


Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease that often results in right ventricular (RV) failure and death. During disease progression, structural and electrical remodeling of the right ventricle impairs pump function, creates proarrhythmic substrates, and triggers for arrhythmias. Notably, RV failure and lethal arrhythmias are major contributors to cardiac death in patients with PAH that are not directly addressed by currently available therapies. Ranolazine (RAN) is an antianginal, anti-ischemic drug that has cardioprotective effects in experimental and clinical settings of left-sided heart dysfunction. RAN also has antiarrhythmic effects due to inhibition of the late sodium current in cardiomyocytes. We therefore hypothesized that RAN could reduce the maladaptive structural and electrical remodeling of the right ventricle and could prevent triggered ventricular arrhythmias in the monocrotaline rat model of PAH. Indeed, in both in vivo and ex vivo experimental settings, chronic RAN treatment reduced electrical heterogeneity (right ventricular-left ventricular action potential duration dispersion), shortened heart-rate corrected QT intervals in the right ventricle, and normalized RV dysfunction. Chronic RAN treatment also dose-dependently reduced ventricular hypertrophy, reduced circulating levels of B-type natriuretic peptide, and decreased the expression of fibrotic markers. In addition, the acute administration of RAN prevented isoproterenol-induced ventricular tachycardia/ventricular fibrillation and subsequent cardiovascular death in rats with established PAH. These results support the notion that RAN can improve the electrical and functional properties of the right ventricle, highlighting its potential benefits in the setting of RV impairment.
Introduction
Pulmonary arterial hypertension (PAH) is a progressive and fatal disease characterized by vascular remodeling and vasoconstriction of the pulmonary arterial circulation that often results in right ventricular (RV) failure and death. The progressive vasculopathy leads to intraluminal narrowing and obstruction of the resistance vasculature, leading to sustained increases in pulmonary vascular resistance and pulmonary arterial pressure. In an attempt to compensate for the increased afterload and wall stress, the right ventricle undergoes structural and electrical remodeling (Malenfant et al., 2013; Simon, 2013), creating proarrhythmic substrates and triggers for arrhythmias (Benoist et al., 2011, 2012; Rocchetti et al., 2014). Although the extent of the vascular pathology in PAH can determine morbidity and mortality, RV dysfunction is now recognized as the key determinant of disease outcome (Voelkel et al., 2012; Archer et al., 2013; Rain et al., 2013, 2014). The central role of RV pathology in disease progression is underscored by evidence showing that RV failure is the primary cause of death in patients with PAH, and by reports that lethal arrhythmias are a major contributor to sudden cardiac death (SCD) in these patients (Rajdev et al., 2012; Voelkel et al., 2012; Olsson et al., 2013; Rain et al., 2013, 2014; Rich et al., 2013).
Current PAH therapies, consisting mainly of prostanoids, phosphopdiesterase type-5 inhibitors, and endothelin receptor antagonists, reduce pulmonary vascular resistance through preferential vasodilation of the pulmonary circulation (Abraham et al., 2010; van de Veerdonk et al., 2011). This strategy of targeting the vasoconstrictive component of PAH improves symptoms and slows disease progression, but mortality rates remain high and functional impairment of RV function remains a significant problem (van de Veerdonk et al., 2011; Archer et al., 2013; Guihaire et al., 2013; Malenfant et al., 2013; Rain et al., 2013). There is no current PAH therapy that directly targets the structural and electrical consequences of RV remodeling (Voelkel et al., 2012; Archer et al., 2013; Olsson et al., 2013).
Ranolazine (RAN) is a Food and Drug Administration–approved antianginal drug with anti-ischemic activity that has also been shown to have antiarrhythmic properties due to inhibition of the late sodium current (INa) in cardiomyocytes (Hale et al., 2008; Antzelevitch et al., 2014). As a result of extensive clinical and experimental studies, the cardioprotective effects of RAN in settings of left-sided heart dysfunction are well established (Rastogi et al., 2008; Aistrup et al., 2013; Maier et al., 2013). Although the left and right ventricles have notable and well defined differences (i.e., structural, functional, electrical, and embryological), common mechanisms such as ischemia, Ca2+ overload, oxidative stress, and fibrosis are implicated in the pathology of both left- and right-sided heart failure (Bogaard et al., 2009; Borgdorff et al., 2013; Simon, 2013; Toischer et al., 2013). Pathologic processes that are known to increase late INa (Voelkel et al., 2012; Freund-Michel et al., 2013; Shryock et al., 2013; Toischer et al., 2013) are also implicated in the progression of RV failure in PAH (Voelkel et al., 2012; Freund-Michel et al., 2013; Shryock et al., 2013; Toischer et al., 2013). It was recently reported that RAN reduced RV mass, improved RV performance, and increased exercise capacity in 10 patients with World Health Organization group 1 PAH and symptoms of angina (Shah et al., 2012). It has also been reported that RAN can reduce RV hypertrophy in a rat model in which RV pressure overload is induced independently from changes in pulmonary arterial pressures by pulmonary artery banding (Fang et al., 2012). In addition, a recent study in a 3-week monocrotaline (MCT) rat model showed that RAN, when initiated 2 days after MCT injection, prevented RV structural and electrical remodeling by suppressing late INa (Rocchetti et al., 2014). MCT is known to consistently induce pulmonary vascular remodeling in the rat within the first 4–7 days after injection, with peak increases in pulmonary pressure occurring at 4 to 5 weeks (Bruner et al., 1983; Rosenberg and Rabinovitch, 1988; Hoorn and Roth, 1992; Lappin and Roth, 1997; Guihaire et al., 2013).
In our study, RAN treatment was started at day 7 after MCT injection to allow for the vasculopathy to develop before intervention, and all end points were assessed 4 weeks after MCT administration. We used a combination of in vivo and ex vivo approaches to determine whether treatment with RAN could halt the development of the PAH phenotype in a model that reliably reproduces RV impairment and major pathologic features of the human disease, and to extend recent findings by determining whether RAN treatment prevents the induction of arrhythmias/SCD in rats with established PAH.
Materials and Methods
Animals.
Male Sprague-Dawley rats (250–300 g; Charles River, Hollister, CA) were used in this study. Experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of Gilead Sciences (Association for Assessment and Accreditation of Laboratory Care International accreditation no. 001135). Animal use conformed to the National Institutes of Health guidelines (publication no. 85-23, revised 1996). Animals received a single subcutaneous injection of MCT (60 mg/kg body weight) to induce PAH within 28 days, and control rats received an equal volume of solvent (vehicle, 1 ml/kg body weight). All animals had free access to standard rodent chow (Purina 5001; Nestle Purina Pet Care Co., Fairburn, GA) and water for the first week after MCT injection; thereafter, subsets of rats were switched to a diet containing either 0.25% RAN or 0.5% RAN by weight to determine the effect of chronic RAN administration during PAH development (Research Diets, New Brunswick, NJ). Plasma levels of RAN achieved in this study using chronic oral administration (0.25%, 1 to 2 μM; 0.5%, 4–7 μM) are within the therapeutic range (6–8 μM). At these concentrations, RAN selectively inhibits late INa versus other targets (Hale et al., 2008; Zhao et al., 2011).
For the in vivo studies, rats were divided into four groups that received the following: 1) vehicle injection and normal diet (sham), 2) MCT injection and normal diet (PAH), 3) MCT injection and diet containing 0.25% RAN, and 4) MCT injection and diet containing 0.5% RAN.
Rats used in the ex vivo studies (isolated heart) were separated into four treatment groups: 1) vehicle injection and normal diet (sham), 2) vehicle injection and diet containing 0.5% RAN (sham plus 0.5% RAN), 3) MCT injection and normal diet (PAH), and 4) MCT injection and diet containing 0.5% RAN (PAH plus 0.5% RAN). For all studies, Purina 5001 (Nestle Purina Pet Care Co.) was used as the standard rodent chow, and the only difference between diets was the concentration of RAN (0% for sham and 0.25 or 0.5% by weight for respective PAH experiments).
In Vivo Measurements.
At 4 weeks after MCT administration, rats were anesthetized using isoflurane (Sigma-Aldrich, St. Louis, MO), and they were then intubated and ventilated. Body temperature was measured using a rectal thermometer and was maintained at 37°C using a heating pad. After a stabilization period, pulmonary hemodynamics were assessed using a Millar catheter (SPR-839; Millar Instruments, Houston, TX) inserted into the right ventricle and advanced into the pulmonary artery. Thereafter, the same SPR-839 catheter was slowly pulled back into the right ventricle to also acquire RV hemodynamics. A separate Millar catheter (SPR-869) was inserted into the right common carotid artery for measurement of systemic arterial pressure. After allowing for stabilization from surgical preparation, data were continuously recorded on a personal computer using a pressure-volume unit (model MPVS-300; ADInstruments, Colorado Springs, CO). In addition, an electrocardiogram (ECG) was recorded using subdermal needle electrodes (25 gauge; ADInstruments) in the lead II configuration.
B-type natriuretic peptide (BNP) (AbCam Inc., Cambridge, MA) protein analysis in plasma was performed according to the manufacturer’s instructions. In brief, a 1:4-fold dilution and a 1:10-fold dilution of plasma for BNP and tissue inhibitor of metalloproteinase-1, respectively, were incubated for 2 hours at room temperature using the manufacturer’s assay reagents. Both BNP and tissue inhibitor of metalloproteinase-1 protein concentrations were calculated from their assay standard curve optical density values.
Tissue Collection and Morphologic Measurements.
At the conclusion of hemodynamic procedures, rats were euthanized so that blood and organs could be collected for further evaluation. The heart was dissected into atria, right ventricle, and left ventricle (including the septum), plotted dry with gauze, and weighed. RV hypertrophy was determined by the ratio of RV weight divided by tibia length (RV/TL). This measurement has the advantage of being unaffected by body weight and left ventricular (LV) mass changes (Yin et al., 1982).
Measurements of Fibrosis and Inflammation Markers.
Paraffin-embedded cross sections (5 μm) of the right ventricle were prepared from paraformaldehyde-fixed tissues and were stained with Masson’s trichrome for tissue collagen and then imaged. The mRNA gene expression of collagen 1α1, connective tissue growth factor (CTGF), and transforming growth factor-β (TGF-β) in RV tissue was determined using Luminex xMap technology according to the manufacturer’s instructions (Luminex Inc., Austin, TX). Briefly, approximately 50 mg tissue was lysed in homogenizing buffer (Affymetrix Inc., Santa Clara, CA) and then incubated with an mRNA detection probe panel (Affymetrix Inc.) at 54°C overnight. On the following day, tissue lysates and mRNA detection probe cocktail were hybridized with amplifier reagents in accordance with the manufacturer’s instructions. Data were collected using laser-detected Luminex SD equipment and were analyzed by Masterplex CT software (Luminex Inc.).
Langendorff Perfusion.
At 28 days after MCT injection, rats were anesthetized (60 mg/kg ketamine/xylazine i.p.), and hearts were isolated, mounted on a Langendorff perfusion system, and perfused at constant pressure (70 mm Hg) with Krebs-Henseleit buffer containing the following: 118 mM NaCl, 25 mM NaHCO3, 11 mM glucose, 4.2 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 0.5 mM EDTA, and 1.5 mM CaCl2. The buffer was continuously gassed with 95% O2 and 5% CO2 (pH 7.4) at 37°C. The effluent of the Thebesian veins was drained via polyethylene tubing (PE-50) placed at the LV apex. Isovolumic contractile performance was measured using a water-filled balloon inserted into the left ventricle (for more information, see the Supplemental Data and Supplemental Table 1). Hearts were paced at 5 Hz using a bipolar electrode placed on the epicardial wall of the right ventricle and connected to a stimulator (S88 stimulator; Grass Technologies, Warwick, RI).
Measurements of Electrophysiological Parameters in the Isolated Heart.
Pseudo ECGs were obtained with Ag-AgCl monopolar ECG electrodes (Harvard Apparatus, Holliston, MA) and the heart-rate corrected QT (QTc) intervals were calculated according to the Bazett formula. Monophasic action potentials (MAPs) were recorded at the epicardial surfaces of the left and right ventricles using pressure-contact mini MAP electrodes (Harvard Apparatus). Monophasic action potential duration (MAPD) was measured at 30, 50, and 90% repolarization. The interventricular dispersion of repolarization was defined as RV – LV MAPD90. The ventricular effective refractory period (ERP) was determined as the shortest S1 to S2 interval generating an MAP by stimulating hearts using an S1 to S2 protocol: S1 pulses were continuously applied at an interval of 200 milliseconds, and an S2 stimulus was introduced at various coupling intervals that were reduced from 100 to 50 milliseconds at 10-millisecond decrements and from 48 to 30 milliseconds at 2-millisecond decrements.
To trigger ventricular arrhythmia, hearts paced at 6 Hz underwent burst pacing (50 Hz for 1 second), at which time the electrical pacing was stopped to allow the heart to resume its intrinsic rate. Stimulus intensity during burst pacing was increased until tachycardia or fibrillation was induced (Benoist et al., 2011).
Induction of Arrhythmias in PAH Rats and Isolated Hearts during Acute RAN Treatment.
In in vivo experiments, cardiac arrhythmia was induced with isoproterenol (ISO, 0.1 mg/kg i.v.; ISO challenge) in a group of rats 49–60 days after MCT or vehicle injection. To determine the acute antiarrhythmic effect of RAN, a subgroup rats with PAH received RAN (8 mg/kg bolus, followed by 10 mg/kg per hour infusion i.v.) 10 minutes before the ISO challenge.
In isolated hearts from rats with established PAH, arrhythmia was provoked with the above-mentioned burst pacing protocol (50 Hz, 1 second, increasing stimulus intensity). Ten rat hearts were treated with 10 µM RAN 10 minutes before and during the burst pacing protocol.
Data were acquired on a PowerLab 8/30 device (ADInstruments) attached to a computer and were analyzed with LabChart Pro7 software (ADInstruments).
Chemicals.
RAN was provided by Gilead Sciences Inc. (Foster City, CA). All other reagents, including MCT, were obtained from Sigma-Aldrich.
Statistical Analyses.
All data are expressed as means ± S.E.M. Statistical analyses were performed using one-way analysis of variance, followed by either the Newman–Keuls post hoc test or Tukey post hoc test for multiple comparisons. The Fisher’s exact test was used for analyzing the differences in the incidences of arrhythmias (Prism 6.00; GraphPad Software, La Jolla, CA). A P value of <0.05 was considered statistically significant.
Results
Chronic Administration of RAN Reduces MCT-Induced RV Hypertrophy and Normalizes RV Performance.
The effects of 0.25 and 0.5% RAN in diet on RV hypertrophy, RV developed pressure (in millimeters of mercury), and plasma levels of BNP (in picograms per milliliter) in MCT rats are summarized in Fig. 1. At 4 weeks, as expected, MCT administration caused RV hypertrophy (9.8 ± 0.8 versus 6.2 ± 0.1 mg/mm, RV/TL), increased RV developed pressure (68 ± 7 versus 24 ± 0.3 mm Hg), and increased plasma BNP levels 3-fold (527 ± 113 versus 173 ± 89 pg/ml) compared with sham controls (Fig. 1). Chronic treatment with RAN (0.25 and 0.5%), starting at 1 week after the administration of MCT, dose-dependently reduced RV hypertrophy (7.8 ± 0.7 and 5.7 ± 0.3 mg/mm), RV developed pressure (51 ± 6 and 34 ± 2 mm Hg), and BNP plasma levels (266 ± 114 and 57 ± 17 pg/ml). RAN had no effect on systemic blood pressure or heart rate (Table 1).

Effect of chronic administration of RAN to rats with PAH. At 4 weeks after MCT injection, the rats developed significant RV hypertrophy (A), increased RV developed pressure (B), and increased plasma BNP levels (C) compared with sham animals. Chronic administration of RAN starting 1 week after the injection of MCT dose-dependently decreased RV hypertrophy, RV developed pressure, and plasma BNP levels. Data are presented as means ± S.E.M. (one-way analysis of variance followed by Newman–Keuls post hoc analysis with all pairwise comparisons; n = 6–10). *P < 0.05 versus sham; †P < 0.05 versus MCT; ‡P < 0.05 versus 0.25% RAN.
Effect of chronic treatment with RAN on systemic blood pressure and heart rate in animals with MCT-induced PAH
Measurements were performed at 28 days after injection of MCT (60 mg/kg body weight). One week after MCT injection, two groups of rats were switched to a diet containing either 0.25 or 0.5% RAN by weight.
Chronic Administration of RAN Reduces MCT-Induced PAH.
Pulmonary hemodynamic measurements were acquired directly from the proximal pulmonary artery using a solid-state catheter (SPR-839) at 4 weeks after MCT injection. MCT administration induced significant increases in systolic pulmonary arterial pressure (68 ± 6 versus 23 ± 1 mm Hg), mean pulmonary arterial pressure (40 ± 3 versus 17 ± 1 mm Hg), and diastolic pulmonary arterial pressure (22 ± 1 versus 10 ± 1 mm Hg) compared with sham controls (Fig. 2). Chronic treatment with RAN (0.25 and 0.5%), starting at 1 week after the injection of MCT, dose-dependently reduced systolic pulmonary arterial pressure (53 ± 5 and 34 ± 1 mm Hg), mean pulmonary arterial pressure (32 ± 2 and 24 ± 1 mm Hg), and diastolic pulmonary arterial pressure (18 ± 1 and 15 ± 1 mm Hg; Fig. 2).

Effect of chronic administration of RAN to rats with MCT-induced PAH. Systolic (A), mean (B), and diastolic (C) pulmonary arterial pressures were measured at 4 weeks after MCT injection. PAH caused significant increases in systolic, mean, and diastolic pulmonary arterial pressures compared with control (sham) animals. One week after MCT injection, two groups of rats were switched to a diet with either 0.25 or 0.5% RAN by weight, which dose-dependently decreased pulmonary arterial pressures. Data are presented as means ± S.E.M. (one-way analysis of variance followed by Newman–Keuls post hoc analysis with all pairwise comparisons; n = 6–10). *P < 0.05 versus sham; †P < 0.05 versus MCT; ‡P < 0.05 versus 0.25% RAN. PAP, pulmonary arterial pressure.
Chronic Administration of RAN Reduces RV Collagen Deposition and Fibrotic Gene Expression.
To evaluate the effect of RAN treatment on the development of cardiac fibrosis and remodeling, RV samples were collected 4 weeks after the injection of MCT for histology and gene expression patterns in the right ventricle (Fig. 3). RV sections stained with Masson’s trichrome showed more pronounced collagen deposition from animals with PAH compared with sham controls and with animals treated with 0.5% RAN (Fig. 3, A–C). Right ventricles from rats with MCT-induced PAH had increased expression of the mRNA transcripts for collagen 1α1, CTGF, and TGF-β (Fig. 3, D–F, respectively). RAN dose-dependently reduced the expression of these fibrotic genes in the right ventricle (Fig. 3, D–F).

Representative images of the right ventricle stained with Masson’s Trichrome from sham (A), MCT plus vehicle (B), and MCT plus 0.5% RAN (C) at 4 weeks after the injection of MCT; fibrosis is colored blue. RAN dose-dependently reduced the PAH-induced increases in mRNA expression of collagen1α1 (D), CTGF (E), and TGF-β (F) in the right ventricle. Data are presented as means ± S.E.M. (one-way analysis of variance followed by a Bonferroni multiple comparisons test; n = 5–9). *P < 0.05 versus sham; †P < 0.05 versus MCT; ‡P < 0.05 versus 0.25% RAN. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; RFU, relative fluorescence unit. Original magnification, 100×.
Chronic Administration of RAN Reduces Electrical Remodeling in Hearts Isolated from Rats with PAH.
Hearts were isolated 4 weeks after the injection of MCT from rats treated with vehicle (standard rodent chow) or 0.5% RAN, or from sham animals with and without RAN treatment. The effect of chronic treatment with RAN on the RV MAPD, the ventricular heterogeneity in repolarization (RV-LV MAPD90), and electrophysiological parameters was determined, and these data are summarized in Figs. 4 and 5. Sham animals on standard chow had RV MAPDs of 17 ± 2.2 milliseconds, 24 ± 2.8 milliseconds, and 50 ± 3.8 milliseconds measured at 30, 50, and 90% repolarization, respectively. RV MAPD30 and MAPD50 increased 2-fold in hearts from animals with PAH compared with sham controls (37 ± 1.7 versus 17 ± 2.2 milliseconds and 47 ± 3.5 versus 24 ± 2.8 milliseconds, respectively) and MAPD90 increased 1.6-fold (83 ± 6.0 versus 50 ± 3.8 milliseconds; Fig. 4B). The prolongation of the MAPDs observed at 4 weeks after the injection of MCT was significantly less in hearts from rats with PAH treated with 0.5% RAN (23 ± 1.7, 31 ± 2.1, and 62 ± 3.5 milliseconds for MAPD30, MAPD50, and MAPD90, respectively).

Effect of chronic administration of RAN on the MAP and repolarization abnormalities in the right ventricle of isolated hearts from rats with PAH. (A) Superimposed MAP of hearts from sham (blue), PAH (black), and PAH plus 0.5% RAN (red) animals showing differences in MAP duration. (B) RV MAP duration at 30, 50, and 90% repolarization was significantly prolonged in hearts from rats with PAH and was reduced with 0.5% RAN (0.5% RAN by weight in chow) treatment (n = 5 to 6). (C) Electrical heterogeneity defined as interventricular dispersion (RV-LV APD90) was significantly increased in hearts from rats with PAH and normalized with chronic 0.5% RAN treatment compared with isolated hearts from normal (sham) rats (n = 5 to 6). Data are presented as means ± S.E.M. (one-way analysis of variance followed by Tukey post hoc analysis with all pairwise comparisons). *P < 0.05 versus sham; †P < 0.05 versus PAH only.

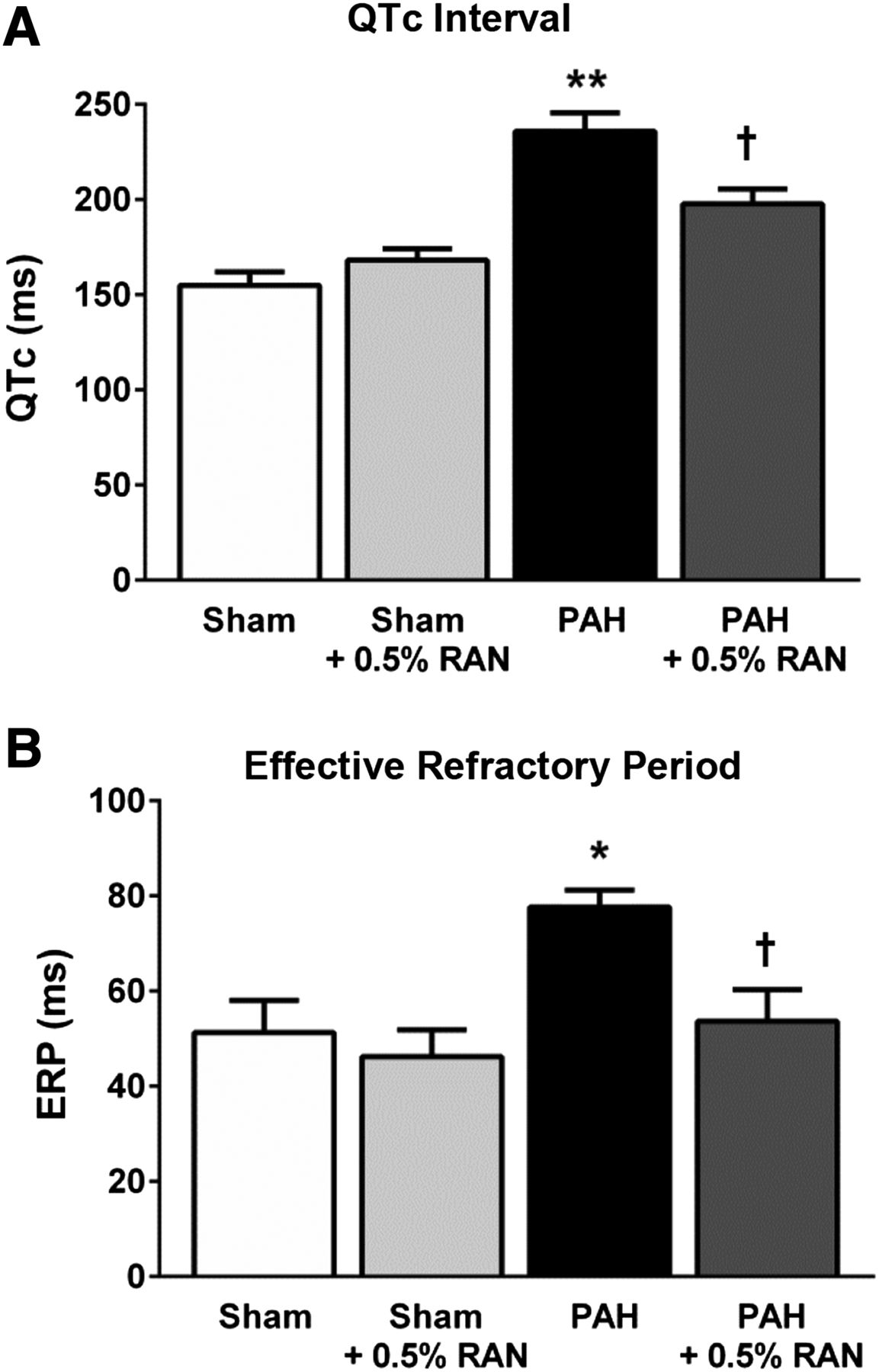
Effect of chronic administration of RAN on electrophysiological parameters. In isolated hearts from rats with PAH, surface ECGs revealed prolonged QTc interval calculated according to the Bazett formula (n = 6 in all groups; A) and increased ERP (n = 4–6; B) compared with normal hearts. Chronic 0.5% RAN treatment, which began at 1 week after the injection of MCT, showed decreased QTc interval and normalized ERP values at 4 weeks after MCT injection. Data are presented as means ± S.E.M. (one-way analysis of variance followed by Tukey post hoc analysis with all pairwise comparisons). *P < 0.05 versus sham; **P < 0.02 versus sham; †P < 0.05 versus PAH only.
In addition, the interventricular dispersion in MAPD90 was markedly increased in hearts from rats with PAH compared with the sham animals (17 ± 5.5 versus −6.1 ± 2.0 milliseconds), whereas RAN treatment normalized dispersion values (−2 ± 2.4 milliseconds; Fig. 4C). Consistent with the MAP data in isolated hearts, MCT-induced prolongation of the QTc interval (236 ± 9.7 versus 155 ± 7.1 milliseconds) was significantly decreased in animals with PAH chronically treated with 0.5% RAN (198 ± 7.8 milliseconds; Fig. 5A). ERP, which was increased in hearts from rats with PAH compared with sham controls (78 ± 3.5 versus 51 ± 6.8 milliseconds), was also normalized in hearts from rats with PAH treated with 0.5% RAN (54 ± 6.6 milliseconds; Fig. 5B). Treatment with 0.5% RAN in sham animals had no (significant) effect on any of these electrophysiological parameters measured.
Effect of Chronic Administration of RAN on Arrhythmogenesis in Hearts Isolated from Rats with PAH.
The effect of RAN on the proarrhythmic potential of PAH was determined using isolated hearts from rats treated with vehicle or 0.5% RAN at 4 weeks after the injection of MCT. Burst pacing for 1 second at 50 Hz induced ventricular tachycardia (VT)/ventricular fibrillation (VF) in 50% of hearts isolated from rats with PAH (three of six hearts). By contrast, VT/VF was not inducible in hearts isolated from RAN-treated rats (P = 0.2) and was only induced in one of six hearts isolated from sham animals (Fig. 6A). Representative tracings of MAP, ECG, and LV pressure during arrhythmia induction are shown in Fig. 6, B and C. Tracings from hearts isolated from sham and RAN-treated animals with PAH show that the hearts returned to normal sinus rhythm and function almost immediately after burst pacing, and a triggered VF episode is evident in tracings from a heart of a rat with PAH without RAN (Fig. 6, B and C).

Effect of chronic administration of RAN on electrical stimulation–induced arrhythmias in isolated hearts from rats with PAH (4 weeks after the injection of MCT). (A) Summarized data of incidences of VT/VF triggered by burst pacing in hearts isolated from normal, PAH, or PAH plus 0.5% RAN (P = 0.2, Fisher’s exact test). (B and C) Representative tracings of RV MAP, ECG, LVP, and of electrical stimulation depicted before, during, and after burst pacing (1 second, 50 Hz) experiments in isolated hearts from rats with PAH (B) and rats with PAH treated with 0.5% RAN in diet by weight (C). LVP, left ventricular pressure.
Acute Effect of RAN on Arrhythmogenesis in PAH Rats and Isolated Hearts.
Both in vivo and ex vivo experiments were conducted to determine the effect of acute administration of RAN on arrhythmia induction in the setting of established RV remodeling (Table 2), and these data are summarized in Fig. 7. Of note, hemodynamics were continually measured to ensure that no changes occurred with the doses of RAN used in these studies. In anesthetized rats with PAH, arrhythmias were induced by a bolus injection of ISO (0.1 mg/kg i.v.; Fig. 7A). Administration of ISO to animals with established PAH caused VT/VF and subsequent death in 70% of animals (7 of 10), whereas ISO induced arrhythmias in only 17% of sham animals (one of six). Furthermore, RAN (8 mg/kg bolus, 10 mg/kg per hour infusion i.v.) given 10 minutes before the ISO challenge completely prevented ISO-induced arrhythmias and death (zero of five; P=0.026 versus without RAN treatment; Fig. 7A). Fig. 7B shows representative tracings of pulmonary arterial pressure, blood pressure, and an ECG in a rat with PAH 6 minutes after an ISO injection that triggered VF.
Summary of pulmonary hemodynamics, RV hypertrophy, and heart rate data from animals subjected to ISO challenge
All rats with PAH had a similar PAH phenotype before the administration of ISO (0.1 mg/kg i.v.). Of note, ISO induced similar increases in heart rate across groups.

Acute administration of RAN in vivo prevents triggered VT/VF in hearts from rats with PAH. Summary data of VT/VF incidences and subsequent SCD in rats with PAH in the presence or absence of RAN (8 mg/kg bolus, followed by 10 mg/kg per hour infusion, i.v.) are shown (*P < 0.03 versus PAH only, Fisher’s exact test). (A) The administration of ISO (0.1 mg/kg, i.v.) to rats with established PAH caused VT/VF. (B) Depicted are representative tracings of pulmonary arterial pressure (PAP), systemic blood pressure (BP), and ECG at 7 minutes after an ISO bolus injection (0.1 mg/kg, i.v.) inducing VF in a rat with PAH (in vivo). Summary data of incidences of VT/VF in the absence or presence of 10 µM RAN are shown for ex vivo experiments. VT/VF was electrically triggered by burst pacing (1 second, 50 Hz). (C) In the PAH group, one heart exhibited spontaneous VT/VF, shown as gray in the black bar. (D) Representative tracings of RV MAP, ECG, LVP, and electrical stimulation before, during, and after burst pacing in isolated hearts from rats with PAH perfused with 10 µM RAN are depicted (ex vivo). LVP, left ventricular pressure.
Similar results were obtained in hearts isolated from rats at day 28 after the administration of MCT (PAH) when VT/VF was triggered using burst pacing for 1 second at 50 Hz. A total of three of six hearts from the PAH group exhibited VT/VF. The electrical stimulation protocol provoked arrhythmias in two hearts, whereas one heart exhibited spontaneous VT/VF before the electrical stimulation protocol. Acute administration of RAN (10 µM) 10 minutes before the burst stimulus completely prevented the induction of VT/VF (0 of 10) in hearts from rats with PAH (P = 0.09 versus without RAN treatment; Fig. 7C). In the sham group, the same stimulation protocol induced arrhythmias in 2 of 11 hearts (18%). Representative tracings of MAP, LV pressure, and an ECG from one RAN-treated PAH heart are shown in Fig. 7D.
Taking all different experiments together, VT/VF could not be induced in PAH rats treated chronically or acutely with RAN, whereas 50–70% of the PAH rats developed provoked VT/VF in the absence of RAN.
Discussion
This study shows that chronic treatment with RAN reduces maladaptive structural and electrical remodeling of the right ventricle and prevents triggered ventricular arrhythmias in rats with MCT-induced PAH. Chronic administration of RAN initiated 1 week after the injection of MCT led to decreased RV hypertrophy and improved pulmonary hemodynamics in a dose-dependent manner. Circulating levels of BNP, a clinically validated biomarker of RV failure, were also significantly decreased in rats treated with RAN, indicating improved RV function (McLaughlin et al., 2013). Hearts isolated from rats with PAH showed a 50% induction in electrical stimulation–induced VT/VF, whereas arrhythmia was not inducible in hearts isolated from PAH rats treated with RAN (acute and chronic). Furthermore, the prolonged action potential duration (APD), ERP, and QTc intervals measured in the right ventricle of animals with PAH were significantly reduced after chronic RAN treatment. In vivo studies using rats with already established PAH and RV remodeling demonstrated that the acute administration of RAN significantly reduced ISO-induced VT/VF and associated cardiovascular death. The results from these in vivo and ex vivo (isolated heart) experiments support that RAN reduces the arrhythmogenic substrate formation in the right ventricle presumably by decreasing electrical heterogeneity (reduced RV-LV APD dispersion) and by decreasing cardiac fibrosis (structural remodeling), which results in an overall improvement in the electrical properties of the right ventricle.
The Effect of RAN on RV Structural and Functional Remodeling in Rats with PAH.
The present understanding of maladaptive ventricular remodeling was established in experimental and human studies of left-sided heart failure; however, many of the same mechanisms have now been described in right-sided heart failure (Borgdorff et al., 2013; Simon, 2013). Importantly however, there are prominent embryological, structural, and metabolic differences that preclude the extrapolation of data between the two ventricles (Bogaard et al., 2009; Simon, 2013).
This study was designed to investigate the effect of chronic treatment with RAN in an animal model of PAH that led to RV remodeling, electrical abnormalities, and failure. A significant increase in pulmonary arterial pressure after MCT injection was accompanied by RV hypertrophy, increased RV fibrosis, and impaired RV performance compared with sham animals (Figs. 1–3). In the right ventricle of rats with PAH, RAN dose-dependently slowed the development of ventricular hypertrophy and normalized the expression of collagen, CTGF, and TGF-β (Figs. 1 and 3). In addition, in hearts isolated from rats with PAH, cardiac performance of the right ventricle was normalized by RAN treatment (Supplemental Table 1). The results of this study evaluating the effects of chronic treatment with RAN are in good agreement with the results from a small clinical study evaluating RAN in patients with PAH (Shah et al., 2012) and with the recent report that RAN prevents MCT-induced pulmonary hypertension, RV hypertrophy, and RV fibrosis (Rocchetti et al., 2014). Our results are also consistent with the study conducted by Fang et al. (2012), which demonstrate that RAN treatment improves RV performance as measured by exercise capacity and cardiac index in rats subjected to RV pressure overload by pulmonary artery banding.
BNP, a well characterized biomarker that is predictive of RV performance, mortality, and clinical outcomes in patients with PAH (Sztrymf et al., 2010; McLaughlin et al., 2013; Vonk-Noordegraaf et al., 2013), was significantly increased in rats with PAH and was decreased by 0.5% RAN (Fig. 1C). BNP is produced by and released from ventricular cardiomyocytes (Bruneau et al., 1997), and plasma levels of BNP are closely linked to the severity of ventricular remodeling and dysfunction (Sztrymf et al., 2010; McLaughlin et al., 2013). The effect of RAN in reducing BNP in our study is consistent with the benefits of RAN in animal models of left-sided heart disease as well as with the amelioration of RV failure in this animal model (Aistrup et al., 2013; Guihaire et al., 2013; McLaughlin et al., 2013).
The Effect of RAN on RV Electrical Remodeling in Rats with PAH.
Cardiac arrhythmias are major contributors to morbidity and mortality in patients with PAH, can contribute to deteriorations of cardiac function, and are not adequately addressed by current therapies (Rajdev et al., 2012; Rich et al., 2013). Importantly, a recent prospective clinical study reported that the restoration of sinus rhythm using antiarrhythmic therapy was associated with a reduction in circulating BNP levels and improved survival (Olsson et al., 2013).
In the present study, the hearts from rats that received MCT had electrophysiological changes in the right ventricle that were proarrhythmic (e.g., prolongation of QTc interval and heterogeneity of APD) and that had been reported in humans with PAH (Rajdev et al., 2012; Rich et al., 2013; Tanaka et al., 2013). As shown in Figs. 4 and 5, chronic treatment with RAN reduced the PAH-associated electrophysiological abnormalities. In addition, ventricular heterogeneity in repolarization (RV-LV APD90), a major contributor to the electrophysiological substrate (i.e., re-entry) leading to the occurrence of lethal arrhythmias, was also decreased by RAN.
It was recently demonstrated that the preventative administration of RAN blocks late INa enhancement in RV myocytes and partially prevents the initiation of MCT-induced RV remodeling (Rocchetti et al., 2014). In that study, RAN administration was associated with improvements in Ca2+ handling, decreases in t-tubule disarray, and reduced incidence of delayed afterdepolarizations in RV myocytes (Rocchetti et al., 2014). Our data extend these findings by demonstrating that treatment with RAN decreases the proarrhythmic substrate of the remodeled right ventricle and results in decreased incidences of provoked arrhythmias and protection from ISO-induced VT/VF. Our results from studies using chronic and acute RAN treatment in a model of RV dysfunction are also consistent with the well described antiarrhythmic effects of RAN (Aistrup et al., 2013; Shryock et al., 2013) and support the therapeutic strategy of normalizing the QTc interval and APD to reduce the arrhythmogenesis of diseased hearts.
Although reports of documented VT/VF are relatively rare in patients with PAH (approximately 8%) compared with the reported incidence of bradycardia (45%), electromechanical dissociation (28%), and asystole (15%), VT/VF remains a clinically relevant risk for many patients with PAH (Hoeper et al., 2002; Vonk-Noordegraaf et al., 2013). In this study, a higher susceptibility to provoked VT/VF was observed in vivo (induced in 70%) and in isolated hearts from rats with PAH (induced in 50%) compared with normal hearts (induced in approximately 18%; Fig. 6). VT/VF inducibility was not observed with chronic RAN treatment and was prevented by acute administration of RAN to animals with established RV remodeling (Fig. 7, A and B). Of note, a single incident of SCD due to an episode of spontaneous VT/VF was captured during telemetric recording in a conscious rat several weeks after the injection of MCT (Supplemental Fig. 1). These data are in good agreement with those of Benoist et al. (2011), who demonstrated that MCT administration resulted in a higher incidence of provoked arrhythmias and sustained VT/VF compared with normal rat hearts.
Limitations.
Late INa was not directly measured in this study; however, there is extensive clinical and experimental evidence to support late INa inhibition as the primary mechanism for the cardioprotective effects of RAN (Maier et al., 2013; Shryock et al., 2013). Notably, Rocchetti et al. (2014) recently demonstrated that late INa is enhanced in the right ventricle of MCT rats and is blocked by RAN. In addition to inhibiting late INa, RAN has been reported to inhibit adrenergic receptors and to partially inhibit fatty acid oxidation at high concentrations (>100 μM; Minotti, 2013). Although these mechanisms cannot be completely excluded, it is unlikely that they are major contributors to the efficacy of RAN in MCT-induced PAH, because the plasma levels of RAN achieved in our study have been shown to selectively inhibit late INa versus all other targets (Hale et al., 2008; Zhao et al., 2011). Of note, steady-state plasma levels of RAN (≤10 μM) had no effect on ISO-induced increases in heart rate in our acute studies, indicating the lack of antiadrenergic effects.
In this study, RAN, an antianginal, anti-ischemic drug known to have beneficial effects in models of LV dysfunction, was efficacious in reducing both structural and electrical remodeling of the right ventricle in an animal model of PAH. In in vivo experiments, chronic administration of RAN dose-dependently decreased pulmonary artery pressure, RV hypertrophy, RV dysfunction, and the expression of fibrosis markers (collagen 1α1, CTGF, and TGF-β) in the right ventricle. These findings were supported with ex vivo experiments showing that RAN decreased electrical remodeling of the right ventricle, as indicated by reduced repolarization abnormalities and by decreases in the prolonged ERP and QTc intervals. In in vivo experiments, we showed that acute RAN treatment prevented triggered arrhythmias in hearts of rats with PAH (Fig. 7). In conclusion, the administration of RAN during PAH development decreased adverse structural remodeling and reduced the electrophysiological abnormalities of the diseased right ventricle, providing evidence that the inhibition of late INa may have beneficial effects in the setting of RV impairment due to PAH.
Acknowledgments
The authors thank Dr. Sarah Fernandes for expert assistance with the telemetry study.
Authorship Contributions
Participated in research design: Liles, Hoyer, Chi, Dhalla, Belardinelli.
Conducted experiments: Liles, Hoyer, Oliver.
Performed data analysis: Liles, Hoyer, Oliver.
Wrote or contributed to the writing of the manuscript: Liles, Hoyer, Chi, Dhalla, Belardinelli.
Footnotes
- Received December 8, 2014.
- Accepted March 12, 2015.
J.T.L. and K.H. contributed equally to this work and are co-first authors.
This research was supported by Gilead Sciences, Inc. All of the authors are employees of Gilead Sciences.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- APD
- action potential duration
- BNP
- B-type natriuretic peptide
- CTGF
- connective tissue growth factor
- ECG
- electrocardiogram
- ERP
- effective refractory period
- INa
- sodium current
- ISO
- isoproterenol
- LV
- left ventricular
- MAP
- monophasic action potential
- MAPD
- monophasic action potential duration
- MCT
- monocrotaline
- PAH
- pulmonary arterial hypertension
- QTc
- heart-rate corrected QT
- RAN
- ranolazine
- RV
- right ventricular
- SCD
- sudden cardiac death
- TGF-β
- transforming growth factor-β
- VF
- ventricular fibrillation
- VT
- ventricular tachycardia
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}