





Abstract
Limited literature sources implicate mast-cell mediator chymase in the pathologies of pulmonary hypertension and pulmonary fibrosis. However, there is no evidence on the contribution of chymase to the development of pulmonary hypertension associated with lung fibrosis, which is an important medical condition linked with increased mortality of patients who already suffer from a life-threatening interstitial lung disease.
The aim of this study was to investigate the role of chymase in this particular pulmonary hypertension form, by using a bleomycin-induced pulmonary hypertension model.
Chymase inhibition resulted in attenuation of pulmonary hypertension and pulmonary fibrosis, as evident from improved haemodynamics, decreased right ventricular remodelling/hypertrophy, pulmonary vascular remodelling and lung fibrosis. These beneficial effects were associated with a strong tendency of reduction in mast cell number and activity, and significantly diminished chymase expression levels. Mechanistically, chymase inhibition led to attenuation of transforming growth factor β1 and matrix-metalloproteinase-2 contents in the lungs. Furthermore, chymase inhibition prevented big endothelin-1-induced vasoconstriction of the pulmonary arteries.
Therefore, chymase plays a role in the pathogenesis of pulmonary hypertension associated with pulmonary fibrosis and may represent a promising therapeutic target. In addition, this study may provide valuable insights on the contribution of chymase in the pulmonary hypertension context, in general, regardless of the pulmonary hypertension form.
Abstract
Chymase plays an important role in pathology of pulmonary hypertension associated with lung fibrosis http://ow.ly/MVYW6
Introduction
Pulmonary hypertension (PH) associated with lung fibrosis represents an important factor linked with augmented mortality of patients who already suffer from severe interstitial lung disease [1]. PH is a relatively frequent medical condition in patients with idiopathic pulmonary fibrosis (IPF) and, therefore, it should receive considerable attention [1]. The underlying pathological mechanisms responsible for PH development in IPF are still poorly understood and there are limited therapeutic options available for this particular form of PH [1].
We have shown, previously, that mast cells (MCs) may play a role in the pathogenesis and progression of both PH and lung fibrosis [2, 3]. Their number and activation were significantly augmented in the lungs of patients with IPF and idiopathic pulmonary arterial hypertension (IPAH) [2, 3]. MCs produce different mediators involved in inflammatory response [4]. Among MC mediators, the chymase may be of particular interest in the context of PH and IPF, owing to its function in activation and processing of transforming growth factor (TGF)-β, endothelin (ET)-1, angiotensin II, interleukins (IL)-1 and IL-18, matrix metalloproteinases (MMP)-2 and 9, and potential involvement in collagen synthesis [5–16]. Following this knowledge, it is worth noting that most of the above mentioned molecular mediators/signalling pathways are suggested to be important culprits in PH and IPF pathologies, thus indicating the potential link between chymase and these diseases [5, 17–27]. Furthermore, it has been described that MCs expressing chymase are indeed altered in PH [28–30]. In addition we recently reviewed the role of this MC mediator in the context of IPF with the conclusion that chymase may also have potential involvement in lung fibrosis [5]. The aim of this study was to analyse and assess the potential role of chymase in PH development associated with pulmonary fibrosis, by using the well-established model of bleomycin-induced lung fibrosis in hamsters. We have chosen the hamster model as there is evidence that hamster chymase displays similar substrate specificity as the human chymase [31]. In contrast, mouse and rat chymase differ in their structure and substrate specificity from human chymase [13, 14]. Although these species are widely used for pulmonary fibrosis research, we used a model which expresses an enzymatic profile closer to that of a human, being aware that there are limitations in the molecular tools for hamster. To address the role of the chymase in PH associated with pulmonary fibrosis (PF), we investigated the effects of chymase inhibition on: 1) haemodynamics, right ventricular (RV) hypertrophy and remodelling, as well as pulmonary vascular remodelling; 2) lung fibrosis; 3) MC activation and expression of chymase in the lungs; and 4) TGF-β1, MMP-2 and ET-1 signals.
Methods
Study design and physiological measurements
PH was induced in adult Syrian hamsters by intratracheal instillation of bleomycin. The healthy control group only received NaCl. Bleomycin-instilled hamsters were randomised into two groups and treated orally by gavage either with chymase inhibitor BCEAB (4-[1-([bis-(4-methyl-phenyl)-methyl]-carbamoyl)3-(2-ethoxy-benzyl)-4-oxo-azetidine-2-yloxy]-benzoic acid) (Bleo-BCEAB) or placebo (Bleo-placebo). Haemodynamic and right heart hypertrophy (RHH) measurements were performed similarly as previously described [32].
Histological analysis
Pulmonary vascular remodelling, fibrosis, MC analysis and right heart remodelling were assessed as previously described [2, 20, 33]. Immunohistochemistry was performed to analyse the chymase. To count the capillaries the STEPanizer stereological tool was used [34].
Analysis of ET-1
Effects of chymase inhibition on big-ET-1-induced vasoconstriction were analysed in isolated pulmonary artery rings. Furthermore, plasma and lung tissue levels of ET-1 and big-ET-1 were measured by radioimmunoassay and enzyme-linked immunoassay.
Analysis of TGF-β1 and MMP-2
Immunohistochemistry for TGF-β1 and MMP-2 with subsequent quantification was performed on lung tissue sections.
Immunohistochemistry of chymase and TGF-β1 in human lung tissues
Human lung tissues were obtained from patients with IPAH and IPF undergoing lung transplantation. Lung tissues were formalin-fixed and paraffin-embedded, as previously reported [2]. Double-immunostaining with anti-chymase and anti-TGF-β1 antibodies was performed and the three-dimensional (3D) effect of the photomicrographs was achieved as previously described [30].
Further details can be found in the online supplementary materials.
Statistics
All numerical data in the manuscript were presented as mean±sem. The results among different experimental groups were statistically compared by one-way ANOVA and Newman–Keuls multiple comparison test.
Study approval
Animal studies were approved by the local authority (Regierungspraesidium Gießen). The study protocol for human lung tissue donation was approved by the ethics committee of the University Hospital Giessen (Giessen, Germany). Each patient or the patient's next kin were asked for a written informed consent [2].
Results
Inhibition of chymase attenuated PH and RHH
Bleomycin instillation in hamsters resulted in the development of PH, as evident from significantly increased RV systolic pressure (RVSP) in the placebo group (40.4±3.2 mmHg), as compared to the healthy control group (22.4±2.6 mmHg) (fig. 1a). The chymase inhibitor BCEAB significantly reduced the RVSP (23.7±1.5 versus 40.4±3.2 mmHg in placebo). Systemic arterial pressure did not differ between experimental groups (fig. 1b). Pulmonary vascular haemodynamic alteration in the bleomycin model of PH was associated with the development of RV hypertrophy, as evident from significantly increased Fulton's index in the placebo group (0.35±0.01), as compared to the healthy control group (0.27±0.01) (fig. 1c). Inhibition of chymase resulted in the significant reduction of Fulton's index (0.28±0.02 versus 0.35±0.01, Bleo-BCEAB versus Bleo-placebo).

Effects of chymase inhibition on haemodynamics and right heart hypertrophy in the bleomycin model of pulmonary hypertension. Bleomycin-instilled hamsters were randomised into two groups and treated orally by gavage either with chymase inhibitor (Bleo-BCEAB) or placebo (Bleo-placebo). Healthy control group received only NaCl. At the end of the treatment animals underwent the haemodynamic and right ventricular (RV) hypertrophy measurements, as described in the methods section. a) Right ventricular systolic pressure (RVSP), b) systemic arterial pressure (SAP) and c) the weight ratio of the RV wall and left ventricle (LV) wall with septum (S) (RV/(LV+S)) are shown. Bars represent mean±sem. One-way ANOVA with Newman–Keuls multiple comparison test was performed for statistical analysis. *: p<0.05 Bleo-placebo versus healthy control. #: p<0.05 Bleo-placebo versus Bleo-BCEAB.
Inhibition of chymase reduced pulmonary vascular remodelling and lung fibrosis
The degree of muscularisation revealed a significant reduction of non-muscularised and augmentation of fully-muscularised pulmonary vessels in the lungs of animals instilled with bleomycin, compared to the healthy control (fig. 2a–c). Treatment with the chymase inhibitor BCEAB resulted in a significant reduction of fully-muscularised pulmonary vessels and an increase in non-muscularised pulmonary vessels, in comparison with placebo. Additionally, medial-wall thickness was noticeable higher in the placebo group (22.6±1.1%), as compared to the healthy control (16.8±1.0%) (fig. 2e). Inhibition of chymase resulted in a significant reduction of the medial-wall thickness (Bleo-BCEAB versus Bleo-placebo, 19.9±0.6% versus 22.6±1.1%). Bleomycin caused a development of lung fibrosis in the placebo group, as evident by the analysis of the Ashcroft score (3.63±0.36) (fig. 2i). Treatment with BCEAB reduced the level of lung fibrosis (Bleo-BCEAB versus Bleo-placebo, 1.98±0.24 versus 3.63±0.36) (fig. 2i).

Effects of chymase inhibition on pulmonary vascular remodelling and lung fibrosis in the bleomycin model of pulmonary hypertension. a–c) The lung tissue sections of hamsters were double immunostained for α-smooth muscle actin and von Willebrand factor in order to analyse d) the degree of muscularisation: none (N), partially (P) or fully (F) muscularised vessels. Representative photomicrographs from different experimental groups are shown: a) healthy control, b) Bleo-placebo, and c) Bleo-BCEAB. Arrows indicate pulmonary vessels. Scale bars=20 μm. Additionally, the lungs were stained with elastica van Gieson to measure e) medial wall thickness. f–h) For histological assessment of lung fibrosis, the lungs were initially stained with haematoxylin and eosin and representative photomicrographs from different experimental groups are shown: f) healthy control, g) Bleo-placebo, and h) Bleo-BCEAB. Scale bars=20 μm. i) Fibrosis scoring, as described in the methods section. Bars represent mean±sem. One-way ANOVA with Newman–Keuls multiple comparison test was performed for statistical analysis. *: p<0.05 Bleo-placebo versus healthy control; #: p<0.05 Bleo-placebo versus Bleo-BCEAB.
Lung MCs and chymase
MC number did not change between healthy control and placebo group (fig. 3a). Interestingly, there was a strong tendency to decrease the number of MCs in animals treated with chymase inhibitor. Although the MC number did not alter, the MCs in the placebo group were more activated, as evident by significant reduction of the index of granulation, in comparison with healthy control (fig. 3b). The chymase inhibitor BCEAB increased the index of granulation, suggesting a reduction of MC activation, but the value did not reach statistical significance. Finally, we analysed the chymase expression (fig. 3c), and found a significant augmentation of chymase in the placebo group (807±285 chymase-positive immunoreactivity per mm2 %), as compared to healthy animals (100±42 chymase-positive immunoreactivity per mm2 %). Chymase inhibition with BCEAB significantly reduced the chymase-positive signal in the bleomycin model (Bleo-BCEAB versus Bleo-placebo, 226±93 versus 807±285 chymase-positive immunoreactivity per mm2 %).

Effects of chymase inhibition on lung mast cells and chymase expression in the bleomycin model of pulmonary hypertension. a) Lung tissue sections were stained with toluidine-blue for detection and quantification of mast cells and b) their activation was evaluated. c) Immunohistochemistry was performed for chymase detection and measured. Bars represent mean±sem. One-way ANOVA with Newman–Keuls multiple comparison test was performed for statistical analysis. *: p<0.05 Bleo-placebo versus healthy control; #: p<0.05 Bleo-placebo versus Bleo-BCEAB.
Chymase inhibition decreased RV remodelling
We have analysed the cardiomyocyte size and found a significant augmentation in the cross-sectional area of the placebo group (270.70±8.94 μm2), as compared to the healthy state (179.20±10.68 μm2) (fig. 4m). Chymase inhibition resulted in a significant reduction of the cardiomyocyte hypertrophy (217.80±14.79 versus 270.70±8.94 μm2 in the placebo group). Also, we have analysed the capillary length and there was no difference in this parameter among experimental groups (fig. 4n). Additionally, RV hypertrophy was associated with the development of RV fibrosis, as reflected by markedly increased collagen content in the right hearts of placebo group (3.14±0.16%), in comparison with healthy animals (1.11±0.17%) (fig. 4o). Chymase inhibition led to a significant reduction of collagen content (Bleo-BCEAB versus Bleo-placebo, 1.54±0.04% versus 3.14±0.16%).

Effects of chymase inhibition on right ventricular remodelling in the bleomycin model of pulmonary hypertension. Right ventricles (RVs) were stained with fluorescein isothiocyanate (FITC)-conjugated wheat germ agglutinin (WGA) to outline the cell membrane, tetramethylrhodamine-5-(and 6)-isothiocyanate (TRITC)-conjugated isolectin B4 (IB4) to outline the capillary endothelial cells, and diamidino phenylindole (DAPI) for nuclear staining. Representative photomicrographs from different experimental groups: healthy control (a, d, g and j), Bleo-placebo (b, e, h and k) and Bleo-BCEAB (c, f, i and l); and stainings: WGA-FITC (a–c), IB4-TRITC (d–f), DAPI (g–i) and merged (j–l). Scale bars=20 μm. The quantification was performed for assessment of cardiomyocyte size and capillary length, and the values expressed as m) cross sectional area and n) capillary length. o) RV tissue sections were additionally stained with sirius red in picric acid for collagen content assessment. The quantification of collagen formation was performed and the values of the collagen content (%) in different experimental groups are given. Bars represent mean±sem. One-way ANOVA with Newman–Keuls multiple comparison test was performed for statistical analysis. *: p<0.05 Bleo-placebo versus healthy control; #: p<0.05 Bleo-placebo versus Bleo-BCEAB.
Chymase inhibition prevented the big-ET-1-induced vasoconstriction in isolated pulmonary artery rings
Big-ET-1 induced a concentration-dependent vasoconstriction in the isolated pulmonary artery rings of hamsters (fig. 5a). This effect on pulmonary arteries was prevented by neutral endopeptidase (NEP)/endothelin converting enzyme inhibitor phosphoramidon used alone or in combination with chymase inhibitor chymostatin (fig. 5a). However, a clear rightward shift of the curve was observed in the presence of both inhibitors, when compared to the vehicle and phosphoramidon alone.
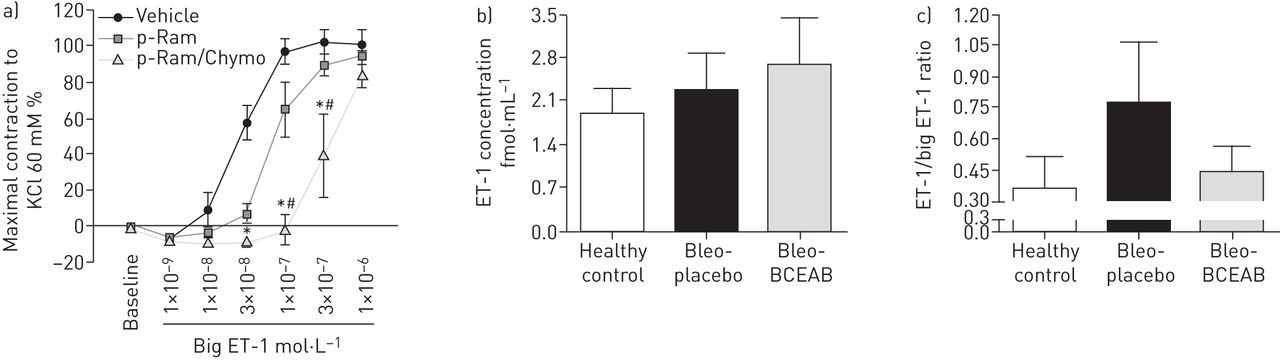
Effects of chymase inhibition on big endothelin (ET)-1 induced pulmonary artery vasoconstriction and on ET-1/big-ET-1 content in the bleomycin model of pulmonary hypertension. a) The concentration-response curves for big-ET-1 in the absence or presence of phosphoramidon (p-Ram) or combination of phosphoramidon and chymase inhibitor chymostatin (p-Ram/Chymo) in isolated pulmonary artery rings are given. b) The ET-1 level was measured in the plasma of different experimental groups and ET-1 concentration (fmol·mL−1) is shown. c) Additionally, the ET-1 and big-ET-1 levels were measured in the lung tissues and ET-1/big-ET-1 ratio is given. Bars represent mean±sem. One-way ANOVA with Newman–Keuls multiple comparison test was performed for statistical analysis. *: p<0.05 p-Ram/Chymo versus vehicle; #: p<0.05 p-Ram/Chymo versus p-Ram.
Chymase inhibition and ET-1/big-ET-1 content
ET-1 concentration in the plasma did not differ significantly between experimental groups (fig. 5b). Although the changes of ET-1/big-ET-1 ratio in the lungs of experimental groups did not reach statistically significant levels, there was a strong tendency to increase in placebo group (0.78±0.29), compared to the healthy controls (0.37±0.15) (fig. 5c). The chymase inhibitor BCEAB caused a strong tendency to decrease the ET-1/big-ET-1 ratio (Bleo-BCEAB versus Bleo-placebo, 0.44±0.13 versus 0.78±0.29).
Chymase inhibition reduced TGF-β1 content
The lungs of animals from different experimental groups were immunostained to detect TGF-β1 (fig. 6a–c). The TGF-β1 content was quantified (fig. 6d) and results revealed a significant increase of TGF-β1 signal after bleomycin instillation (0.66±0.17 score value %), as compared to healthy control (0.10±0.03 score value %). Chymase inhibition significantly reduced the TGF-β1 content (Bleo-BCEAB versus Bleo-placebo, 0.06±0.02 versus 0.66±0.17 score value %).

Effects of chymase inhibition on lung transforming growth factor (TGF)-β1 and matrix metalloproteinase (MMP)-2 contents in the bleomycin model of pulmonary hypertension. The lungs of animals were immunostained to detect and quantify the d) TGF-β1 and h) MMP-2 contents, and representative photomicrographs from different experimental groups, with focus on pulmonary vasculature, are given: healthy control (a and e), Bleo-placebo (b and f) and Bleo-BCEAB (c and g). Arrows indicate the TGF-β1 (a–c) and MMP-2 (e–g) positive immunostaining (red). Scale bars=20 μm. The three-dimensional effect of the photomicrographs was achieved by using the special prisms for microscopes, as described in the methods section. The TGF-β1 (d) and MMP-2 (h) signals were quantified and results expressed as score value. Bars represent mean±sem. One-way ANOVA with Newman–Keuls multiple comparison test was performed for statistical analysis. *: p<0.05 Bleo-placebo versus healthy control; #: p<0.05 Bleo-placebo versus Bleo-BCEAB.
Chymase inhibition reduced MMP-2 content
The lungs of hamsters from different experimental groups were immunostained to detect MMP-2 (fig. 6e–g). The quantification of the MMP-2 was performed (fig. 6h) and results revealed a significant increase of MMP-2 signal after bleomycin instillation (0.70±0.08 score value %), in comparison with healthy control (0.05±0.02 score value %). Chymase inhibition led to a marked reduction in MMP-2 content (Bleo-BCEAB versus Bleo-placebo, 0.18±0.05 versus 0.70±0.08 score value %).
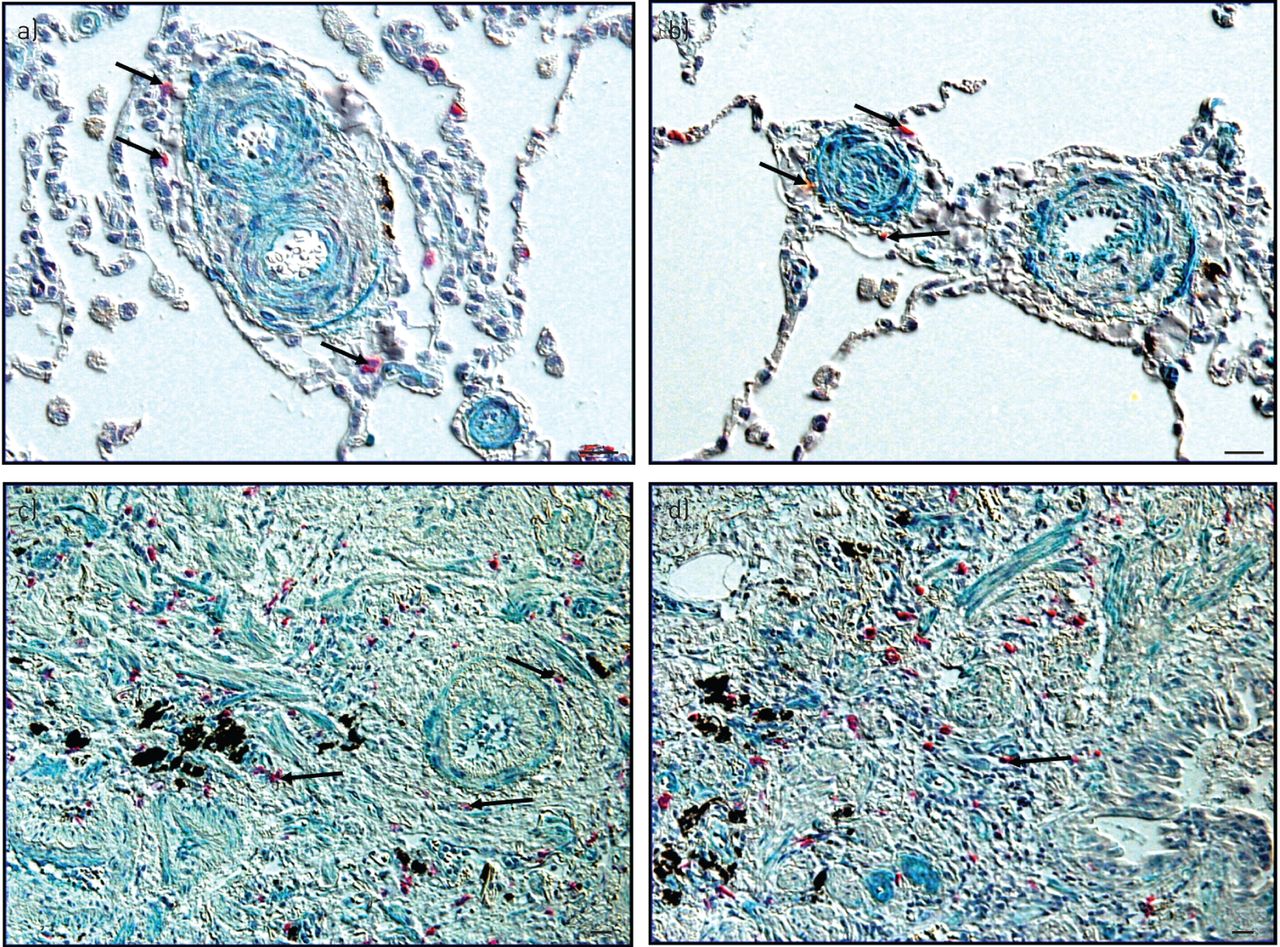
Chymase-positive MC accumulation in lungs of patients with PH and lung fibrosis
The lungs of patients with IPAH (fig. 7a and b) and IPF (fig. 7c and d) were double immunostained for detection of chymase-positive MCs (red-stained cells) and TGF-β1 (green-stained structures). Regarding IPAH histopathology, the chymase-expressing MCs were scattered throughout the lungs including the perivascular regions of remodelled vessels. Importantly, these vessels were positive to TGF-β1 signal, suggesting the close proximity of the MCs that express chymase to the regions stained with TGF-β1. In the patients with IPF, chymase-expressing MCs were scattered throughout the lung tissue, including the fibrotic and perivascular regions. Also, the TGF-β1 signal was present throughout the fibrotic lungs. Similarly to the IPAH situation, the chymase-producing MCs were located in close proximity to the TGF-β1 signal.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Chymase-positive mast cells accumulation in lungs of patients with pulmonary hypertension and lung fibrosis, the clinical relevance. Human lung tissues from patients with idiopathic pulmonary arterial hypertension (a and b) and idiopathic pulmonary fibrosis (c and d) were double-immunostained for chymase (red cells) and transforming growth factor β1 (green structures), and photomicrographs were taken, using special prisms for microscopes that resulted in a three-dimensional effect. Arrows indicate chymase-positive mast cells. Scale bars=20 μm.
Discussion
The findings from our study revealed that chymase inhibition in bleomycin-induced PH resulted in: 1) improved haemodynamics and attenuated pulmonary vascular remodelling and lung fibrosis; 2) reduced RV hypertrophy associated with decreased collagen content and cardiomyocyte size; 3) high tendency for preventing the MCs presence and even more importantly their activation, and significantly reducing expression of chymase in the lungs.
In recent years we demonstrated that augmentation of the MC number and their activity were characteristics of both IPAH and IPF pathologies [2, 3]. Activated MCs release a variety of mediators, and among them chymase may have great importance in the context of PH and lung fibrosis development [5, 28–30]. There is evidence from the literature, although very limited, that chymase inhibition led to attenuation of lung fibrosis in bleomycin models of mice and hamsters and suppression of silica-induced PF in mice [16, 35–37]. It is worth mentioning that MC chymase has a potential role in the regulation of collagen synthesis, suggesting one of the possibilities to be involved in the pathology of fibrosis [7]. In accordance to the literature, we found that the chymase inhibitor BCEAB significantly reduced lung fibrosis, as seen from histological scoring analysis. Importantly, in the two studies focusing on the role of chymase in a bleomycin model of PF the treatments with chymase inhibitors were done in the preventive approach manner, while our study revealed that chymase inhibition reduced lung fibrosis even in the therapeutic approach, which is actually more clinically relevant [16, 35]. There is limited evidence on the in vivo effects of chymase inhibition in the field of PH and our study is one of the rare to systematically investigate this important scientific issue [38, 39]. Chymase inhibition resulted in significant reduction of RVSP and RV hypertrophy, without any effects on the systemic circulation. Importantly, haemodynamic and RV hypertrophy beneficial effects were associated with diminished pulmonary vascular remodelling. Reduced RV hypertrophy was associated with significantly decreased collagen content and cardiomyocyte size in the hearts of animals treated with the chymase inhibitor. These novel findings clearly suggest the potential involvement of chymase in the pathology of PH associated with lung fibrosis and, even more, we can extend this knowledge to the PH field in general, as augmented inflammation and particularly increased accumulation of MCs are indeed recognised pathological features of other PH forms, such as pulmonary arterial hypertension (PAH). Therefore, future studies focusing on the role of this MC mediator in different well-established models of PH are needed.
After activation/degranulation, caused by different stimuli, MCs release a variety of mediators including chymase [4]. Chymase activity was noticeably increased in the lungs of animals following bleomycin instillation, as shown previously [16, 35]. In line with the literature, we demonstrated a significant augmentation of MC activation/degranulation, although the number did not change, and prominent expression of chymase in the lungs of bleomycin instilled hamsters, compared to healthy controls. Chymase inhibition resulted in strong tendency to decrease the MC number and degranulation and, even more, there was a significant reduction of chymase expression, in comparison with the placebo group. Thus, our findings indicated that chymase inhibition might interfere with general MC function, affecting the MC count, activation and chymase expression, and suggested the potential existence of self-perpetuating events during bleomycin-induced pathogenesis. We observed similar phenomena previously, in the context of monocrotaline-induced PAH, where MC stabiliser retained the progressive MC accumulation and enhanced degranulation during development of severe pulmonary vascular disease [2]. One of the possible explanations may be due to fact that chymase is also able to increase levels of the stem cell factor, which is known to be involved in different cellular processes related to general MC biology, such as maturation and activation, migration, survival and proliferation [40–42].
In order to unravel the underlying mechanisms responsible for PH associated with lung fibrosis (fig. S1), we focused on the link between chymase and three different signalling pathways that are known to be potentially involved in both PH and PF pathologies, such as ET-1, TGF-β and MMP-2 [18–23, 25, 26, 43]. MC chymase converts big-ET-1 into 31 amino-acid ET-1(1-31), which can be further processed into ET-1(1-21) by neutral NEP, suggesting the role of this MC mediator in maturation of ET-1 [5, 8–10]. In this context, the ET-1 system is indeed implicated as an important culprit in PH and lung fibrosis [19–21]. The concentration of ET-1 in plasma of hamsters treated with BCEAB was slightly increased compared to the placebo. Similar phenomena of augmented circulating ET-1 levels were observed in patients treated with non-selective ET antagonists and were explained due to an altered ET-B-mediated pulmonary clearance [44]. Therefore, one could expect that similar effects were achieved in our study where the interference with chymase-ET-1 axis might also affect the ET-1 clearance. However, further research should be performed to address this issue. Lung ET-1/Big-ET-1 ratio tended to be increased in disease condition, compared to healthy controls and BCEAB treated animals, suggesting the possibility that more of the big-ET-1 was used for procession to biologically active form. In addition, we demonstrated that chymase inhibition prevented the big-ET-1 induced pulmonary artery vasoconstriction ex vivo. However, future investigation by using of selective chymase inhibition and ET-1 antagonists in experimental models is crucial for final understanding if chymase-ET-1 axis is involved in disease. Controversially, it has to be taken into consideration the fact that chymase may even exert ET-1 degrading activities [10, 45]. Altered TGF-β signalling plays a role in development of PH and PF [5, 18, 24, 25, 39, 43, 46]. In this line, it was demonstrated that chymase is capable of TGF-β1 activation and maturation, by processing latent TGF-β-binding protein to latent TGF-β, and further this inactive form to biologically active TGF-β form [5, 15, 16]. Our results revealed that chymase inhibition led to significant reduction of TGF-β1 expression in the lungs of hamsters from bleomycin PH model and findings are in agreement with literature [15, 16, 39]. Therefore, we can conclude that chymase-TGF-β1 axis may contribute to pathogenesis of PH associated with lung fibrosis. Additionally, impaired function of the MMP system is also implicated in the context of PH and lung fibrosis, as recently reviewed [22, 23]. Tchougounova et al. [6] showed a key role of chymase in the activation of inactive pro-MMP-2 and -9. Following this knowledge, we successfully demonstrated that chymase inhibition resulted in a significant decrease of MMP-2 content in the lungs of animals, suggesting a role of chymase-MMP-2 axis in development of PH associated with PF.
Conclusions
In the end, our study revealed that chymase inhibition attenuated PH associated with lung fibrosis bleomycin animal model. The beneficial physiological and morphological effects were associated with diminished levels of TGF-β1 and MMP-2, reduced ET-1/Big-ET-1 ratio and prevented big-ET-1-induced vasoconstriction of the pulmonary arteries. Finally, there was a prominent accumulation of chymase-expressing mast cells in IPAH and IPF lungs, and their presence was in close proximity to the regions with marked TGF-β1 expression, suggesting the relevance of animal model findings to the human situation. As a conclusion, chymase plays a role in PH associated with lung fibrosis and therefore, may represent a promising therapeutic option in the future. Although the accumulation of MCs and increased chymase expression were described in the pulmonary vascular disease many years ago, our study provided useful and novel insights on the contribution of this mediator to the pathogenesis of, not only the PH associated with lung fibrosis, but also PH in general, as many of the pathological features are indeed shared between different PH types [47, 48].
Acknowledgements
We acknowledge Christina Vroom and Ewa Bieniek for their valuable technical assistance and we thank Stefanie Hezel and Ingrid Henneke, from the group of Prof. Andreas Günther, for indispensable technical assistance regarding bleomycin application (all: Universities of Giessen and Marburg Lung Center (UGMLC), Member of the German Center for Lung Research, Giessen, Germany). We also thank Walter Klepetko from the University of Vienna (Vienna, Austria) for providing human lung tissues and Pauline Loriette-Steiner from Actelion Pharmaceuticals Ltd (Allschwill, Switzerland) for her valuable technical assistance regarding ex vivo study.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: This study was funded by a TI Pharma grant. Funding information for this article has been deposited with FundRef.
Conflict of interest: None declared.
- Received February 2, 2015.
- Accepted April 9, 2015.
- Copyright ©ERS 2015
References










