


Abstract
Urate-lowering therapy is indispensable for the treatment of gout, but available drugs do not control serum urate levels tightly enough. Although the uricosurics benzbromarone and probenecid inhibit a urate reabsorption transporter known as renal urate transporter 1 (URAT1) and thus lower serum urate levels, they also inhibit other transporters responsible for secretion of urate into urine, which suggests that inhibiting URAT1 selectively would lower serum urate more effectively. We identified a novel potent and selective URAT1 inhibitor, UR-1102, and compared its efficacy with benzbromarone in vitro and in vivo. In human embryonic kidney (HEK)293 cells overexpressing URAT1, organic anion transporter 1 (OAT1), and OAT3, benzbromarone inhibited all transporters similarly, whereas UR-1102 inhibited URAT1 comparably to benzbromarone but inhibited OAT1 and OAT3 quite modestly. UR-1102 at 3–30 mg/kg or benzbromarone at 3–100 mg/kg was administered orally once a day for 3 consecutive days to tufted capuchin monkeys, whose low uricase activity causes a high plasma urate level. When compared with the same dosage of benzbromarone, UR-1102 showed a better pharmacokinetic profile, increased the fractional excretion of urinary uric acid, and reduced plasma uric acid more effectively. Moreover, the maximum efficacy of UR-1102 was twice that of benzbromarone, suggesting that selective inhibition of URAT1 is effective. Additionally UR-1102 showed lower in vitro potential for mechanisms causing the hepatotoxicity induced by benzbromarone. These results indicate that UR-1102 achieves strong uricosuric effects by selectively inhibiting URAT1 over OAT1 and OAT3 in monkeys, and could be a novel therapeutic option for patients with gout or hyperuricemia.
Introduction
Gout has become a global burden in recent years because of its increasing prevalence, extensive comorbidity associated with disability, and increased mortality (Robinson and Dalbeth, 2015). Because the clinical manifestations of gout are associated with the formation of monosodium urate crystals in the joints, controlling serum urate is important when treating gout. Several major guidelines, including the 2012 American College of Rheumatology Guidelines for Management of Gout (ACR guidelines), recommend that serum urate level should be lowered sufficiently to improve signs and symptoms of gout, with a target minimum of 6 mg/dl, or in many cases 5 mg/dl (Khanna et al., 2012). Moreover the lower the serum urate level, the faster and more significant the reduction in the size of tophi, which are solid deposits of monosodium urate crystals (Perez-Ruiz et al., 2002, 2007). This is ordinarily achieved with either a urate synthesis inhibitor (USI) or a uricosuric (Shahid and Singh, 2015). The ACR guidelines recommend the use of a USI, such as allopurinol or febuxostat, as the first-line approach for pharmacologic urate-lowering therapy and, because the urate-lowering efficacy of currently available USI drugs is insufficient, that this be combined with a uricosuric when the serum urate target is not met by the USI. Typical uricosurics, namely benzbromarone or probenecid, increase the urate excretion by inhibiting urate transporter 1 (URAT1), the transporter responsible for urate reabsorption in the renal proximal tubules (Enomoto et al., 2002; Shin et al., 2011).
URAT1 is a member of the SLC22A—or organic anion transporter (OAT)—family, and its mutations can cause hypouricemia (Ichida et al., 2004; Dinour et al., 2011). Transporters in the SLC22A family play important roles in the reabsorption and excretion of anionic substances, including urate and other endogenous substances, xenobiotics, and drugs in the kidney (Burckhardt, 2012). In addition to URAT1, OAT1, and OAT3 are also SLC22A family members and transport urate in the kidney; however, these are thought to be responsible for the secretion of urate into the urine, whereas URAT1 is responsible for reabsorption (Bakhiya et al., 2003; Ichida et al., 2003; Choi et al., 2005). In fact, inhibition of OAT3 is thought to be one reason why some angiotensin II receptor blockers, such as candesartan or valsartan, can cause hyperuricemia (Sato et al., 2008). Probenecid, which is considered to have insufficient clinical efficacy in lowering urate is known to inhibit not only URAT1 but also OAT1 and OAT3 (Takeda et al., 2001; Shitara et al., 2005; Kusuhara et al., 2013). Although the precise contribution of the two secretory transporters to urate excretion remains unclear, selective inhibition of URAT1 would potentially achieve stronger urate excretion. Benzbromarone lowers serum urate more effectively than probenecid; however, it is only available in a few countries outside the United States, owing to safety concerns, which include the risk of hepatitis (Lee et al., 2008). In addition, benzbromarone also reportedly inhibits OAT3 (Kimura et al., 2000), which suggests that its uricosuric efficacy could be surpassed by an inhibitor that is selective only for URAT1. In summary, a safe and potent uricosuric drug that can achieve target serum urate levels of less than 5 mg/dl is required to treat patients with gout, especially those with tophi, and a selective URAT1 inhibitor could be one approach for realizing this concept.
It is well known that there are marked species differences in purine metabolism and urinary uric acid excretion between humans and most experimental animals, such as rats, rabbits, dogs, and cynomolgus monkeys (Roch-Ramel and Peters, 1978). The enzyme uricase in these animals can degrade uric acid to allantoin, and as a result their serum urate levels are about 1/10th of human values (Choi et al., 2005), which makes it difficult to assess the uricosuric effect. However, New World monkeys in general have relatively low uricase activity compared with other mammals and show relatively higher levels of serum uric acid (Simkin, 1971). One subgroup in particular, Cebus monkeys, has relatively high serum urate levels, and the renal tubular transport of urate is similar to that of humans (Fanelli et al., 1970; Roch-Ramel and Weiner, 1973), which makes this animal quite valuable for assessing uricosurics (Dan et al., 1989; Fannelli and Weiner, 1975).
Here, we identified UR-1102 as a novel URAT1 inhibitor that showed high selectivity to URAT1 over OAT1 and OAT3 in vitro when compared with benzbromarone. Then, by comparing these two compounds in vivo using tufted capuchin monkeys (Cebus apella), we examined the importance of the selective inhibition of URAT1 for the treatment of gout.
Materials and Methods
Chemicals
UR-1102 [(3,5-dibromo-4-hydroxyphenyl)(2,3-dihydro-4H-pyrido[4,3-b][1,4]oxazin-4-yl)methanone; Fig. 1] was synthesized at JW Pharmaceutical Corp. (Seoul, Republic of Korea). UR-1102 was synthesized using the condensation reaction of benzoyl chloride with pyrido-oxazine. 4H-Pyrido[4,3-b][1,4]oxazin-3-one was converted to the corresponding pyrido-oxazine by reaction with 1.0 mol/l lithium aluminum hydride. The resulting pyrido-oxazine was then reacted with 3,5-dibromo-4-methoxybenzoyl chloride, followed by demethylation under acidic conditions. Details of synthesis are included in Example 4 in Ahn et al. (2013). [14C]UR-1102 and [14C]benzbromarone were synthesized at Korea RadioChemicals Center (Suwon, Republic of Korea). Benzbromarone was purchased from Sigma-Aldrich/MerckMillipore (St. Louis, MO). [8-14C]Uric acid ([14C]uric acid) and [ring-14C]acetamidophenol ([14C]acetaminophen) were purchased from American Radiolabeled Chemicals (St. Louis, MO). p-[Glycyl-2-3H]-aminohippuric acid ([3H]PAH) was obtained from PerkinElmer Inc. (Waltham, MA). p-Aminohippuric acid (PAH) and acetaminophen were purchased from Wako Pure Chemical Industries, Ltd. (Tokyo, Japan). All other chemicals were of the highest purity available.

Structure of UR-1102 and benzbromarone.
In Vitro Transporter Inhibition Assays
Cell Culture and Transfection.
An expression plasmid containing human SLC22A12 (URAT1) (GenBank Accession No. NM_144585) was prepared by digesting the fragments from URAT1-pME18SFL3 vector (Toyobo Co., Tokyo, Japan) and ligating them into the multicloning site of pcDNA3.1(+) (Life Technologies/Thermo Fisher Scientific, Waltham, MA). Expression plasmids containing human SLC22A6 (OAT1) and SLC22A8 (OAT3) (GenBank Accession Nos. NM_153276, NM_004254) were purchased from Origene Technologies (Rockville, MD).
Human embryonic kidney (HEK)293 cells (American Type Culture Collection, Manassas, VA), a transformed cell line derived from human embryonic kidney, were cultured in complete medium consisting of Dulbecco’s modified Eagle’s medium (Sigma-Aldrich/MerckMillipore) with 10% fetal calf serum in an atmosphere of 5% CO2, and 95% air at 37°C. Cells were seeded in 100-mm cell culture dishes. When they reached confluency, they were transfected with URAT1 plasmid, OAT1 plasmid, OAT3 plasmid, or control vector [pcDNA3.1(+) for URAT1 and pCMV6-XL5 (Origene Technologies) for OAT1/3], using Lipofectamine 2000 transfection reagent (Life Technologies/Thermo Fisher Scientific) according to the manufacturer’s instructions. After 4–6 hours of transfection, cells were subcultured in poly-d-lysine–coated 24-well plates at a density of 2.0 × 105 cells/well. At 48 hours after transfection, the cells were used for uptake experiments.
Uptake Experiment in HEK293 Cells.
Cells expressing URAT1, OAT1, OAT3, or control vector were used to measure the amount of specific substrate (uric acid for URAT1, PAH for OAT1 and OAT3) taken up at various concentrations of UR-1102 or benzbromarone. The composition of the incubation medium was as follows: 140 mM sodium gluconate, 2.7 mM potassium gluconate, 1.8 mM KH2PO4, and 10 mM Na2HPO4 for URAT1; 137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, and 8.1 mM Na2HPO4 for OAT1 and OAT3. The cells were incubated with incubation medium containing UR-1102 or benzbromarone for a certain period of time (30 minutes for URAT1, 15 minutes for OAT1 and OAT3) at 37°C. A certain amount medium containing [14C]uric acid or [3H]PAH was added to initiate the uptake. The medium was aspirated at the end of the incubation period, and the monolayers were rapidly washed twice with 1 ml of ice-cold incubation medium. The cells were solubilized in 0.4 ml of 0.5 N NaOH, and then the radioactivity in the aliquots was determined by liquid scintillation counting. On the basis of the measured data, the Ki values of both UR-1102 and benzbromarone for URAT1, OAT1, and OAT3 were calculated. The velocity of substrate uptake (v) that depends on URAT1, OAT1, or OAT3 was calculated by subtracting the uptake in the mock-transfected cells from that in cells expressing URAT1, OAT1, or OAT3. The Lineweaver-Burk plot was created by plotting the inverse velocity (1/v) as a function of the inverse of the substrate concentration (1/s). The slope of the least squares regression line of the Lineweaver-Burk plot was calculated, and then it was defined as Si for each concentration of test compound (i). S0 was defined as the slope of the regression line for i=0. Following that, S′ was calculated as the slope of the least squares regression line of the plot that was created by plotting test compound concentration (i) as a function of Si. Finally, the Ki value was calculated by dividing S0 by S′. The average and standard deviation (S.D.) of each Ki value were calculated from the data of three independent studies.
In Vivo Evaluation in Tufted Capuchin Monkeys
Animals.
Four male and two female tufted capuchin monkeys 6- to 13-years-old at Shin Nippon Biomedical Laboratories, Ltd. (Kagoshima, Japan) were used. The animals were housed under a 12-hour light/dark cycle (lights on 7:00 AM to 7:00 PM), at a controlled room temperature (23–29°C) and humidity (35–75%). Approximately 80 g of solid food for monkeys {New World Primate Diet 5040; Purina Mills, LLC, St. Louis, MO) and approximately 100 g of apple as a supplement were provided to each animal once daily between 13:00 and 14:00. Water was available ad libitum from an automatic supply. This study was approved by the Institutional Animal Care and Use Committee (Approval No. IACUC036-120) and was performed in accordance with the animal welfare bylaws of Shin Nippon Biomedical Laboratories Drug Safety Research Laboratories, which is fully accredited by AAALAC International.
Effect of UR-1102 and Benzbromarone on Renal Fractional Excretion of Uric Acid and Plasma Uric Acid.
UR-1102 at 3, 10, and 30 mg kg−1 day−1, benzbromarone at 3, 10, 30, and 100 mg kg−1 day−1, or vehicle (1% methylcellulose and 0.01 mol/l HCl solution) were administered by oral gavage to the monkeys. Compounds or vehicle were administered by once-daily dosing for 3 days to complete each phase, with 2-week intervals between each of eight phases (crossover design with eight groups). Clinical signs and body weight were checked throughout the study period. Urine and plasma were collected to measure uric acid or creatinine concentration at the stated times. Blood samples were collected from the saphenous vein immediately before dosing (0 hours) and at 2, 4, 8, and 24 hours after every dosing, and plasma samples were obtained. Urine was collected from 8 hours to immediately before dosing, and 0 to 8 hours after every dosing. The concentration levels of uric acid and creatinine were measured by the uricase F-DAOS method and the creatinase F-DAOS method, respectively, using an automatic analyzer (JCA-BM6070; JEOL Co., Ltd., Tokyo, Japan).
In this monkey model, as shown in the control group, oral administration induced a transient increase in plasma uric acid; the concentration returned to normal at 24 hours. Although the precise mechanism of this increase remains unknown, it might be the result of transient stimulation of urate synthesis, because the increase was suppressed by febuxostat at 3 mg/kg, which is equivalent to the clinical dose. After confirming in a preliminary study that this increase in plasma uric acid concentration was reproducible and was suppressed by both a USI (febuxostat) and a uricosuric (benzbromarone), we concluded that this monkey model would be suitable for evaluating urate-lowering drugs. Because the plasma uric acid concentration was unstable, we used the average plasma uric acid concentration as the parameter. The averages for plasma uric acid and creatinine were calculated from the areas under the plasma uric acid and creatinine concentration-versus-time curves from 0 to 8 hours after dosing (calculated according to the linear-trapezoidal rule) divided by the collection time (8 hours). Urinary uric acid fractional excretion (FEUA) was calculated using the equation below:
 (1)
(1)Pharmacokinetics of UR-1102 and Benzbromarone.
Plasma samples on the first day were obtained at six time points (0.5, 1, 2, 4, 8, and 24 hours after dosing) and the pharmacokinetic (PK) parameters were calculated. UR-1102 and benzbromarone were extracted from plasma by adding acetonitrile and were measured with a liquid chromatography–tandem mass spectrometry system [high-performance liquid chromatography (Shimadzu 20A; Shimadzu Corporation, Kyoto, Japan) and tandem quadrupole mass spectrometer (API 4000; Applied Biosystems, Foster City, CA)]. Pharmacokinetic parameters (Cmax, Tmax, AUC0–inf, t1/2) were obtained from the quantitative values of each animal by using WinNonlin version 5.2.1 (Pharsight Corporation, Mountain View, CA).
In Vitro Safety Assessments
Mitochondrial Toxicity Study.
The human hepatoma cell line HepG2 cells (American Type Culture Collection) was maintained in minimum essential medium (MEM) containing 10% fetal bovine serum supplemented with MEM nonessential amino acids, sodium pyruvate, HEPES buffer, and penicillin streptomycin (all from Life Technologies/Thermo Fisher Scientific) in an atmosphere of 5% CO2 and 95% air at 37°C. To assess the mitochondrial toxicity, we evaluated the mitochondrial membrane potential (MMP) as a marker of mitochondrial integrity and function by using an MMP-dependent fluorescent probe. HepG2 cells were plated onto a 96-well plate (20,000 cells/well) and treated with UR-1102 or benzbromarone for 4 hours in MEM plus the supplements listed above, except for fetal bovine serum. After incubation, 0.2 μmol/l of MitoTracker Red CMXRos (Life Technologies/Thermo Fisher Scientific) and 10 μg/ml of Hoechst 33342 (Life Technologies/Thermo Fisher Scientific) was added to the cells, which were then incubated for 30–40 minutes at 37°C. Automated live-cell multispectral images were acquired on a Cellomics ArrayScan VTI HCS Reader using a 20× objective (Thermo Fisher Scientific, Waltham, MA) and an XF93 filter (Omega Optical, Inc., Brattleboro, VT). The images were analyzed using vHCS software (Spot Detector V4 algorithm; Thermo Fisher Scientific), and the MMP was analyzed as “Mean Object Spot Total Intensity”. Cell viability was evaluated by measuring intracellular lactate dehydrogenase (LDH) activity using an LDH-Cytotoxic Test (Wako Pure Chemical Industries, Richmond, VA). Both compounds were tested in duplicate at each concentration and the test was repeated four times.
Reactive Metabolite Formation.
The formation of reactive metabolites in human liver microsomes by UR-1102 and benzbromarone was investigated. Briefly, 1 mg/ml of human liver microsomes (XenoTech, LLC, Lenexa, KS) was incubated at 37°C for 30 minutes with [14C]UR-1102 or [14C]benzbromarone at a concentration of 10 μmol/l in the presence or absence of 1 mmol/l NADPH. The remaining radioactivity in the microsomal protein after washing with 0.1% sulfuric acid/methanol solution was measured using a liquid scintillation counter (Tri-Carb 3110TR; PerkinElmer Inc.). The protein content was assayed using a BCA protein assay kit (Pierce Biotechnology Inc., Rockford, IL) according to the manufacturer’s instructions. The formation of reactive metabolites by [14C]acetaminophen was also measured as a positive control.
Binding on Various Receptors, Channels, and Transporters
The selectivity of UR-1102 for various molecular targets (70 receptors, 5 ion channels, 3 transporters) was examined by Cerep (Celle L’Evescault, France) using standard in vitro radioligand binding assays (Supplemental Table S1). UR-1102 was tested at 10 μM. All studies were internally controlled with reference ligands, and further details of the methodologies for each assay can be found at http://www.cerep.fr.
Statistical Analysis
Average plasma uric acid concentrations and FEUA at each analysis point were taken as fixed effects and animals as random effects, and these were analyzed by pairwise comparison between the control group and the UR-1102 groups on the basis of a mixed-effects model. Dose dependency was investigated by applying an Emax-based nonlinear model (Dheeraj and Gomathi, 2012). The above-stated analyses were performed to compare UR-1102 and benzbromarone groups with the control group to investigate the efficacy of UR-1102 or benzbromarone and their dose dependency.
To compare the maximum efficacy of UR-1102 and benzbromarone in the tufted capuchin monkeys, average plasma uric acid concentrations and FEUA from 0 to 8 hours after dosing at 10 or 30 mg kg−1 day−1 UR-1102-01 and at 100 mg kg−1 day−1 benzbromarone on day 3 were analyzed in pairwise comparison by t test (two-tailed at 5%).
Above analyses were performed using SAS System for Windows, version 9.2 (SAS Institute Inc., Cary, NC).
For the mitochondrial toxicity study, IC50 values for MMP in each test were calculated using sigmoidal fittings with the Dose Response model in OriginPro 8J SR4 v8.0951 (OriginLab Corp., Northampton, MA). The 95% confidence interval (CI) for IC50 value was calculated using the analysis of variance (ANOVA) test in SAS version 8.2 (SAS Institute).
Results
In Vitro URAT1, OAT1, and OAT3 Inhibition.
To identify a compound that is more highly selective against URAT1 than OAT1 or OAT3 and is highly soluble for a good PK profile, we screened 79 compounds modified from the benzbromarone structure at 0.1, 1, and 10 μmol/l and finally obtained UR-1102. The structure of UR-1102 and benzbromarone are shown in Fig. 1. UR-1102 inhibited urate and PAH uptake by HEK293 cells transiently expressing URAT1, OAT1, or OAT3. Analysis using Lineweaver–Burk plots showed that both UR-1102 and benzbromarone inhibited urate uptake in a mixed and a noncompetitive inhibition manner (Fig. 2, A and B). In addition, these compounds also inhibited PAH uptake by OAT1 and OAT3 (Fig. 2, C–F), but with different levels of efficacy, as the Ki values for UR-1102 and benzbromarone show (Table 1). Although the Ki value for UR-1102 against URAT1 was comparable to that of benzbromarone (0.057 ± 0.036 μmol/l versus 0.052 ± 0.017 μmol/l), the Ki values for UR-1102 against OAT1 and OAT3 just exceeded 10 times the Ki values of benzbromarone (OAT1: 7.2 ± 0.8 μmol/l versus 0.22 ± 0.03 μmol/l; OAT3: 2.4 ± 0.2 μmol/l versus 0.11 ± 0.05 μmol/l). As a result, the Ki ratios for URAT1 over OAT1 and OAT3, which show the selectivity of UR-1102, were 130 and 42, respectively, whereas those of benzbromarone were only 4.2 and 2.1 (Table 1).

Inhibition of URAT1, OAT1, and OAT3 by UR-1102 (A, C, E) and by benzbromarone (B, D, F). Urate or PAH uptake was measured in HEK293 cells transiently expressing URAT1, OAT1, or OAT3 in the presence or absence of UR-1102 or benzbromarone at various concentrations of each substrate. Velocity (v) was calculated and used for Lineweaver–Burk plots against 1/[s]. Experiments were performed three times.
Inhibitory effects of UR-1102 and benzbromarone on URAT1, OAT1, and OAT3. Figures in parentheses represent the level of URAT1 selectivity on the basis of the Ki value.
Pharmacokinetics of UR-1102 and Benzbromarone in the Monkey Model.
To compare the uricosuric efficacy of UR-1102 with that of benzbromarone, we referred to the preliminary PK results and the in vitro URAT1 inhibitory activity and set the dose at 3, 10, and 30 mg kg−1 day−1 for UR-1102 and at 3, 10, 30, and 100 mg kg−1 day−1 for benzbromarone. To confirm the validity of the dose selection and to examine the PK profiles of UR-1102 and benzbromarone, plasma concentrations of both compounds were measured and the PK parameters were analyzed for 24 hours after the first dosing on day 1 (Table 2). After oral administration, UR-1102 was rapidly absorbed into the blood and reached a maximum plasma concentration at 0.5 to 0.8 hours, while benzbromarone showed a lower Cmax and a longer tmax at the same dose. Plasma concentration of UR-1102 then decreased with a longer t1/2 than that of benzbromarone, resulting in higher exposure of UR-1102 than that of benzbromarone at the same dose. The plasma exposure (AUC0−inf) of a 3-fold lower dose of UR-1102 was comparable to that of the benzbromarone dosage, indicating that the selected dose range was valid for further comparison of these two compounds.
PK of UR-1102 and benzbromarone in tufted capuchin monkeys on the 1st day of dosing
Urate-Lowering Effects of UR-1102 and Benzbromarone in the Monkey Model.
In the monkey study described above we compared the plasma urate-lowering efficacy of UR-1102 and benzbromarone. In this monkey model, the control group showed that oral administration of vehicle induced a transient increase in the concentration of plasma uric acid for 8 hours, which returned to normal within 24 hours, and a similar response was observed on days 2 and 3 (Fig. 3). Both UR-1102 and benzbromarone dose dependently suppressed the increased plasma uric acid concentration from 0 to 8 hours after dosing over 3 days (Fig. 3 and Supplemental Fig. S1). On day 3, the average plasma uric acid concentration from 0 to 8 hours compared with vehicle dosing was significantly reduced by UR-1102 at 3, 10, and 30 mg/kg and by benzbromarone at 10, 30, and 100 mg/kg, but not by 3 mg/kg benzbromarone (Fig. 4). Comparing 30 mg/kg of UR-1102 and 100 mg/kg of benzbromarone (doses with comparable plasma exposure, Table 2), average plasma uric acid concentration with UR-1102 was significantly lower than that with benzbromarone (1.8 ± 0.2 versus 2.7 ± 0.2 mg/dl, p < 0.01, Fig. 4). Moreover, the urate-lowering effect in plasma achieved by UR-1102 at 3 mg/kg was comparable to that of benzbromarone at 100 mg/kg (2.5 ± 0.3 versus 2.7 ± 0.2 mg/dl, Fig. 4).
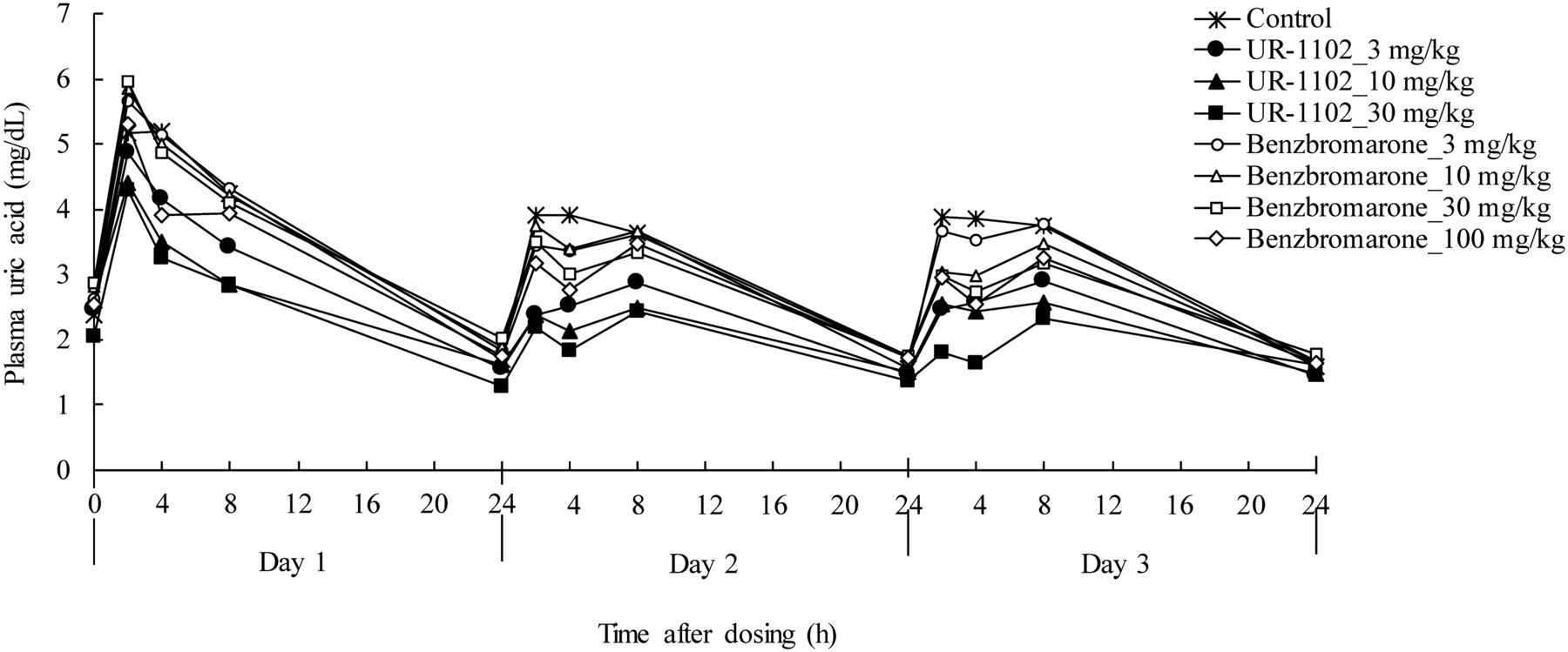
Chronological changes in plasma uric acid. UR-1102 (3, 10, or 30 mg/kg), benzbromarone (3, 10, 30, or 100 mg/kg), or vehicle was administered to tufted capuchin monkeys by oral gavage on 3 consecutive days. Blood was collected at 0, 2, 4, 8, and 24 hours after dosing. Data represents mean (n = 6).

Average plasma uric acid concentration from 0 to 8 hours after dosing on day 3. UR-1102 (3, 10, or 30 mg/kg), benzbromarone (3, 10, 30, or 100 mg/kg), or vehicle was administered to tufted capuchin monkeys by oral gavage on 3 consecutive days. Blood was collected at 0, 2, 4, 8, and 24 hours after dosing. Average plasma uric acid was calculated as described in Materials and Methods. Data represents mean + S.E. (n = 6). *p < 0.05, **p < 0.01, significant difference from control group (paired t test on the basis of a mixed-effects model). #p < 0.01, significant difference between UR-1102 at 30 mg/kg and benzbromarone at 100 mg/kg (paired t test).
Uricosuric Effects of UR-1102 and Benzbromarone in the Monkey Model.
In the above monkey model, we also assessed the effects on urinary excretion of uric acid as FEUA. Both UR-1102 and benzbromarone dose dependently increased FEUA from 0 to 8 hours after dosing on all 3 days (Supplemental Fig. S2). Compared with vehicle dosing, FEUA on day 3 was significantly increased by UR-1102 at 3, 10, 30 mg/kg and by benzbromarone at 10, 30, and 100 mg/kg, but not by 3 mg/kg benzbromarone treatment (Fig. 5). Interestingly, FEUA was significantly higher at 30 mg/kg UR-1102 than at 100 mg/kg benzbromarone on day 3 (42.1 ± 4.4% versus 20.6 ± 2.3%, p < 0.05). Furthermore, the increase in FEUA achieved by 3 mg/kg UR-1102 was comparable to that achieved by benzbromarone at 100 mg/kg (22.6 ± 3.6% versus 20.6 ± 2.3%) (Fig. 5).

Fractional excretion of uric acid from 0 to 8 hours after dosing on day 3. UR-1102 (3, 10, or 30 mg/kg), benzbromarone (3, 10, 30, or 100 mg/kg), or vehicle was administered to tufted capuchin monkeys by oral gavage on 3 consecutive days. Blood was collected at 0, 2, 4, and 8 hours after dosing. Urine was collected from 0 to 8 hours after dosing. FEUA was calculated as described in Materials and Methods. Data represents mean + S.E. (n = 6). *p < 0.05, **p < 0.01, significant difference from control group (paired t test on the basis of a mixed-effects model). #p < 0.05, significant difference between UR-1102 at 30 mg/kg and benzbromarone at 100 mg/kg (paired t test).
We also used the Emax-based model to assess FEUA of UR-1102 and benzbromarone on day 3. The Emax-based model showed that the compounds had comparable ED50 (Fig. 6 and Table 3), and the simulated Emax values of FEUA for UR-1102 and benzbromarone at the highest doses were similar to the actual FEUA. Most importantly, the simulated Emax for UR-1102 was twice as high as that of benzbromarone, the effect of which reached its maximum at a lower dose (30 mg/kg).

Emax model of FEUA at day 3 from oral treatment with (A) UR-1102 or (B) benzbromarone. Solid line shows the estimated dose-response curve. Dots represent actual values (n = 6 at each dose). Parameters are listed in Table 3.
Parameters estimated from the Emax model of FEUA at day 3
Emax model: Response = Emax × Doseγ / (Doseγ +  )
)
General Condition and Changes in Renal Function in the Monkey Model.
We also assessed the safety of UR-1102 in the monkey model during the dosing and observation periods. The monkeys exhibited no compound-related adverse changes in their general condition. Additionally, regarding the effects on renal function, no significant changes in plasma creatinine or urinary creatinine excretion by either UR-1102 or benzbromarone were observed during the dosing period (Supplemental Tables S2 and S3).
In Vitro Safety Assessments.
The hepatic toxicity of benzbromarone is a known safety concern. Although the detailed mechanisms underlying its hepatic toxicity are still unknown, mitochondrial toxicity and reactive metabolite formation have been proposed as potential mechanisms (Kaufmann et al., 2005; McDonald and Rettie, 2007; Felser et al., 2014).
Therefore, we evaluated MMP in HepG2 cells to assess the potential of UR-1102 to induce these mechanisms in comparison with benzbromarone. Although both UR-1102 and benzbromarone showed concentration-dependent decreases in MMP, their intracellular LDH activity did not change in a viability assay (Fig. 7), which indicates that the decreases in MMP were not caused by cell loss but by the effects on mitochondrial function. Although benzbromarone decreased MMP almost completely at 89 μmol/l (Fig. 7B), UR-1102 showed no effect at the same concentration (Fig. 7A). Furthermore, the 95% CI of the IC50 values for MMP in UR-1102-treated cells was 258–1830 μmol/l, which was 27.6–1649 times that in cells exposed to benzbromarone (95% CI: 1.11–9.34 μmol/l) (Supplemental Table S4). These results suggest that UR-1102 probably has a lower potential for mitochondrial toxicity than benzbromarone.

Effects of UR-1102 and benzbromarone on mitochondrial membrane potential and cell viability in HepG2 cells. The graph shows representative example of four experiments. MMP and intracellular lactate dehydrogenase activity (cell viability) were measured in the cells treated with UR-1102 (A) or benzbromarone (B) for 4 hours. Each assay was performed in duplicate. UR, UR-1102; BZ, benzbromarone.
We then assessed the potential of UR-1102 and benzbromarone to form reactive metabolites by using radiolabeled compounds that were incubated with human liver microsome extracts in the presence of NADPH. While the amount of covalent binding to [14C]acetaminophen (as a positive control) and to [14C]benzbromarone was 134.5 and 698.1 pmol/mg human microsomal protein, respectively, that of [14C]UR-1102 was significantly lower (13.6 pmol/mg human microsomal protein) (Fig. 8). These results suggest a lower potential for reactive metabolite formation in the liver.

Covalent binding assessment of UR-1102 and benzbromarone. Each bar represents the amount of covalent binding of [14C]UR-1102 or [14C]benzbromarone in the presence of NADPH (open bar) or absence of NADPH (closed bar) (mean value of duplicate analyses).
Discussion
In the present study we identified a novel compound, UR-1102, as a potent URAT1 inhibitor that is selective over OAT1 and OAT3. In the monkey model, UR-1102 showed a better PK profile and reduced the levels of urate in plasma to a greater extent through its stronger uricosuric effect compared with benzbromarone, even when the plasma exposures were comparable, and achieved a maximum efficacy twice that of benzbromarone at a lower dose. Additionally, UR-1102 showed lower in vitro toxic potential in every mechanism that has been proposed for the hepatic toxicity of benzbromarone.
OAT1 and OAT3 are nonspecific anion transporters reported to be involved in urate excretion. Because they are both expressed in the basolateral membrane of renal proximal tubules and transport urate from the blood to inside the cells (Bakhiya et al., 2003; Ichida et al., 2003; Choi et al., 2005), inhibiting these transporters would decrease the secretion of urate into the urine. As mentioned earlier, the inability of probenecid to sufficiently lower urate (Kydd et al., 2014) suggests the hypothesis that if URAT1 could be selectively inhibited, greater urate-lowering efficacy could be achieved. However, separating the inhibitory action of a particular compound on URAT1 from that on OAT1 and OAT3 was challenging, because URAT1, OAT1, and OAT3 all belong to the SLC22A family, and the amino acid sequence homology between human URAT1 and human OAT1 or OAT3 is relatively high (45% and 41%, respectively). From our extensive compound screening on the basis of the modification of benzbromarone, we identified a potent and selective URAT1 inhibitor, UR-1102, with an inhibitory effect on URAT1 almost equivalent to that of benzbromarone and an inhibitory effect on OAT1 and OAT3 significantly weaker than that of benzbromarone (about 30- and 20-fold higher Ki, respectively).
By using UR-1102 in a monkey model, we evaluated the effect of selective URAT1 inhibition on the urate-lowering efficacy in vivo by comparing the compound with the non-selective URAT1 inhibitor benzbromarone. The PK analyses showed that the plasma exposure (AUC0–inf) of UR-1102 was similar to that of a 3-fold higher dose of benzbromarone; in addition, the inhibitory activity of UR-1102 on URAT1 in vitro was similar to that of benzbromarone, with the protein binding of both compounds to plasma of tufted capuchin monkeys at about 98% (data not shown). These results suggested that a dose of UR-1102 one-third that of benzbromarone would have the same URAT1 inhibitory activity in monkeys; in fact, both the uricosuric and urate-lowering effects of UR-1102 at 3 mg/kg, the lowest dose, were comparable to those of benzbromarone at 100 mg/kg, the highest dose, with maximum efficacy. Moreover, the maximum efficacy of UR-1102 at 30 mg/kg was significantly higher than that of benzbromarone at 100 mg/kg. Evaluation using the FEUA Emax model showed that the Emax for UR-1102 was twice that of benzbromarone. When protein binding is considered, the free concentration of plasma UR-1102 at 10–30 mg/kg or benzbromarone at 100 mg/kg is estimated to be about 2–4 μmol/l. At this concentration, benzbromarone significantly inhibited OAT1 and OAT3 but UR-1102 did not, since the Ki values of benzbromarone and UR-1102 for OAT1 are 0.22 μmol/l and 7.2 μmol/l and those for OAT3 are 0.11 μmol/l and 2.4 μmol/l, respectively. Thus, the high dose of benzbromarone would compromise the uricosuric effect induced by URAT1 inhibition, but this is probably not the case for UR-1102, which inhibits URAT1 more selectively and which showed incremental uricosuric effects even at high doses. In summary, selectively inhibiting URAT1 over OAT1 and OAT3 appears to be a valuable approach for effectively lowering urate.
In the monkey experiment, the plasma exposure of UR-1102 was clearly better than that of benzbromarone on day 1. One reason for this better PK would be the good solubility of this compound [UR-1102 can be solubilized in phosphate buffer (pH 6.8) at 970 μg/ml, whereas benzbromarone can only be solubilized at 18 μg/ml (data not shown)]. The better PK profile would also contribute to better efficacy, especially at the lower dose, and UR-1102 at 3 mg/kg exhibited both better systemic exposure and significantly greater plasma urate–lowering than benzbromarone at 3 mg/kg.
There are significant differences between species in terms of urate metabolism. In humans, as mentioned earlier, URAT1 mutations cause severe hypouricemia, and most gout patients are classified as under-excretors (Choi et al., 2005); therefore, URAT1 inhibition would be expected to cause significant urate-lowering in gout patients. On the other hand, Cebus monkeys have a certain level of uricase, and additionally their rate of urate synthesis is 7–8 times that in humans (Scott et al., 1969; Simkin, 1971). Taken together, these facts would suggest that the contribution of URAT1 to urate metabolism is smaller in tufted capuchin monkeys than in humans; that is to say, the effect of UR-1102 or benzbromarone might be slighter in this animal. Actually, benzbromarone shows a more significant uricosuric effect in patients with gout than the results obtained in this tufted capuchin monkey study; likewise, UR-1102 would be expected to show a much more significant effect in patients with gout than the greater decrease in plasma urate level it achieved in these monkeys. From these predictions and the above discussions, UR-1102 would potentially provide significant uricosuric efficacy in the clinical setting.
Recently, several transporters other than URAT1, OAT1, and OAT3 have been proposed as also involved in the control of urate metabolism, particularly by the results of genome-wide association studies of hyperuricemia (Chiba et al., 2015; Matsuo et al., 2015). Although the data shown here and the results of our global assessment of enzymes and transporters (Supplemental Table S1) show that UR-1102 is selective to URAT1, the assessment does not cover all transporters and enzymes involved in urate metabolism; therefore, it is possible that the significant effect of UR-1102 shown here may be attributable to inhibition of proteins other than URAT1, OAT1, and OAT3.
UR-1102, which is derived from the chemical structure of benzbromarone, was designed to not only improve URAT1 selectivity and solubility but also to avoid the hepatic toxicity concern of benzbromarone, which is known to be associated with hepatic injury and to have the potential to cause fulminant hepatitis in humans (Lee et al., 2008). The mechanism causing the hepatotoxicity of drugs is often unknown, but in some instances (especially in the case of idiosyncratic hepatotoxicity) the pathogenesis of toxic reactions may be the formation of a reactive metabolite that induces an immune-mediated reaction through immunogenic conjugates. One mechanism that has been proposed for the hepatotoxicity caused by benzbromarone is the formation of drug–protein adducts through a reactive metabolite (McDonald and Rettie, 2007). The propensity for a reactive intermediate to form is generally thought to be revealed by a covalent binding assessment (Ju and Uetrecht, 2002), and quantitatively, a compound with a binding affinity of over 50 pmol equivalent/mg microsomal protein is considered to have a potentially toxic profile (Evans et al., 2004). In our evaluation using the standard format, the covalent binding of UR-1102 was only 13.6 pmol equivalent/mg equivalent microsomal protein, whereas that of benzbromarone and acetaminophen was much higher (698.1 and 134.5 pmol equivalent/mg human microsomal protein, respectively) (Fig. 8). In addition to the formation of reactive metabolites, the hepatotoxicity associated with benzbromarone is also considered to be caused in part by hepatocyte mitochondrial dysfunction (Kaufmann et al., 2005; Felser et al., 2014). Although UR-1102 and benzbromarone both showed a concentration-dependent decrease in MMP other than that caused by cell injury, the 95% CI of the IC50 values of UR-1102 (258–1830 μmol/l) was much higher than that of benzbromarone (1.11–9.34 μmol/l) (Fig. 7). Taken together, these data imply that UR-1102 has a lower risk of hepatotoxicity from the standpoint of covalent binding and mitochondrial dysfunction.
In conclusion, UR-1102 is a highly selective and potent inhibitor of URAT1 and showed more potent serum urate–lowering efficacy than benzbromarone. Being a selective URAT1 inhibitor, it would contribute to meeting an unmet medical need in current gout treatment. UR-1102 is now being evaluated in Phase II clinical trials, and its pharmacological effect in patients with hyperuricemia will be revealed soon.
Acknowledgments
We thank Hitoshi Hagita (employee of Chugai Research Institute for Medical Science, Inc.) and Miho Fukasawa (employee of Chugai Pharmaceutical Co., Ltd.) for their assistance in evaluating the pharmacological profiles in the in vitro experiments; Kiyonori Kuromaru, Yasuharu Kato (employees of Chugai Pharmaceutical Co., Ltd.), Chan Hee Park, Jun Hwan Im, Soon Ok Lee, Kwang Seok Ko, Min Jung Kong, Ji Eun Lee and Yu Mi Song (employees of C&C Research Laboratories) for their assistance with overall execution of the study; Do Kyu Pyun (employee of JW Pharmaceutical Corp.) for his assistance in synthesizing UR-1102; Sally Matsuura (employee of Chugai Pharmaceutical Co., Ltd.) for her assistance with English usage; and Takaki Koga and Fumihiko Ichikawa (employees of Chugai Pharmaceutical Co., Ltd.) for helpful discussions.
Authorship Contributions
Participated in research design: Horiba, Ohtomo, Kiyokawa, Nakagawa, Lee, Ahn.
Conducted experiments: Ohtomo, Kiyokawa, Yamane, Nakagawa, Tanaka, KH Kim, BH Kim, Horiba.
Contributed new reagents or analytic tools: Ahn.
Performed data analysis: Horiba, Ohtomo, Kiyokawa, Yamane, Nakagawa, and Tanaka.
Wrote or contributed to the writing of the manuscript: Horiba, Ohtomo, Kiyokawa, Nakagawa, Ahn, Kawabe.
Footnotes
- Received December 21, 2015.
- Accepted February 11, 2016.
Primary laboratory of origin: C&C Research Laboratories.
Results of this study were presented in part as: Ahn SO, Horiba N, Ohtomo S, Lee KJ, Kim KH, Kim BH, Kiyokawa J, Yamane M, Ikegami H, Nakagawa T, et al. (2013) The therapeutic efficacy of the novel uricosuric agent UR-1102 for hyperuricemia and gout. Annual European Congress of Rheumatology; June 2013; Madrid, Spain. Abstr. no. SAT0356, The European League Against Rheumatism (EULAR), Kilchberg, Switzerland; published as Ann Rheum Dis 72 (suppl. 3):704.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- FEUA
- fractional excretion of uric acid
- HEK
- human embryonic kidney
- LDH
- lactate dehydrogenase
- MEM
- minimum essential medium
- MMP
- mitochondrial membrane potential
- OAT
- organic anion transporter
- PAH
- p-aminohippuric acid
- PK
- pharmacokinetic
- URAT1
- urate transporter 1
- USI
- urate synthesis inhibitor
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}