


Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease defined by a chronic elevation in pulmonary arterial pressure with extensive pulmonary vascular remodeling and perivascular inflammation characterized by an accumulation of macrophages, lymphocytes, dendritic cells, and mast cells. Although the exact etiology of the disease is unknown, clinical as well as preclinical data strongly implicate a role for serotonin (5-HT) in the process. Here, we investigated the chronic effects of pharmacological inhibition of tryptophan hydroxylase 1 (TPH1), the rate-limiting enzyme in peripheral 5-HT biosynthesis, in two preclinical models of pulmonary hypertension (PH), the monocrotaline (MCT) rat and the semaxanib (SUGEN, Medinoah, Suzhou, China)-hypoxia rat. In both PH models, ethyl (S)-8-(2-amino-6-((R)-1-(5-chloro-[1,1′-biphenyl]-2-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylate and ethyl (S)-8-(2-amino-6-((R)-1-(3′,4′-dimethyl-3-(3-methyl-1 H-pyrazol-1-yl)-[1,1′-biphenyl]-4-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylate, novel orally active TPH1 inhibitors with nanomolar in vitro potency, decreased serum, gut, and lung 5-HT levels in a dose-dependent manner and significantly reduced pulmonary arterial pressure, and pulmonary vessel wall thickness and occlusion in male rats. In the MCT rat model, decreases in lung 5-HT significantly correlated with reductions in histamine levels and mast cell number (P < 0.001, r2 = 0.88). In contrast, neither ambrisentan nor tadalafil, which are vasodilators approved for the treatment of PAH, reduced mast cell number or 5-HT levels, nor were they as effective in treating the vascular remodeling as were the TPH1 inhibitors. When administered in combination with ambrisentan, the TPH1 inhibitors showed an additive effect on pulmonary vascular remodeling and pressures. These data demonstrate that in addition to reducing vascular remodeling, TPH1 inhibition has the added benefit of reducing the perivascular mast cell accumulation associated with PH.
Introduction
Pulmonary arterial hypertension (PAH) is a progressive and incurable disorder characterized by increases in pulmonary vascular resistance, which partly results from endothelial dysfunction leading to pulmonary arterial smooth muscle cell (PA-SMC) proliferation, decreased apoptosis, and vasoconstriction of the small pulmonary arteries (Tuder et al., 2013; Huertas et al., 2014; Rabinovitch et al., 2014; Galiè et al., 2015; Guignabert et al., 2015). The present treatment of patients with PAH includes the use of prostanoids, phosphodiesterase type-5 inhibitors, and endothelin receptor antagonists (ERAs), which target the prostacyclin, endothelin-1, and nitric oxide pathways aimed at inducing relaxation of the pulmonary arteries and decreasing pulmonary vascular remodeling (Humbert et al., 2004, 2014). These current medical treatments only partially improve symptoms, and lung transplantation remains an important treatment in eligible patients with severe PAH refractory to medical management (Humbert et al., 2010a,b).
Although pulmonary vascular remodeling is characterized by complex interactions of multiple cell types and signaling molecules (Huertas et al., 2014; Guignabert et al., 2015), serotonin (5-HT) has long been associated as a major factor in the development of PAH. Indeed, numerous preclinical studies have demonstrated genetic, molecular, functional, and pathologic links between increased 5-HT levels and the development of the disease, supporting the concept that the serotoninergic system could open new avenues for targeted therapy in PAH. However, the wide and often overlapping distribution of the 15 known receptors, coupled with either synergistic or opposing action upon receptor activation by 5-HT, underlines the complexity of the serotoninergic system and the difficulty in developing effective 5-HT receptor antagonists. In addition, circulating 5-HT levels are also regulated by the cell surface 5-HT transporter (SERT) (Rhodes et al., 2009; Thomas et al., 2013; Tu and Guignabert, 2013), which activates a number of intracellular signaling cascades linked to the pathogenesis of PAH. Despite the wealth of evidence that links 5-HT signaling to many of the pathologic pathways and processes characteristic of PAH, the trials undertaken with 5-HT receptor inhibitors have been discouraging. Given the sheer volume of studies implicating multiple 5-HT receptors in the different areas of PAH pathology and the potential adverse consequences of displacing 5-HT from one receptor, thereby increasing the availability of 5-HT to another, it is perhaps not surprising that the clinical data generated thus far with 5-HT receptor antagonists is less than inspiring.
To circumvent these limitations, our approach seeks to interfere with the systemic 5-HT pathway by reducing the ligand through the use of new small molecules targeting specifically the rate-limiting enzyme in peripheral 5-HT biosynthesis, called L-tryptophan hydroxylase 1 (TPH1). Tryptophan hydroxylases are members of a family of pterin-dependent aromatic amino acid hydroxylases that catalyze a biopterin-dependent mono-oxygenation reaction using (6R)-L-erythro-5,6,7,8,-tetrahydropterin as a cofactor and molecular oxygen to hydrolyze L-tryptophan or tryptophan to 5-hydroxytryptophan. Tryptophan hydroxylases exist as two isoforms, TPH1 and tryptophan hydroxylase 2, which are encoded by separate genes. TPH1 is expressed mainly in the enterochromaffin cells of the intestinal mucosa, whereas tryptophan hydroxylase 2 is only expressed in neurons. Since 5-HT does not cross the blood-brain barrier, the brain and systemic 5-HT systems are separated and have their own distinct tryptophan hydroxylase isoforms (Walther et al., 2003). Other researchers have previously demonstrated that both genetic and chemical TPH1 ablation prevents hypoxia-induced lesions in murine models of PAH (Abid et al., 2012; Ciuclan et al., 2013). Here, we investigated the chronic effects of pharmacological inhibition of TPH1 in two preclinical models of pulmonary hypertension (PH), the monocrotaline (MCT) rat and the semaxanib (SUGEN, Medinoah, Suzhou, China)-hypoxia rat.
Materials and Methods
Recombinant TPH1 Enzyme Assay.
Full-length human recombinant tryptophan 5-hydroxylase enzyme-1 with a maltose-binding protein tag was expressed in Escherichia coli BL21 and purified using an amylose resin column. The inhibitory effects of two ester prodrugs, ethyl (S)-8-(2-amino-6-((R)-1-(5-chloro-[1,1′-biphenyl]-2-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylate (KAR5585) and ethyl (S)-8-(2-amino-6-((R)-1-(3′,4′-dimethyl-3-(3-methyl-1 H-pyrazol-1-yl)-[1,1′-biphenyl]-4-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylate (KAR5416), and their active forms, (S)-8-(2-amino-6-((R)-1-(5-chloro-[1,1′-biphenyl]-2-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylic acid (KAR5417) and (S)-8-(2-amino-6-((R)-1-(3′,4′-dimethyl-3-(3-methyl-1 H-pyrazol-1-yl)-[1,1′-biphenyl]-4-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylic acid (KAR5395), respectively, against the purified recombinant enzyme were tested in a continuous fluorescence assay at compound concentrations ranging from 0.0001 to 20 µM using protein-free incubation media containing tryptophan (substrate) and methyltetrahydropterin (cofactor) at their Km concentrations (60 and 72 µM, respectively). Fluorescence was measured over time on a SpectraMax 5 fluorometer and the IC50 value for each compound was determined. Competition assays against the TPH1 substrate, L-tryptophan, were determined using a [3H]-H2O release assay described by Vrana et al. (1993). The concentration of methyltetrahydropterin (cofactor) was held constant at 100 µM and the substrate was diluted in an assay buffer containing 50 mM 2-(N-morpholino)ethanesulfonic acid, 200 mM ammonium sulfate, 7 mM dithiothreitol, 50 µg/ml catalase, and 25 µM iron ammonium sulfate hexahydrate in water. Compound was added and the reaction was initiated by adding 0.5 µCi of L-[5-3H(N)]-tryptophan to each well. After proceeding at room temperature for 60 minutes the reaction was quenched with 300 µl of 7.5% activated charcoal in 1 N HCl. The reaction mixture was centrifuged at 13,000 rpm for 20 minutes at room temperature. Supernates were collected and the production of [3H]-H2O was measured on a liquid scintillation counter.
Cell-Based Assays.
Flp-in human embryonic kidney cells (Flp-in293) (Life Technologies, Carlsbad, CA) stably transfected with human TPH1-expression plasmid (pcDNA5/FRT/HuTPH-1) were incubated in Dulbecco’s modified eagle medium containing 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, and 150 µg/ml hygromycin for 24 hours in 96-well poly-d-lysine plates. The medium was changed to opti-MEM (ThermoFisher Scientific, Illkirch, France) containing 200 µM methyltetrahydropterin with either dimethylsulfoxide or TPH1 inhibitors for 24 hours. Incubations were conducted in the presence and absence of 1% bovine serum albumin. Following overnight incubation, plates were centrifuged and 100 µl of medium was extracted in 100 µl of high-performance liquid chromatography (HPLC) homogenization buffer for measurement of 5-hydroxytrpyptophan via HPLC fluorometric detection as previously described (Liu et al., 2008).
Pulmonary Arterial Smooth Muscle Cell Proliferation.
Lung specimens were obtained at the time of lung transplantation from patients with idiopathic PAH or obtained from patients without any evidence of pulmonary vascular disease who underwent lobectomy or pneumonectomy for localized lung cancer with the normal tissue collected at a distance from the tumors (Marie Lannelongue Hospital, Le Plessis-Robinson, France). The samples were de-identified and may have included tissues from both male and female patients. This study was approved by the local ethics committee (CPP Ile-de-France VII, Paris) and all patients signed a written, informed consent. Human pulmonary endothelial cells (P-ECs) and PA-SMCs were isolated and cultured as previously described (Tu et al., 2011, 2012; Ricard et al., 2014; Huertas et al., 2015; Le Hiress et al., 2015). P-ECs cultured in MCDB131 (ThermoFisher Scientific) medium were seeded in 6-well plates at a density of 150,000 cells/well and allowed to adhere. After being starved overnight in MCDB131 medium containing 0.3% fetal calf serum, the cells were treated with vehicle (dimethylsulfoxide), 0.1, 0.5, or 1 μM KAR5417 for 24 hours at 37°C. Conditioned media were collected, snap frozen, and stored at −80°C. Human PA-SMCs cultured in Dulbecco’s modified eagle medium supplemented with 10% fetal calf serum were seeded in 96-well plates at a density of 4000 cells/well and allowed to adhere. After being subjected to growth arrest for 48 hours in medium lacking fetal calf serum, the PA-SMCs were treated overnight with 100 µl of the conditioned P-EC media. PA-SMC proliferation was assessed by measuring 5-bromo-2-deoxyuridine (BrdU) incorporation using a DELFIA kit (Perkin Elmer, Shelton, CT). Briefly, PA-SMCs were labeled with 10 µl of BrdU labeling medium for 2 hours at 37°C. After the cells were fixed in 100 µl of fixing solution for 30 minutes at room temperature, they were incubated with 100 µl of an anti-BrdU/5-ethynyl uridine working solution for 2 hours at room temperature. Cells were washed in buffer and incubated with 200 µl of a DELFIA inducing agent on a plate shaker for 15 minutes at room temperature. BrdU incorporation was determined by measuring 5-ethynyl uridine fluorescence in a time-resolved fluorometer.
Animals.
Male Sprague-Dawley rats (175–200 g, Harlan, Frederick, MD) were maintained on a 12-hour light/dark cycle with free access to water and an irradiated rodent diet with 18% protein (Envigo, Indianapolis, IN). Rats were orally dosed with vehicle (0.5% methyl cellulose), KAR5585 or KAR5416, tadalafil (Cialis, Eli Lilly, Indianapolis, IN, 10 mg/kg), a phosphodiesterase type-5 inhibitor, ambrisentan (Letairis, Gilead Sciences, Foster City, CA, 10 mg/kg), an ERA, or a combination of KAR5585 with either tadalafil or ambrisentan. Body weight was recorded weekly in all studies and demonstrated no significant compound effect compared with vehicle (data not shown). Blood and tissues were collected at time of sacrifice for biochemical and histologic measurements. Study protocols were approved by the Institutional Animal Care and Use Committee and all animals received humane treatment according to the guidelines of the National Institutes of Health (Publication No. 85-23, revised 1996; https://grants.nih.gov/grants/olaw/Guide-for-the-Care-and-Use-of-Laboratory-Animals_Prepub.pdf).
PAH Induction.
PAH induction in the MCT prevention rat model was accomplished with a single 60 mg/kg subcutaneous injection of MCT (Oakwood Products, Estill, SC) at which time oral administration of compound was initiated and continued for 28 days. A treatment modality for PAH was conducted in the SUGEN-hypoxia rat model. PAH induction was accomplished with a single 20 mg/kg subcutaneous injection of semaxanib (SUGEN) in conjunction with continuous exposure to 10% oxygen in a hypobaric chamber that simulated 17,000 feet of altitude for 3 weeks. Animals were removed from chambers and a subgroup was sacrificed for baseline measurements of disease induction. The remaining animals were orally administered compound for an additional 5 weeks under normoxic conditions.
5-HT, 5-Hydroxyindoleacetic Acid (5-HIAA), and Histamine.
At time of sacrifice, rats were anesthetized with isofluorane administered 2% in oxygen continuous via inhalation. Blood was collected and serum isolated by centrifugation at 6000 rpm for 20 minutes. Following whole body perfusion in situ with 10 ml phosphate-buffered saline, intestine, brain, and lung tissue samples were collected. Serum (50 µl) and tissue (50–75 g) were extracted in 100 and 300 µl, respectively, of HPLC homogenization buffer containing 0.1 M sodium acetate, 20 mM sodium bisulfate, 0.3 M trichloroacetic acid, 10 mM EDTA, and 50 mM ascorbic acid. Homogenates were centrifuged at 13,000 rpm for 30 minutes at 4°C. 5-HT and 5-HIAA was measured in the supernates using a Perkin Elmer Flexar HPLC system equipped with a fluorescent detector and a Brownlee validated aqueous C18 column (3 µm, 50 × 2.1 mm; Perkin Elmer) as described previously (Liu et al., 2008). The mobile phase used was 100 mM sodium acetate, pH 3.5. Detection was achieved at an excitation wavelength of 280 nm and an emission wavelength of 330 nm. Lung histamine was measured in lung tissue by enzyme-linked immunosorbent assay (Immuno-Biologic Laboratories, Minneapolis, MN) and the absorbance was read on a spectrophotometer at 450 nm.
Urinary 5-HIAA.
MCT-treated rats orally dosed once daily for 28 days with either vehicle or KAR5585 were placed in metabolic cages on day 26. Urine was collected over an 18-hour period into tubes containing 200 µl of 6 N HCl. Rats were allowed free access to food and water ad libitum during the collection period. Urinary 5-HIAA was measured by enzyme-linked immunosorbent assay (ALPCO, Salem, NH) and normalized to creatinine, which was measured by colorimetric assay (R&D Systems, Minneapolis, MN).
Mean Pulmonary Arterial Blood Pressure.
Rats were anesthetized with isofluorane administered 2% in oxygen continuous via inhalation and the trachea was intubated. Mean pulmonary arterial blood pressure measurements were made by inserting a Millar Mikro-tip pressure catheter (Millar, Houston, TX) into a small hole in the right ventricle 1–2 mm below the inception of the pulmonary artery. The catheter was advanced into the pulmonary artery and continuous pulmonary blood pressure measurements were recorded, using an ADInstruments (Colorado Springs, CO) Powerlab 8/35 data collection device with LabChart software, and were determined as the top of the pressure recording over a 1- to 3-minute period during which pressure recordings remained stable.
Echocardiography.
Rats were anesthetized with isofluorane administered 2% in medical air continuous via inhalation and the thorax and abdominal areas were shaved. Micro-ultrasound (Visual Sonics, Toronto, Canada) was used to assess pulmonary hemodynamics. Pulse-wave Doppler of pulmonary outflow was recorded in the parasternal short-axis view at the level of the aortic valve to measure the pulmonary artery velocity-time integral and pulmonary valve acceleration time.
Histologic Measurements.
Whole body perfusion was accomplished with phosphate-buffered saline and the left lobe of the lung was removed, perfusion fixed in 10% neutral buffered formalin, processed, embedded in paraffin, and sectioned at 5 µm for histologic analysis. For each animal, a representative cross section through the central portion of the left lobe including the main pulmonary artery was stained with Elastica van Gieson (Polyscientific, Bay Shore, NY). A digital camera attached to a light microscope was then used to capture 50 pulmonary vessels at a magnification of 40×. The outer and inner perimeters of the vessel walls were delineated using Image-Pro software (Rockville, MD) and the relative vessel wall thickness was calculated as [(outer vessel wall circumference) − (inner vessel wall circumference)]/(outer vessel wall circumference). The percent of vessels that were occluded (relative vessel wall thickness ≥70%) for each animal was also calculated. Representative cross sections were also stained for mast cells with toluidine blue (Sigma, St. Louis, MO). The number of mast cells per mm2 area was determined using Image-Pro software in 25 images (10× magnification) of the lung captured with a digital camera attached to a light microscope.
TPH1 mRNA Expression.
RNA isolated from 20 to 30 mg of tissue from the right lung was homogenized in 20× volume of T-PER (Thermo Scientific, Waltham, MA) with 1× HALT protease/phosphatase Inhibitor (Thermo Scientific) using the PureLink RNA Mini kit (Ambion, Rockford, IL). One microgram of RNA was subjected to reverse transcription using SuperScript III First-Strand Synthesis Super Mix (Life Technologies). Transcript quantitation was performed using Taqman polymerase chain reaction primer/probe sets from Life Technologies [TPH1 (Rn01476867_m1) and glyceraldehyde-3-phosphate dehydrogenase endogenous control (4352338E)]. Relative gene expression was determined using glyceraldehyde-3-phosphate dehydrogenase as the endogenous control and the 2−ΔΔCT method.
Statistical Analysis.
Analysis of variance was used to test for significant differences between groups. Post-hoc Bonferroni multiple comparison test analysis was used to determine significant differences among the mean values. All statistical analyses were accomplished using Graph Pad Prism 5 software (La Jolla, CA).
Results
Inhibition of Recombinant TPH1.
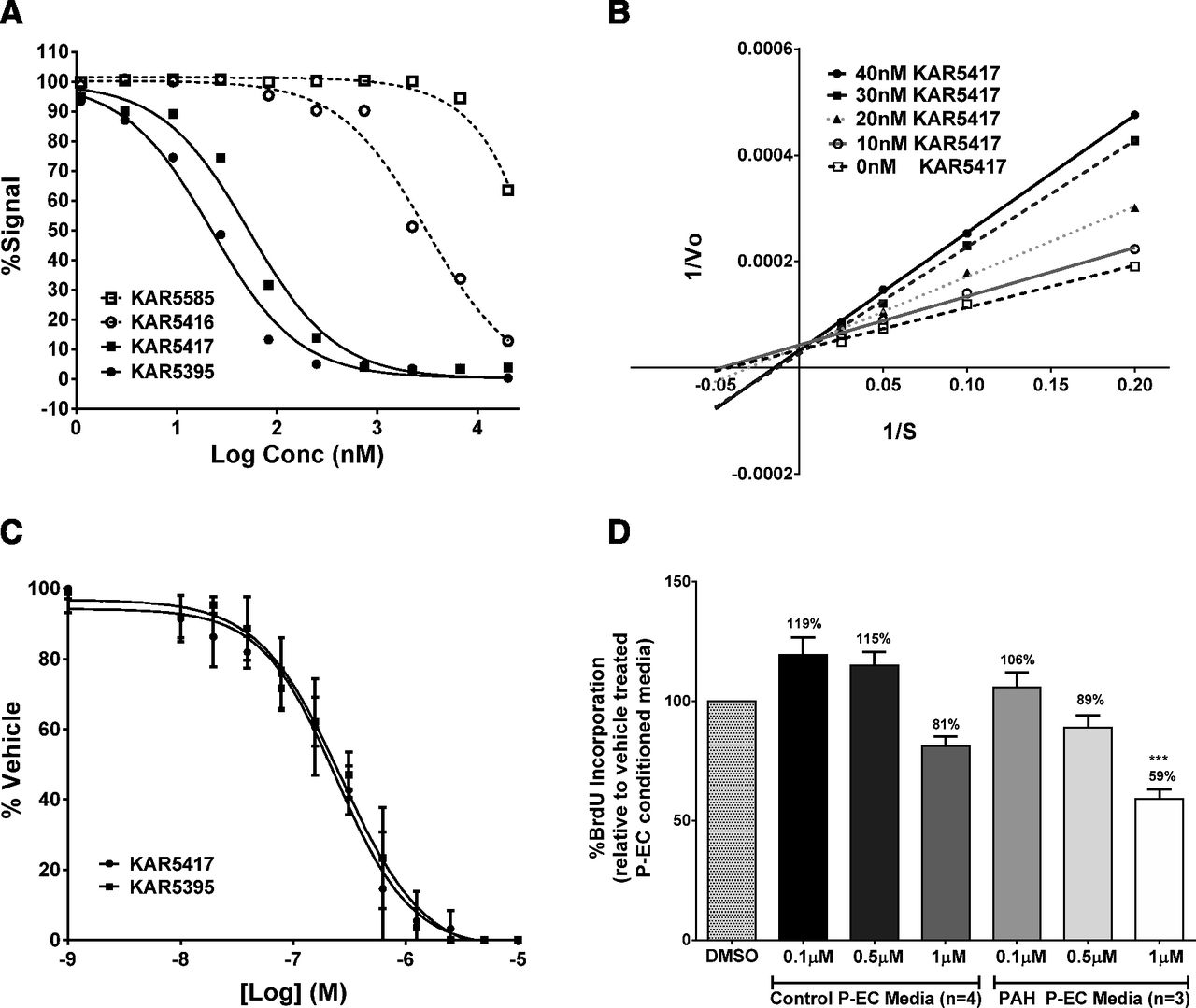
The small molecule TPH1 inhibitors used in these studies are prodrugs designed to have increased bioavailability and absorption. Once absorbed, KAR5585 and KAR5416 are rapidly converted in vivo through endogenous esterase activity to their active forms, KAR5417 and KAR5395, respectively. The potency, selectivity, pharmacokinetic properties, and chemical structures have been previously reported (De Lombaert et al., 2015b, 2016). Although the prodrugs show no activity against human recombinant TPH1 (Fig. 1A), as shown in Fig. 1B, the active form of KAR5585 (KAR5417) is competitive with tryptophan. The IC50 values for KAR5417 and KAR5395 were 52.4 and 22.1 nM, respectively. In a cell-based system using human embryonic kidney cells (Flp-in 293) stably overexpressing human TPH1, the IC50 values were 296 ± 75 and 223 ± 46 nM for KAR5417 and KAR5395, respectively (Fig. 1C).

(A) Inhibition of purified human recombinant TPH1 by KAR5585, KAR5417, KAR5416, and KAR5395 was tested in a continuous fluorescence assay using human recombinant TPH1. (B) A [3H]-H2O release assay was used to determine the competitiveness of KAR5417 against L-tryptophan. (C) 5-Hydroxytrpyptophan was measured in the medium of F293hTPH1 cells incubated 18 hours without bovine serum albumin in the presence of various concentrations of KAR5417 and KAR5395. The data represent three different experiments and samples were run in duplicate. (D) Human P-ECs isolated from normal control patients or patients with idiopathic PAH were treated with dimethylsulfoxide (DMSO) or 0.1, 0.5, and 1 µM KAR5417 for 24 hours. Conditioned P-EC media were removed and added to human PA-SMCs isolated from normal control patients. Following incubation, PA-SMC proliferation was assessed by BrdU incorporation and calculated as a percent of DMSO vehicle-treated P-EC conditioned media. Data are expressed as mean ± S.E.M. and samples from each patient were run in duplicate. ***P < 0.001 significance relative to conditioned media.
Inhibition of Human PA-SMC Proliferation.
Direct effects of endothelial TPH1 inhibition on human PA-SMC proliferation were evaluated using P-ECs derived from PAH patients. Human P-ECs were cultured for 24 hours with or without KAR5417 and the conditioned media collected after culture was incubated with human PA-SMCs obtained from control patients. BrdU incorporation was used as a measure of cell proliferation. When serum-free medium from cultured P-ECs treated with KAR5417 (from either control subjects or patients with PAH) was added to PA-SMCs, a dose-dependent inhibition of smooth muscle cell proliferation was observed. As shown in Fig. 1D, the response to KAR5417 was more pronounced using conditioned media from P-ECs from PAH patients compared with those from control patients. These data demonstrated that KAR5417, the active form of KAR5585, inhibits TPH1 in human P-ECs, thereby reducing the mitogenic paracrine effects of P-EC-produced 5-HT on PA-SMCs. These data are also consistent with literature reporting higher expression of TPH1 in P-ECs from patients with idiopathic PAH (Eddahibi et al., 2006).
Serum, Brain, and Gut 5-HT.
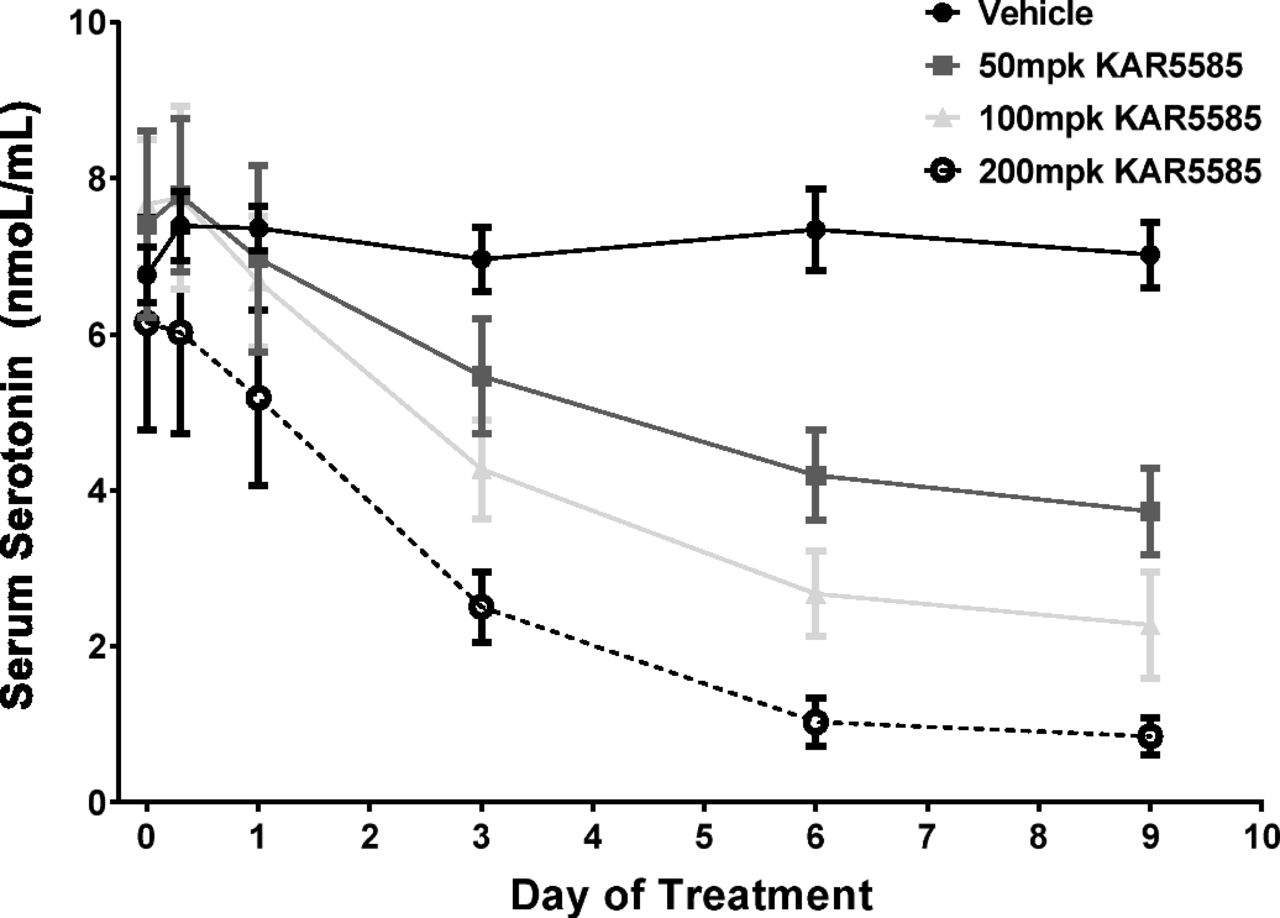
Changes in circulating 5-HT levels following TPH1 inhibition are not only dependent on the production of 5-HT through TPH1, but are also affected by the release of 5-HT stored in the gut as well as by the uptake and clearance of 5-HT by platelets that have a half-life of 3–5 days (Siegal et al., 1989). A time course to monitor changes in circulating levels of 5-HT following administration of KAR5585 was performed. As shown in Fig. 2, KAR5585 reduced serum 5-HT in both a time- and dose-dependent manner, with the maximum inhibition occurring after 7 days of treatment.

Serum 5-HT was measured in blood samples collected from naive rats at T = 0 and 8 hours, and 1, 2, 3, 6, and 9 days after oral dosing with vehicle or KAR5585 (50, 100, or 200 mg/kg) (n = 8/group) once daily for 14 days. Data are expressed as mean ± S.E.M.
To determine the effects of TPH1 inhibition on systemic and central 5-HT metabolism in rat models of PAH, steady-state levels of 5-HT and its metabolite, 5-HIAA, were measured in intestine and brain tissue from MCT-treated rats orally dosed with KAR5585 for 28 days. The significant (P < 0.05) 78% reduction in serum 5-HT (Fig. 3A) corresponded to a 40% decrease in the systemic production of 5-HT, as measured by 24-hour 5-HIAA urinary output (Fig. 3B). In mucosal tissue where the majority of systemic 5-HT is synthesized, reductions in both 5-HT (Fig. 3C) and 5-HIAA were observed following oral administration of KAR5585. The altered 5-HIAA/5-HT ratio indicates that the increase in the ratio of the product, 5-HIAA, divided by the substrate, 5-HT, was largely due to a 90% reduction in intestinal 5-HT levels (Fig. 3D). In contrast, no changes in either 5-HT (Fig. 3E) or 5-HIAA (Fig. 3F) levels were observed in brain tissue. The lack of detectable changes in central 5-HT metabolism is consistent with the inability of these large molecular weight TPH1 inhibitors (molecular weight >550 Da) to penetrate the blood-brain barrier. These data were also consistent with the lack of any detectable levels of active compound in brain tissue following oral and intravenous administration (data not shown).

Serum, mucosa, and brain tissue were obtained from rats that received a subcutaneous 60 mg/kg injection of MCT. Daily oral administration of vehicle or KAR5585 (100 or 200 mg/kg) was initiated at the time of PAH induction and continued for 28 days. 5-HT and 5-HIAA were measured in tissue samples obtained at sacrifice. Urinary 5-HIAA corrected for creatinine was collected over 24 hours between days 26 and 28. Data represent mean ± S.E.M. **P < 0.01 and ***P < 0.001 significance relative to MCT-treated vehicle controls.
TPH1 Inhibition in the SUGEN-Hypoxia Rat Model of PH.
To evaluate the ability of TPH1 inhibition to treat established PH, we compared KAR5585, tadalafil, and ambrisentan in a SUGEN-hypoxia rat model. Oral administration of compounds was initiated 21 days after induction of PH by a combination of treatment with the vascular endothelial growth factor receptor inhibitor, semaxanib (SUGEN), and chronic exposure to hypoxia (11% O2) in hypobaric chambers. At the end of the 21-day PH progression period, rats were removed from hypoxic conditions and orally administered KAR5585, ambrisentan alone, KAR5585 + ambrisentan, or ambrisentan + tadalafil for an additional 28 days at Denver, CO, altitude.
As shown in Fig. 4, baseline hemodynamic and histologic measurements taken at 3 weeks in a subgroup of SUGEN-hypoxia rats demonstrated severe induction of the disease as exhibited by increases in mean pulmonary arterial pressure (mPAP) and pulmonary arterial pulse pressure and marked reductions in the velocity-time integral indicating deterioration of pulmonary hemodynamics. The hemodynamic and histologic parameters were significantly improved following treatment with a suboptimal dose of 100 mg/kg KAR5585 in combination with 10 mg/kg ambrisentan. The combination of KAR5585 with ambrisentan significantly reduced elevations in mPAP (Fig. 4A) and mean pulmonary pulse pressure (Fig. 4B), and improved the velocity-time integral (Fig. 4C). As a single agent, KAR5585 demonstrated a dose-responsive reduction in pulmonary vascular remodeling with a maximal response occurring at 200 mg/kg, and when combined with ambrisentan at a suboptimal dose of 100 mg/kg it showed significant improvement compared with treatment with the gold standard combination of ambrisentan and tadalafil (Fig. 4D).
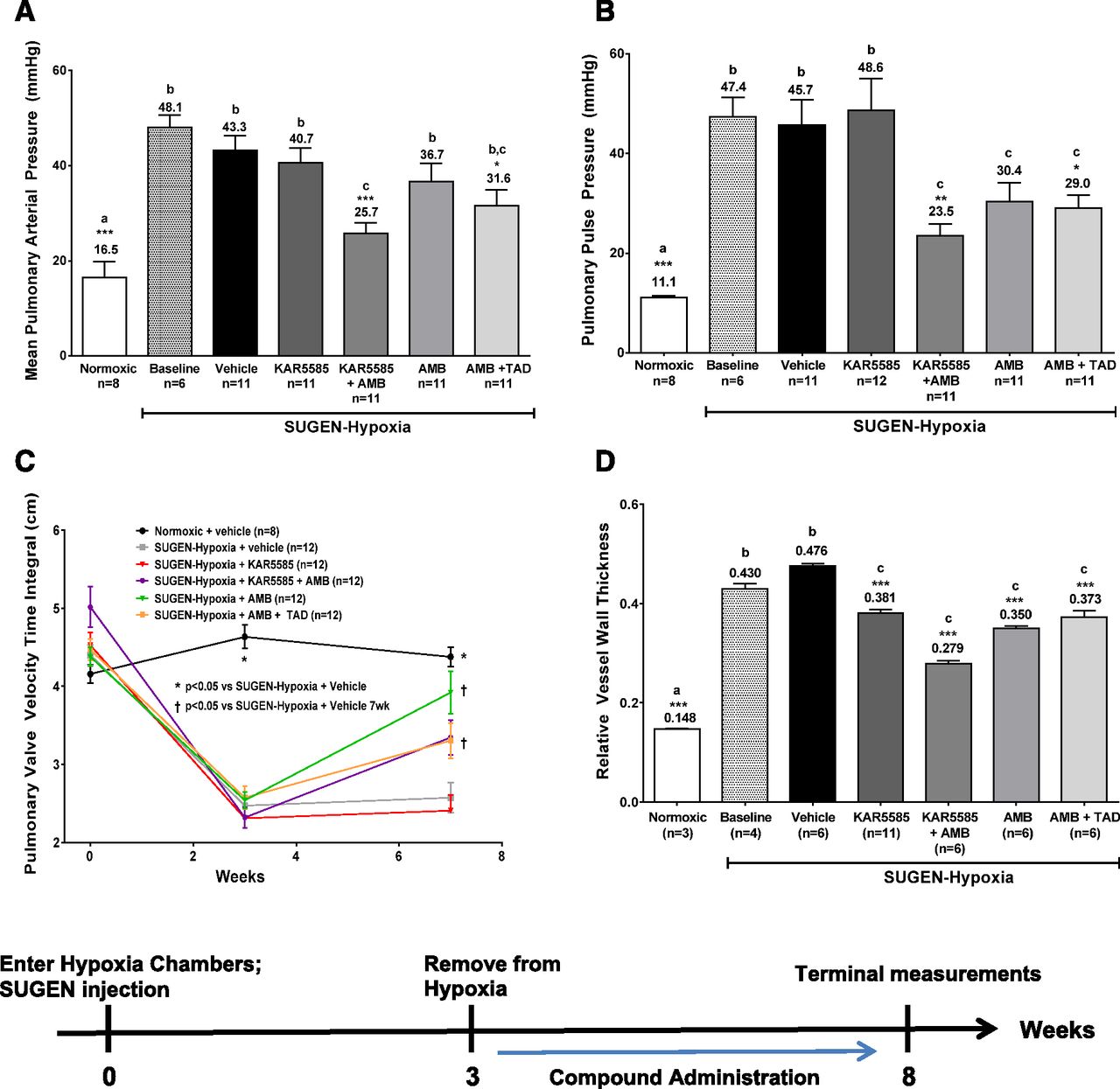
Measurements of (A) pulmonary vessel occlusions, (B) mean pulmonary arterial blood pressure (mPAP), (C) pulmonary pulse pressure, and (D) pulmonary valve velocity-time integral were determined in a PAH treatment model. PAH induction was accomplished with a single 20 mg/kg subcutaneous injection of SUGEN in conjunction with continuous exposure to 10% oxygen in a hypobaric chamber simulating 17,000 feet of altitude for 3 weeks. Administration of 100 mg/kg KAR5585, 10 mg/kg ambrisentan (AMB), 10 mg/kg AMB + 10 mg/kg tadalafil (TAD), or a combination of 100 mg/kg KAR5585 + 10 mg/kg AMB was initiated at baseline (week 3) and continued once daily for an additional 5 weeks under normoxic conditions. *P < 0.05, **P < 0.01, and ***P < 0.001 significance relative to SUGEN hypoxia–treated vehicle controls. Lower-case letters (a, b, and c) represent the mean values, which are significantly different (P < 0.01) from each other.
TPH1 Inhibition in the MCT Rat Model of PH.
The MCT rat model of PH was used to further differentiate the mechanism of TPH1 inhibition from other PAH treatments. TPH1 inhibition demonstrated its advantages on affecting the inflammatory response, which precedes the onset of cell proliferation in the development of the disease. Treatment with KAR5585 markedly limited the remodeling and occlusion of the pulmonary microvasculature in a dose-dependent manner (Fig. 5). In this model, the extent of pulmonary vessel wall remodeling was less in rats treated with KAR5585 compared with rats treated with ambrisentan (ERA) alone, apparently due to proliferation and hypertrophy of the medial layers in the pulmonary microvasculature (Fig. 5).

Analyses of (A) vessel wall thickness and (B) occluded vessels were determined in a prevention rat model of PAH. Daily oral administration of vehicle; 50, 100 and 200 mg/kg KAR5585 (n = 5); 10 mg/kg ambrisentan [(AMB) n = 5]; or a combination of 100 mg/kg KAR5585 + AMB (n = 5) was initiated at the same time as induction of PAH by MCT injection and continued for 28 days. (C) Cross sections of perfused, formalin-fixed lung tissue were stained with Elastica van Gieson (Polyscientific) in MCT-treated rats orally dosed with i) vehicle, ii) 200 mg/kg KAR5585, or iii) untreated, normal controls (40× magnification; scale = 20 µM). ***P < 0.001 significance to MCT-treated vehicle controls.
A second MCT rat study examined the effects of a suboptimal dose of 100 mg/kg KAR5585 alone or in combination with the phosphodiesterase type-5 inhibitor, tadalafil. Reductions in lung 5-HT (Fig. 6A) following treatment with KAR5585 were associated with decreases in both pulmonary mast cell number (Fig. 6B) and occlusions of the pulmonary microvasculature (Fig. 6C). In contrast, tadalafil had no effect on pulmonary mast cell number and lung 5-HT levels. The combination of KAR5585 and tadalafil appeared to have an additive effect on limiting pulmonary vascular remodeling (Fig. 6D). Reductions in toluidine-blue positive mast cells (Fig. 6E) were significantly correlated (P < 0.05) with the reductions in lung 5-HT.

Decreases in (A) 5-HT, (B) mast cell number, (C) vessel occlusion, and (D) vessel wall thickness were observed in a prevention rat model of PAH. Daily oral administration of vehicle, 100 mg/kg KAR5585, 10 mg/kg tadalafil (TAD), or a combination of 100 mg/kg KAR5585 + 10 mg/kg TAD was initiated at the time of PAH induction by MCT injection and continued for 28 days. The number of mast cells per mm2 area was determined in 25 fields (10× magnification scale = 100 µM) within a single lung cross section. Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.01, and ***P < 0.001 significance relative to MCT vehicle controls. Lower-case letters (a, b, and c) represent the mean values, which are significantly different (P < 0.01) from each other. (E) Photomicrographs showing mast cell staining in lung tissue from MCT-treated rats. Mast cells are indicated by dark violet staining in rats treated with i) vehicle, ii) 100 mg/kg KAR5585, iii) 10 mg/kg TAD, or iv) 100 mg/kg KAR5585 + 10 mg/kg TAD (10× magnification; scale = 100 µM).
To compare the relative efficacy of TPH1 inhibition on development and treatment of PAH, the effect of TPH1 inhibition was examined in the MCT progression model and the SUGEN-hypoxia treatment model using a second orally active TPH1 inhibitor, KAR5416. In the MCT model, rats were orally dosed 1× daily for 21 days with 100 mg/kg KAR5416 at the time of MCT induction. In the SUGEN-hypoxia model, dosing with KAR5416 began 1 week after rats were removed from hypoxic chambers and dosing continued for 28 days under normoxic conditions. In both models, administration of KAR5416 resulted in significant (P < 0.05) improvements in hemodynamic measurements of mPAP and total pulmonary vascular resistance (Fig. 7). These changes were associated with significant reductions in ventricular hypertrophy and pulmonary vascular occlusion.

Significant decreases in mPAP, total pulmonary vascular resistance, right ventricular hypertrophy, and vessel wall thickness were observed in both the (A) semaxanib (SUGEN)-hypoxia treatment rat model and (B) a MCT prevention rat model following oral administration of 200 mg/kg KAR5416. Data are expressed as mean ± S.E.M. *P < 0.05, *P < 0.01, and ***P < 0.001 significance relative to vehicle controls.
Lung TPH1 Expression.
The pulmonary vascular endothelium, a metabolically active tissue, serves as an important site of injury in experimental models of PH. There are multiple mechanisms by which circulating 5-HT and TPH1 activity in pulmonary tissue could predispose rats to PH. Increases in TPH1 expression were first observed on day 14 after induction of PH by MCT administration and paralleled the accumulation of lung 5-HT (Fig. 8A) and histamine levels (Fig. 8B). Treatment with a TPH1 inhibitor resulted in reductions in both toluidine-blue positive-stained mast cells (Fig. 8C) and 5-HT levels in the lung, whereas TPH1 mRNA expression levels remained elevated (Fig. 8D). These data are consistent with previous reports of increased TPH1 expression in pulmonary vascular cells of PAH patients (Eddahibi et al., 2006). In the SUGEN-hypoxia rat model, we found no elevation in lung 5-HT levels, mast cell numbers, or the expression of genes associated with the 5-HT system (data not shown).

Time-dependent changes in lung (A) 5-HT, (B) histamine, (C) TPH1 mRNA expression, and (D) mast cell numbers were observed in lung tissue collected from MCT-treated rats following oral administration of 200 mg/kg KAR5585 for 7, 14, and 28 days. Data are expressed as mean ± S.E.M. (n = 4/group). ***P < 0.001 significance relative to MCT vehicle controls at day 28.
Discussion
PAH exhibits a complex pathobiology with many factors influencing both vascular remodeling and reactivity. PA-SMC hyperplasia leads to arterial wall thickening and progressive narrowing of the lumen, a common pathophysiological feature found clinically in human PAH and preclinically in experimental animal models of induced PH (Rabinovitch, 2012; Guignabert and Dorfmuller, 2013; Guignabert et al., 2015). Mechanisms leading to pulmonary vascular remodeling and cell proliferation are not well understood and vary among cell types and species. Our approach was to induce PH by using either a single pathologic insult of MCT or multiple insults of SUGEN and chronic hypoxia to simulate different aspects of the disease.
Female sex hormones and their metabolites have been postulated to be responsible for the protective effect against the disease in females who have a higher survival rate compared with males. This contradiction is known as the estrogen paradox (de Jesus Perez, 2011). Preclinically, gender differences in the progression and severity of PH has also been established in rodent models (White et al., 2011); however, male rats are more commonly used in PH research with various agents. Therefore, to facilitate integration of our findings with those from the literature, we chose to employ male rats in the models included in this study, recognizing that hormonal factors affect the serotoninergic system’s impact on PH (White et al., 2011).
In our studies, reductions in systemic 5-HT levels by TPH1 inhibition provide an alternative mechanism of action to current PAH therapies. In two rat models of PAH, TPH1 inhibition treated the disease as a stand-alone therapy with a unique mechanism of action differing from the common standard of care. When our TPH1 inhibitors were administered in combination with an ERA or a phosphodiesterase type-5 inhibitor, some histologic and hemodynamic parameters, including vessel wall thickness and mPAP, were improved greater than the individual therapies alone. In contrast, the blockade of mast cell accumulation in tissue collected from MCT-treated rats was only observed with the TPH1 inhibitor alone.
Administration of orally active TPH1 inhibitors produced changes in serum, gut mucosa, and lung 5-HT levels without affecting the brain serotonergic pathway. The time- and dose-dependent reductions in serum 5-HT following oral administration of a selective TPH1 inhibitor are consistent with changes in circulating 5-HT levels that are dependent on platelet turnover and time required for depletion of the 5-HT storage pools (Anderson et al., 1987; Janusonis, 2008).
Plasma 5-HT originates in the gut where it is synthesized from tryptophan by TPH1 in the enterochromaffin cells. Some of the synthesized 5-HT is stored in the gut and some is released into the general circulation where it is rapidly cleared by the liver and lung. A small fraction that is released from the intestines is taken up by platelets, which account for greater than 99% of circulating 5-HT. During the life span of several days, platelets take up 5-HT using SERT; however, because the serotoninergic system is cyclic in nature it does not allow for easy interpretation of how changes in platelet steady-state levels of 5-HT relate to changes in 5-HT production rates. The majority of 5-HT is oxidized to 5-HIAA by monoamine oxidase and is secreted into the urine; therefore, urinary 5-HIAA collected over a 24-hour period can be used to estimate total 5-HT production (Deacon, 1994). In our disease models, a dose of 200 mg/kg KAR5585 showed significant efficacy and decreased urinary 5-HIAA by 50%. These data demonstrate that significant improvements in PAH can be obtained by marginally reducing 5-HT production rates and do not require complete TPH1 blockade.
5-HT involvement in the pathobiology of PAH has been well documented and a significant increase in circulating 5-HT is found in PAH patients; however, its mechanism of action remains unclear. Circulating free and platelet-bound 5-HT synthesized in the gut, released from inflammatory cells, or produced within the pulmonary vasculature can pass into the underlying PA-SMCs through SERT. As a potent inducer of cell proliferation, 5-HT effects pulmonary cell signaling by activation of its associated receptors and transporters and post-translational modification of proteins (Eddahibi et al., 2001; Guilluy et al., 2009; Wei et al., 2012; Tu and Guignabert, 2013; Penumatsa and Fanburg, 2014). PA-SMCs and endothelial cells express mRNA for a number of 5-HT receptors including 5HT1b, 5HT2a, 5HT7, and 5HT2b, and endothelial cells from lung tissue obtained from patients with PAH have increased rates of 5-HT uptake and synthesis (Eddahibi et al., 2001; de Caestecker, 2006).
Although pharmacological blockade of selective 5-HT receptors can prevent experimental PH, these approaches have not been successful clinically due to multiple receptors sharing overlapping pathways and redundancy in 5-HT signaling (Archer et al., 2010). Our approach was to reduce systemic 5-HT tone by reducing the ligand for these multiple receptors through a partial blockade of the rate-limiting enzyme for 5-HT synthesis, TPH1.
Previously, both genetic and pharmacological blockade of TPH1 showed protective effects on the remodeling of the pulmonary vasculature and right ventricular hypertrophy in mice (Izikki et al., 2007; Ciuclan et al., 2013). Of particular note are the differences among the models as they relate to 5-HT metabolism. In the MCT rat model, there are marked increases in pulmonary 5-HT levels and TPH1 mRNA expression. These data are consistent with previous findings in mouse PAH models and PAH patients demonstrating significant intrapulmonary 5-HT synthesis in the diseased lung. Other studies have shown that 5-HT synthesized in the P-ECs can act in a paracrine manner, causing constriction and remodeling of the underlying PA-SMCs (Eddahibi et al., 2006; Morecroft et al., 2012). These effects are mediated through both SERT and 5-HT receptors, and abnormalities in P-EC 5-HT production and transport have been reported in patients with idiopathic PAH (Launay et al., 2002; Guignabert et al., 2005, 2006; Eddahibi et al., 2006; Dempsie and MacLean, 2008). Increases in 5-HT and TPH1 mRNA expression in the lung tissue of MCT rats may be attributed to both intrapulmonary synthesis as well as the local accumulation of mast cells observed in this model upon induction of PAH. Mast cells store 5-HT removed from circulation through SERT and release it upon degranulation. Patients with PAH and MCT rats have mast cell accumulation in their pulmonary vasculature, which may be an important contributor to the inflammatory component of the disease. Kosanovic et al. (2015) demonstrated that reductions in mast cell number and chymase activity were associated with improved hemodynamics and decreased pulmonary vascular remodeling in a bleomycin model of lung fibrosis. In this study, we demonstrated that TPH1 inhibition directly effects pulmonary vascular remodeling and has the capacity to affect inflammatory processes by reducing mast cell infiltration. Although 5-HT acts as a chemoattractant for mast cells in vitro, further experimentation is required to establish the role of TPH1 and/or 5-HT in mast cell proliferation and migration.
Compared with the MCT rat model, the SUGEN-hypoxia–induced rat model has little detectable TPH1 lung mRNA expression and lung 5-HT levels are not increased following PH induction. Nevertheless, TPH1 inhibition positively affected PH in the SUGEN-hypoxia rat. Reductions in mPAP and vascular remodeling in this model cannot be attributed to intrapulmonary synthesis of 5-HT but are more consistent with improvements due to decreases in circulating 5-HT levels. Indeed, alterations in the availability of free or platelet-derived 5-HT have been central to the PAH serotonin hypothesis (MacLean and Dempsie, 2010). Several laboratories have linked platelet-derived 5-HT to pulmonary vascular disease, where others have suggested increased uptake and metabolism of 5-HT by the lung triggers development of idiopathic PAH (Dees et al., 2011). In TPH1- and SERT-deficient mice with plasma 5-HT depletion, development of chronic hypoxia-induced pulmonary vascular remodeling is reduced (Ciuclan et al., 2013). PA-SMCs obtained from patients with PAH have 2- to 3-fold increased rates of 5-HT uptake (Eddahibi et al., 1999). Increased 5-HT uptake and metabolism via monoamine oxidase (Lawrie et al., 2005) and/or nicotinamide adenine dinucleotide phosphate oxidase (Lee et al., 1998; Liu and Folz, 2004) increases the production of reactive oxygen species and attenuates the uncoupling of endothelial nitric oxide synthase. Thus, excess 5-HT metabolism can directly lead to mitochondrial dysfunction causing impaired vascular tone and cell proliferation of the pulmonary microvasculature (Stenmark et al., 2009).
It has been well established that a combination of factors is required to cause PAH. For example, the effects of 5-HT on pulmonary hyperplasia are modified by interaction between the 5-HT pathway and BMPR2 signaling. Furthermore, sustained infusions of 5-HT cause more extensive pulmonary vascular remodeling in BMPR2-haplo insufficient mice compared with wild-type mice (Long et al., 2006). Mice overexpressing SERT have decreased Kv1.5 expression and positively respond to pyruvate dehydrogenase kinase inhibition therapy (Guignabert et al., 2009), suggesting a link between mitochondrial dysfunction, the Kv1.5 channel, and regulation of the 5-HT pathway.
Despite multiple mechanisms contributing to the pathophysiology of PAH, TPH1 inhibition distinguished itself as being more effective than current PAH therapies in reducing both pulmonary vascular remodeling and vessel occlusion in all preclinical models. When our TPH1 inhibitors are combined with therapies directed toward vasodilation, such as an ERA or a phosphodiesterase type-5 inhibitor, the potential of an additive effect on both improving the symptoms and the pathophysiology of the disease might be achieved in the clinic. Together, our data suggest that TPH1 inhibition as monotherapy or used in polypharmacy has the potential to provide a novel treatment of PAH.
In conclusion, preclinical studies with the TPH1 inhibitor KAR5585 demonstrated efficacy in both prevention and treatment models of PH. The complex pathophysiology of idiopathic PAH involves multiple mechanisms that may not be completely predictive by a single preclinical model. However, many of the disease-related pathways including the cellular proliferative processes have been linked to the 5-HT system. The demonstration that TPH1 inhibition can be an effective treatment in several models of PAH with distinct pathophysiologies suggests that it would be a novel, stand-alone therapeutic intervention providing a different mechanism of action from other orally active treatment options currently available. TPH1 inhibition by KAR5585 warrants further clinical testing to provide physicians with a novel option for treating this debilitating disease; clinical evaluation of KAR5585 has commenced (https://clinicaltrials.gov/ct2/show/NCT02746237?term=KAR5585&rank=1).
Acknowledgments
Karos Pharmaceuticals thanks James Valentine, William Zavadoski, Nicole Barucci, and Ron Mays for technical support, and Marco Garcia and Michele de Crescenzo at North East Life Sciences for assistance with compound administration and animal husbandry.
Authorship Contributions
Participated in research design: Aiello, Bourassa, Zhang, Dubins, Paraklar, McKinsey, Cavasin, Humbert, Guignabert
Conducted experiments: Aiello, Bourassa, Zhang, Dubins, McKinsey, Cavasin, Humbert, Guignabert.
Contributed new reagents or analytic tools: Goldberg and De Lombaert.
Performed data analysis: Aiello, Bourassa, Zhang, Dubins, McKinsey, Cavasin, Guignabert.
Wrote or contributed to the writing of the manuscript: Aiello, Bourassa, Dubins, Paraklar, McKinsey, Cavasin, Humbert, Guignabert.
Footnotes
- Received September 27, 2016.
- Accepted December 5, 2016.
INSERM UMR_S 999 received financial support from Karos Pharmaceuticals, New Haven, CT, to conduct the in vitro study.
M.H. has relationships with drug companies (Actelion, Bayer, Gilead, GSK, Karos Pharmaceuticals, and Pfizer) that relate to products or molecules under investigation in pulmonary hypertension. In addition to being investigator in trials involving these companies, relationships include consultancy service and membership of scientific advisory boards. T.A.M. at the Division of Cardiology and Consortium for Fibrosis Research and Translation, Department of Medicine, University of Colorado Denver-Anschutz Medical Campus, received financial support from Karos Pharmaceuticals, New Haven, CT, to conduct in vivo studies.
Abbreviations
- BrdU
- 5-bromo-2-deoxyuridine
- 5-HIAA
- 5-hydroxyindoleacetic acid
- 5-HT
- serotonin
- ERA
- endothelin receptor antagonist
- HPLC
- high-performance liquid chromatography
- KAR5395
- (S)-8-(2-amino-6-((R)-1-(3′,4′-dimethyl-3-(3-methyl-1 H-pyrazol-1-yl)-[1,1′-biphenyl]-4-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylic acid
- KAR5416
- ethyl (S)-8-(2-amino-6-((R)-1-(3′,4′-dimethyl-3-(3-methyl-1 H-pyrazol-1-yl)-[1,1′-biphenyl]-4-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylate
- KAR5417
- (S)-8-(2-amino-6-((R)-1-(5-chloro-[1,1′-biphenyl]-2-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylic acid
- KAR5585
- ethyl (S)-8-(2-amino-6-((R)-1-(5-chloro-[1,1′-biphenyl]-2-yl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)-2,8-diazaspiro[4.5]decane-3-carboxylate
- MCT
- monocrotaline
- mPAP
- mean pulmonary arterial pressure
- PAH
- pulmonary arterial hypertension
- PA-SMC
- pulmonary arterial smooth muscle cell
- P-EC
- pulmonary endothelial cells
- PH
- pulmonary hypertension
- SERT
- serotonin transporter
- TPH1
- tryptophan hydroxylase 1
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}