


Pain is an international health problem that impacts nearly one in five people worldwide. For relief of pain, an estimated 60 million people worldwide use opioids, which can lead to secondary complications of opioid use, abuse, and dependence (https://www.unodc.org/unodc/data-and-analysis/wdr2021.html). Therefore, alternative nonopioid strategies to treat pain are urgently needed.
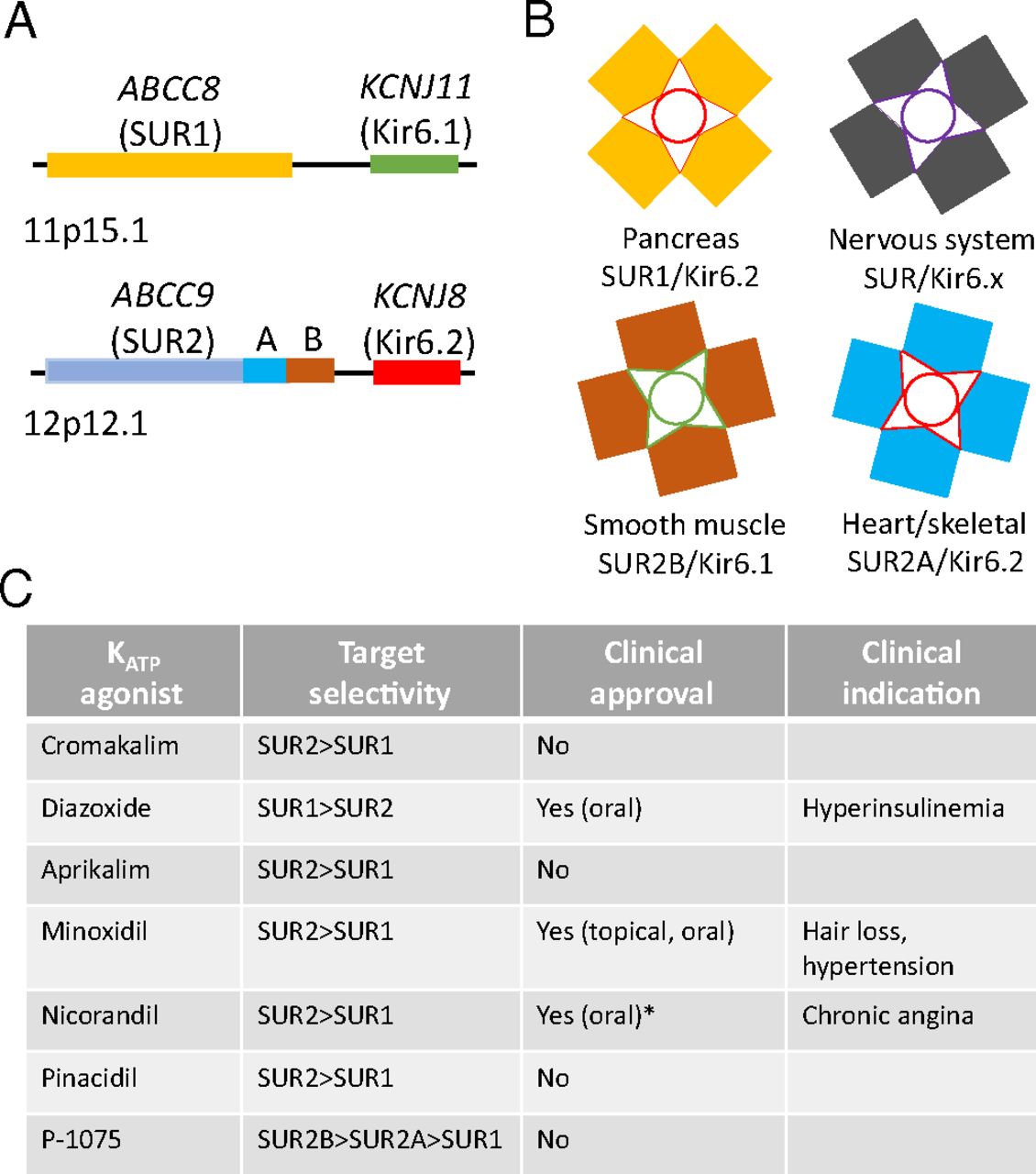
An attractive target to develop nonopioid analgesics is the adenosine triphosphate (ATP) sensitive potassium channel (KATP). The KATP channel, derived from genes encoding the channel on chromosomes 11 and 12, is a homotetramer that is structurally comprised of either Kir6.1 or Kir6.2 subunits, which complex with four sulfonylurea receptor subunits of either SUR1 or SUR2 to form a channel (Fig. 1A). Further, the combination of Kir6.x and SUR, in addition to SUR2 splice variants SUR2A and SUR2B, provide some degree of organ specificity (Fig. 1B). The KATP channel is regulated by ATP, where binding of ATP to any one of the Kir6.x subunits that compose the channel limits KATP channel opening. First discovered in the heart by Noma (1983), the KATP channel has been an interest in cardiovascular research as a therapeutic target to treat hypertension, chronic angina, diabetes, and ischemic injury. Over the years, several classes of KATP channel agonists were developed targeting the SUR subunits, with some approved for clinical use (Fig. 1C).

KATP channel structure. (A) The genes encoding the KATP channel are on chromosome 11p15.1 (ABCC8 and KCNJ11) and chromosome 12p12.1 (ABCC9 and KCNJ8), which encode SUR1, Kir6.1, SUR2, and Kir6.2, respectively. (B) The KATP channel specificity is determined by the composition of Kir6.x and SUR subtypes within the receptor. Pancreatic β cells regulate insulin release through a KATP channel composed of Kir6.2 and SUR1. In the nervous system, the KATP channel is composed of a Kir6.x subunit with SUR1 and SUR2. SUR2 has splice variants (SUR2A or SUR2B) that are differentially expressed within smooth muscle (Kir6.1 and SUR2B) and cardiac/skeletal cells (Kir6.2 and SUR2A). (C) KATP channel agonists target specific KATP channel subtypes by targeting the SUR subunit, leading to a level of selectivity. For example, the KATP channel agonist diazoxide targets SUR1 with greater affinity than SUR2. *Not approved within the United States.
In this issue of The Journal of Pharmacology and Experimental Therapeutics, Doucette et al. (2023) investigated the therapeutic efficacy of cromakalim analogs targeting the KATP channel in rodents subjected to inflammatory, nerve injury, morphine-induced hypersensitivity, and precipitated morphine withdrawal models. Cromakalim is a synthetic KATP channel agonist whose activation results in the hyperpolarization of neurons, preventing the propagation of action potentials and neurotransmitter release. Preclinical studies demonstrate that cromakalim reduces hypersensitivity in mice when delivered intrathecally or intracerebroventricularly (Asano et al., 2000; Du et al., 2011; Niu et al., 2011; Wu et al., 2011). However, cromakalim has limited solubility. In this regard, CKLP1, CKLP2, and CF3-CKLP are prodrugs of cromakalim with improved solubility that are activated by endogenous alkaline phosphatase enzymes present in the peripheral and central nervous systems. Using rodent models of pain, the authors describe that CKLP2 and CKLP1 induce antinociception in a neuropathic pain model caused by spinal nerve ligation. Further, all three compounds attenuate inflammatory mechanical hypersensitivity induced by Complete Freund’s Adjuvant. Interestingly, CKLP1 and CKLP2 reduce morphine-induced hypersensitivity and tolerance to the analgesic effect, whereas CKLP2 limits withdrawal behaviors after naloxone treatment. Importantly, the prodrug cleavage to the active form was confirmed by liquid chromatography analysis of the spinal cord. However, whether the analgesic effect of these highly aqueous cromakalim analogs is preserved when administered through intravenous, oral, or subcutaneous routes remains to be elucidated. Taken together, the manuscript by Doucette could open up fresh opportunities to develop a new line of nonopioid analgesics targeting the KATP channel.
Moreover, to fully understand the druggable nature of this target as an analgesic, the subunit composition of the KATP channel within the central and peripheral nervous system requires further investigation. Within the central nervous system, Kir6.2 and Kir6.1 have been identified, with astrocytes potentially primarily expressing Kir6.1 (Thomzig et al., 2001). However, another study identified that, after traumatic brain injury, Kir6.2 is upregulated in astrocytes, suggesting that cellular injury may change the composition of the KATP channel (Gerzanich et al., 2019). Within the peripheral nervous system, the KATP channels are composed of Kir6.2 with SUR1 or SUR2 receptors within the dorsal root ganglia, peripheral nerve fibers, glia, and Schwann cells. In dorsal root ganglion (DRG) neurons, blocking SUR1 by glybenclamide dose-dependently decreases KATP channel activity assessed by patch-clamping. This suggests that Kir6.2 with SUR1 is the predominant functional KATP channel subtype within DRG (Zoga et al., 2010). However, in a separate study after morphine treatment and spinal nerve ligation, mRNA expression of the Abcc8 gene (SUR1) decreases, whereas the Abcc9 gene (SUR2) increases 10-fold (Fisher et al., 2019), also suggesting that KATP channel expression after injury may lead to changes in KATP channel composition within the DRG. Taken together, more studies are needed to determine how KATP channel expression and function is altered with the DRG under painful stimuli or injury and with analgesic treatment. An additional understanding of the KATP channel subtypes at the single-cell level and with conditional knockouts can lead to the development of drugs selectively targeting specific cell types expressing the KATP channel for pain relief.
As some KATP channel agonists are already approved and used by humans, tremendous opportunity exists where KATP channel agonists could be repurposed as analgesics. Minoxidil is clinically approved within the United States as a topical agent to stimulate hair growth and for oral use to reduce blood pressure with refractory hypertension. If different routes of administration prove to be effective analgesics, the topical formulation could be repurposed to treat musculoskeletal pain with a known side effect of hair growth. Nicorandil, which is a nitrate and KATP channel agonist, has also been approved for nearly 40 years to treat chronic stable angina in several countries, including Japan, Australia, and most of Europe. Although nicorandil is not approved within the United States, the use in patients with cardiovascular disease suggests that nicorandil is relatively cardiac safe. Preclinically, KATP channel agonists, including nicorandil, also provide an added benefit of protecting the myocardium from ischemic injury (Mizumura et al., 1996; Gross et al., 2003). This limits the concern regarding the potential cardiovascular risks of KATP channel agonists as opposed to other analgesic targets (Bally et al., 2017). Besides more well defined roles of KATP channels, recent studies also indicate that KATP channels regulate lymphatic contractions, lymph flow, and gastrointestinal motility, suggesting that more research is needed into understanding the role of KATP channels across each organ system (York et al., 2020; Garner et al., 2021). The upside is that KATP channel agonists are already safely used in humans, which can lead to an opportunity for a rapid translation of KATP channel agonists for general analgesic use.
Footnotes
- Received April 17, 2023.
- Accepted May 25, 2023.
This work is supported by funding from National Institutes of Health National Institute of General Medical Sciences [Grant GM119522] (to E.R.G.) and FAPESP 21/14831-8 and 18/19332-7 (V.O.Z.).
Eric Gross is a consultant for Chiima Therapeutics and an Associate Editor for JPET.
- Copyright © 2023 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}