0.05). On the contrary, benzbromarone and dotinurad did not have any effect on the plasma concentration of adefovir.

Effect of dotinurad and other uricosuric agents on the plasma concentration of adefovir in Sprague-Dawley rats. Hypouricemic agents or their vehicle as control (0.5% MC) was orally administered to Sprague-Dawley rats fasted for 18 hours. After 30 minutes, adefovir was intravenously administered. Blood samples were collected at 0.083, 0.25, 0.5, 1, 2, and 4 hours after adefovir administration. Plasma samples were derivatized, and adefovir concentrations were measured using HPLC. Each value is the mean ± S.D. of six animals. *P < 0.05; **P < 0.01, significant differences from the 0.5% MC control group after Dunnett’s multiple comparison test.
Discussion
The kidneys play an important role in urate elimination. After filtration through the glomerulus, urate undergoes complex bidirectional steps of reabsorption and secretion in the proximal tubules, resulting in a net urinary excretion of <10% of the filtered amount (Chonko and Grantham, 1981). Because renal handling of urate is reabsorption-dominant, the reabsorption process is the target of uricosuric agents. In the renal tubules, several transporters are involved in urate reabsorption. In particular, URAT1 in the brush-border membrane and GLUT9 in the basolateral membrane of renal tubules are the main urate reabsorption transporters. Aberrant function of these transporters caused by single nucleotide polymorphisms (SNPs) results in insufficient urate reabsorption, increased urinary urate excretion, and remarkable hypouricemia (<1 mg/dl) (Kikuchi et al., 2000; Dinour et al., 2010). Uricosuric agents are known to exert their pharmacological effect via URAT1 inhibition, by which almost maximum effect is obtained without affecting other urate reabsorption transporters. On the contrary, urate secretion transporters are also present in the renal tubules: OAT1/2/3 are localized in the basolateral membrane, whereas ABCG2, MRP2/4, and NPT1/4 are localized in the brush-border membrane (Sato et al., 2010; Nigam and Bhatnagar, 2018). SNPs in ABCG2 result in a higher risk of gout caused by dysfunctional urate excretion (extrarenal excretion) from the intestines (Matsuo et al., 2009; Miyata et al., 2016). Furthermore, gene association studies performed in a patient cohort with chronic kidney disease showed that SNPs in ABCG2, MRP4, and OAT3 are associated with increased serum urate levels, although the association of SNPs in ABCG2 with serum levels was modest in the non–chronic kidney disease cohort (Bhatnagar et al., 2016). In humans, the contribution of other urate secretion transporters to serum urate level has not been reported. Our findings suggested that SURIs had superior hypouricemic effects to those of nonselective urate reabsorption inhibitors.
Our previous study showed that in rats treated with topitoxostat, a urate production inhibitor, dotinurad decreases plasma urate levels and increases FEUA in a dose-dependent manner without inhibiting xanthine oxidase or uricase (Matsumoto et al., 2011; Taniguchi et al., 2016). In studies in pyrazinamide-treated rats and brush-border membrane vesicles, dotinurad inhibits urate uptake at the brush-border membrane of the renal tubules with pyrazinecarboxylic acid as an exchange substrate (Taniguchi et al., 2017). It was postulated that this effect is mediated by urate transporters; however, the detailed underlying mechanisms are unknown. Based on these findings, we next evaluated the inhibitory effect of dotinurad on urate uptake by MDCKII or HEK293 cells overexpressing transporters. Dotinurad inhibited urate transport mediated by URAT1 with an IC50 value of 0.0372 μM. The effect of dotinurad was approximately 5.11, 806, and 4440 times more potent than that of benzbromarone, lesinurad, and probenecid, respectively, because the IC50 values of these compounds were 0.190, 30.0, and 165 μM, respectively (Table 1). Dotinurad modestly inhibited urate transport by ABCG2 and OAT1/3 with IC50 values of 4.16, 4.08, and 1.32 μM, which were 112, 110, and 35.5 times higher than its IC50 values for URAT1, respectively (Tables 1 and 2). By contrast, the IC50 ratios of benzbromarone, lesinurad, and probenecid for ABCG2 and OAT1/3 to URAT1 were 1.52–16.5, 0.0357–0.880, and 0.0144–2.62, respectively. These findings showed that dotinurad inhibited URAT1 more selectively than the other uricosuric agents. Several lines of evidence showed the inhibitory effect of benzbromarone, lesinurad, and probenecid on urate transport by URAT1, ABCG2, OAT1, and OAT3 (Ichida et al., 2003; Miner et al., 2016; Miyata et al., 2016), although the inhibitory effects of benzbromarone and probenecid on urate transport by OAT3 are not reported. Our data were consistent with these reports, except for the URAT1 inhibitory effect of lesinurad and probenecid (the IC50 values are 3.53 and 13.23 μM, respectively). We could not evaluate the inhibitory effect of uricosuric agents on MRP4, because urate transport by MRP4-overexpressing cells was not reported. These data also suggested that dotinurad did not affect ABCG2 and OAT1/3 in clinical use because its unbound fraction in plasma appeared to be extremely low (Supplemental Table 3) (US Food and Drug Administration, 2017; Pharmaceuticals, 2019).
Subsequently, we compared the hypouricemic efficacy of dotinurad with that of benzbromarone in Cebus monkeys. Dotinurad at doses of 1, 5, and 30 mg/kg decreased plasma urate levels and increased FEUA in a dose-dependent manner, and its effect at 1 mg/kg was approximately equal to that of 30 mg/kg benzbromarone (Fig. 3, A and B). However, the stringency of URAT1 inhibition (the IC50 values of dotinurad and benzbromarone were 0.0372 and 0.190 μM, respectively) did not sufficiently account for the difference in their hypouricemic effects. We hypothesized that the pharmacokinetic profiles of these drugs possibly contribute to their hypouricemic effects. Indeed, dotinurad had higher Cmax and AUC0–24h values than those of benzbromarone (Table 3). The unbound fractions of maximal plasma concentration of dotinurad at doses of 1, 5, and 30 mg/kg and that of benzbromarone at a dose of 30 mg/kg, which are considered to correlate with their concentration at the renal proximal tubules, were calculated to be 0.0144, 0.0800, 0.750, and 0.564 μg/ml, respectively (Table 3). Because these values are 1.1, 6.0, 56, and 7.2 times higher than the IC50 values of URAT1, respectively, an equal hypouricemic effect was expected to be obtained by 5 mg/kg dotinurad and 30 mg/kg benzbromarone (Supplemental Table 3; Table 3). Considering that 6-hydroxybenzbromarone, a major metabolite of benzbromarone, also inhibited URAT1 and contributed to the hypouricemic effect in addition to benzbromarone itself (Shin et al., 2011), another factor was assumed to explain the difference in hypouricemic effect. Thus, we speculated that property as SURIs, defined as potent URAT1 inhibitors with minimal effect on urate secretion transporters, ABCG2 and OAT1/3, generated the difference of hypouricemic effect apart from nonselective inhibitor.
To estimate the effect of uricosuric agents on urate secretion in vivo accurately, we evaluated whether dotinurad and other uricosuric agents interact with ABCG2 and OAT1 using their probe substrates in Sprague-Dawley rats. Sulfasalazine is known as a typical ABCG2 substrate with low absorption because it is excreted on the intestinal brush-border membrane (Dahan and Amidon, 2009). In our rat study using sulfasalazine as a probe substrate, benzbromarone and the positive control agent febuxostat markedly increased the plasma concentration and F of sulfasalazine (Fig. 4; Table 4), and these increases were assumed to be caused by inhibition of intestinal ABCG2, which appeared to be important for urate excretion (Ichida et al., 2012). Adefovir, a well known OAT1 substrate, is excreted in urine (as adefovir itself), and its fraction of drug elimination in urine (fe) is approximately 90% in humans (Cundy et al., 1995). In our rat study using adefovir as a probe substrate, lesinurad and probenecid increased the plasma concentration of adefovir (Fig. 5); this increase was assumed to be caused by inhibition of renal OAT1, which mediates urate secretion (Wu et al., 2017). Because these drugs showed more potent inhibition on OAT3 than on OAT1, further experiments should be conducted using a probe substrate for OAT3. In our rat studies, dotinurad did not affect the plasma concentration of probe substrates, indicating that dotinurad hardly affected urate secretion transporter in vivo.
We must consider that the probe substrates did not move exactly in the same manner as urate. Substrate movement can be predicted by evaluating the inhibitory effect of a compound on urate and probe substrates. For example, another ABCG2 inhibitor, rifampicin, inhibits both urate and sulfasalazine transport by ABCG2 at a similar concentration (Hosomi et al., 2012; Kosa et al., 2018). In terms of OAT1, probenecid inhibits adefovir transport with an inhibition constant (Ki) of 18.6 μM (Maeda et al., 2014), suggesting a similarity to our result. These reports indicated that sulfasalazine and adefovir were transported in the same manner as urate. Furthermore, species differences between rats and humans have to be taken into account. The AUC0–48h of sulfasalazine was 3-fold higher for functional ABCG2 SNPs, and a similar effect was observed in ABCG2-knockout mice (Gotanda et al., 2015; Miyata et al., 2016). The fe of adefovir in rodents is approximately 80%, which is nearly the same as that for humans (Naesens et al., 1996). Therefore, we concluded that the data obtained from our rat studies could predict urate handling in humans.
Studies on Cebus monkeys showed that the hypouricemic effect of UR-1102 (also called as URC102) [(3,5-dibromo-4-hydroxyphenyl)(2,3-dihydro-4H-pyrido-[4,3-b][1,4]oxazin-4-yl)methanone], a selective inhibitor of URAT1 versus OAT1/3, showed a higher estimated Emax of urinary urate excretion than that shown by benzbromarone (Ahn et al., 2016). In this report, authors discussed that URAT1 selectivity, defined as potent URAT1 inhibition without an effect on OAT1/3, is important for producing a potent hypouricemic effect. In fact, UR-1102, in a dose-dependent manner, lowered serum urate in humans at doses (0.25–2 mg) much lower than that of benzbromarone ((Jun et al., 2017)).
Figure 6 shows a scheme of urate pool reduction by SURIs or nonselective inhibitors. SURIs effectively reduce the urate pool by increasing renal excretion of urate. On the contrary, nonselective inhibitors modestly reduce the urate pool because of their inhibitory effect on urate secretion transporters. For nonselective inhibitors to reduce the urate pool at a similar level as SURIs, they require more URAT1 inhibition at higher doses, which results in excessively elevated renal excretion of urate and poses a risk factor for urolithiasis (Yu and Gutman, 1967). In the present study, benzbromarone did not exert an apparent effect on plasma urate levels in Cebus monkeys (Fig. 3A), although it increased urinary urate excretion more potently than 1 mg/kg dotinurad (Fig. 3C). The phenomenon appeared to be mediated by inhibition of urate secretion transporters, especially ABCG2 in the intestine. These findings suggested that nonselective inhibitors can only reduce the urate pool. Furthermore, our findings proved that SURIs were not associated with a risk of drug-drug interaction under clinical settings.

Comparison of the potency of selective and nonselective urate reabsorption inhibitors in decreasing urate pool and the concomitant effects of nonselective inhibitors. (Upper) The usual state of urate handling showing urate excretion from the renal tubules and the small intestine. (A) SURI increases urate excretion from the renal tubules due to URAT1 inhibition, leading to a potent reduction of urate pool. (B) A nonselective inhibitor decreases urate excretion from the renal tubules and/or the small intestine through inhibition of ABCG2 and/or OAT1/3. For a nonselective inhibitor to obtain the same effect at a level similar to SURI, more potent inhibition of URAT1 and additional renal excretion are required. Arrow width represents the quantity of urate handling, which is affected by inhibition of URAT1, ABCG2, and OAT1/3.
In conclusion, we showed that dotinurad, a SURI, could be an effective therapeutic option for the treatment of hyperuricemia because of its more effective hypouricemic action than that of commercially available uricosuric agents. Furthermore, dotinurad could be a useful agent in studies on urate transporters because of its ability to selectively and potently inhibit URAT1.
Acknowledgments
We appreciate Jo Tanaka, Department of Drug Safety Research Laboratories, Shin Nippon Biomedical Laboratories Ltd., and Dr. Erika Csonka and Dr. Daniel Gliesche, SOLVO Biotechnology, for their assistance with the pharmacological evaluation. We appreciate Kazuma Sekine for his assistance with the pharmacokinetic evaluation. We appreciate Kazumi Miyamoto for her assistance with the pharmacological evaluation.
Authorship Contributions
Participated in research design: Taniguchi, Ashizawa, Matsumoto, Motoki, Iwanaga.
Conducted experiments: Taniguchi, Ashizawa, Matsumoto, Motoki, Sakai, Chikamatsu, Hagihara.
Performed data analysis: Taniguchi, Ashizawa, Matsumoto, Motoki.
Wrote or contributed to the writing of the manuscript: Taniguchi, Ashizawa, Matsumoto, Saito, Motoki, Hashiba, Iwanaga.
Footnotes
- Received April 24, 2019.
- Accepted July 16, 2019.
A part of the Cebus monkey studies in the work was previously presented at the 86th annual meeting of the Japanese Pharmacological Society (2013), Abstract number P2-143, Fukuoka, Japan.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ABCG2
- ATP-binding cassette subfamily G member 2
- AUC
- area under the plasma concentration-time curve
- dotinurad
- (3,5-dichloro-4-hydroxyphenyl)(1,1-dioxo-1,2-dihydro-3H-1λ6-1,3-benzothiazol-3-yl)methanone
- FEUA
- fractional excretion of urate
- HBSS
- Hanks’ balanced salt solution
- HPLC
- high-performance liquid chromatography
- MC
- methylcellulose
- OAT
- organic anion transporter
- SNP
- single nucleotide polymorphism
- SURI
- selective urate reabsorption inhibitor
- URAT1
- urate transporter 1
- Copyright © 2019 The Author(s).
This is an open access article distributed under the CC BY Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}