


Abstract
After treatment of millions of patients suffering from gastroesophageal reflux disease (GERD) and other acid-related ailments with proton pump inhibitors, there are still unmet medical needs such as rapid and reliable pain relief, especially for nocturnal acid breakthrough. In this work, we introduce and characterize the biochemistry and pharmacology of the potassium-competitive acid blocker (P-CAB) soraprazan, a novel, reversible, and fast-acting inhibitor of gastric H,K-ATPase. Inhibitory and binding properties of soraprazan were analyzed together with its mode of action, its selectivity, and its in vivo potency. This P-CAB has an IC50 of 0.1 μM if measured with ion leaky vesicles and of 0.19 μM in isolated gastric glands. With a Ki of 6.4 nM, a Kd of 26.4 nM, and a Bmax of 2.89 nmol/mg, this compound is a highly potent and reversible inhibitor of the H,K-ATPase. Soraprazan shows immediate inhibition of acid secretion in various in vitro models and in vivo and was found to be more than 2000-fold selective for H,K-ATPase over Na,K- and Ca-ATPases. Soraprazan is superior to esomeprazole in terms of onset of action and the extent and duration of pH elevation in vivo in the dog. Rapid and consistent inhibition of acid secretion by soraprazan renders the P-CABs a promising group of compounds for therapy of GERD.
Acid-related diseases of the upper gastrointestinal tract, especially gastroesophageal reflux disease (GERD), continue to be a widespread problem worldwide (Bytzer and Blum, 2004). With the introduction of the histamine H2 receptor antagonists, the first effective, acceptable medical treatment became available, which revolutionized therapy of these ailments. However, although this class of drugs was effective in healing peptic ulcers, it was less effective in treatment of erosive esophagitis. Also, all the H2 receptor antagonists show ∼50% tachyphylaxis after administration for several days and do not inhibit the cholinergic stimulation of acid secretion (Teyssen et al., 2001). The introduction and use of proton pump inhibitors (PPIs), drugs targeted against the gastric acid pump, improved acid control. In addition, these drugs were able to heal erosive esophagitis after 8 weeks of treatment (Klinkenberg-Knoll et al., 2000; Stolte et al., 2000) with significant superiority over the H2 receptor antagonists (DiPalma, 2001). However, these are acid-activated prodrugs, and their mechanism of action requires activity of the ATPase and therefore acid secretion to allow conversion to the active thiophilic derivative that binds covalently to the pump. Therefore, the effectiveness of PPIs is dependent on food intake or other means of stimulation of acid secretion. There is a lag phase before secretory inhibition is achieved and is a delay in and less than full steady-state inhibition (Sachs, 2003, 2006) because not all pumps are active during the residence time of effective drug levels in the blood, and turnover of the pump is ∼25% per day (Gedda et al., 1995). The circadian rhythm of acid secretion shows strong activity in the early morning hours. However, after morning dosing, generally only a variable fraction of the available pumps are stimulated and, therefore, inhibited. Both result in ∼70% inhibition of maximal acid output on morning dosage and relatively poor performance at night (Ang and Fock, 2006). To achieve optimal acid suppression and successful therapy, administration of PPIs for at least 3 days is necessary (Sachs, 2001; Bytzer and Blum, 2004; Sachs et al., 2006).
The gastric H,K-ATPase is a P2-type ATPase. This enzyme is responsible for secretion of H+ into the secretory canaliculus of the parietal cell by electroneutral exchange of H+ for K+ (Ganser and Forte, 1973; Sachs et al., 1976). Inhibition of the enzyme is accepted currently as being the best target for medical treatment of acid-related diseases as it is the final step of acid secretion.
As dephosphorylation of the enzyme is dependent on the presence of potassium (Stewart et al., 1981), an alternative means of inhibition of the H,K-ATPase is to block the access of potassium to the ion binding site, competitively or noncompetitively (Wallmark et al., 1987; Vagin et al., 2002, 2003). The discovery that tertiary amines were K+-competitive inhibitors of the ATPase (Im et al., 1984) led to the elucidation of the mechanism of an imidazo-[1,2α] pyridine, SCH28080, that was a potent inhibitor of this type (Beil et al., 1986; Wallmark et al., 1987). Other imidazo-[1,2α] pyridines, such as pumaprazole (von Büdingen et al., 1996), were synthesized and have structures that provide an alternative means of pump inhibition. These particular imidazopyridines bind to the E2P form of the enzyme and are strictly K+-competitive (Mendlein and Sachs, 1990). This mechanism allows rapid inhibition of the pump without the need for acidity at its luminal surface since the pump is blocked in mid-cycle.
Several structural derivatives were tested as inhibitors of the H,K-ATPase, allowing development of a model of the active conformation of SCH28080, in which the benzene ring was orthogonal to the imidazopyridine. The generation of a bridged structure such as a naphthyridine promised greater efficacy than the more flexible SCH28080. This was achieved with the synthesis of soraprazan [BYK61359, (7R,8R,9R)-2, 3-dimethyl-8-hydroxy-7 (2-methoxyethoxy)-9-phenyl-7,8,9,10-tetrahydro-imidazo-[1,2-h][1,7]-naphthyridine].
Here, we describe the properties of this reversible inhibitor of the H,K-ATPase. Soraprazan is purely K+-competitive and therefore has been named a potassium-competitive acid blocker (P-CAB) (Senn-Bilfinger et al., 2006). A more general term for reversible H,K-ATPase inhibitors would be acid pump antagonists (APAs) because not all of these will be strictly K+-competitive. This nomenclature has been used previously for the naphthyridine discussed here (Shin et al., 2005).
The aim of this study was to characterize the potency, affinity, and mode of action of this naphthyridine inhibitor. This work demonstrates that soraprazan is a selective K+-competitive inhibitor of the H,K-ATPase with convincing in vitro and in vivo potency.
Methods and Materials
Hog Gastric H,K-ATPase Enzyme Preparation. The gastric H,K-ATPase was derived from hog gastric mucosa by a previously published method, which involves differential and density gradient centrifugation (Rabon et al., 1988). Briefly, all operations were carried out at 1 to 4°C. The gastric mucosa was stripped from the stomach fundus and homogenized in a solution of 0.25 M sucrose, 5 mM PIPES/Tris, pH 6.8, 1 mM EDTA, and 1 mM EGTA. The homogenate was centrifuged at 11,000 rpm in a Sorvall GSA rotor for 45 min. The pellet was discarded, and the supernatant was centrifuged at 34,000 rpm in a Beckman type 35 rotor for 1 h. The microsomal membrane pellet was resuspended in a solution of 0.25 M sucrose, 5 mM PIPES/Tris, pH 6.8, 1 mM EDTA, and 1 mM EGTA, and was purified on a Ficoll/sucrose step gradient; composed of 34% (w/v) sucrose, 5 mM PIPES/Tris, pH 6.8, 1 mM EDTA, and 1 mM EGTA overlaid by a solution composed of 7.5% Ficoll, 0.25 M sucrose, 5 mM PIPES/Tris, pH 6.8, 1 mM EDTA, and 1 mM EGTA, using a SW28 rotor at 27,000 rpm for 2 h. The ion-tight vesicle fraction above the 7.5% Ficoll gradient was collected and diluted by adding 3 volumes excess of a solution of 5 mM PIPES/Tris, pH 6.8, 1 mM EDTA, and 1 mM EGTA. The suspension was centrifuged at 100,000g for 1 h, and the pellet was resuspended in a solution of 0.25 M sucrose and 5 mM PIPES/Tris, pH 6.8.
The vesicles obtained have been shown to be >90% cytoplasmic side out (Shin et al., 2005). The Mg2+-dependent activity was approximately 5.7 μmol/mg/h. The ion impermeability of the vesicles was determined by the difference in K+ stimulation of ATPase activity in the presence of KCl alone and in the presence of KCl and the ionophore, nigericin, that allows K+ penetration to the luminal surface of the pump. The activity in the presence of nigericin was 105 μmol of ATP hydrolyzed/mg of protein/h and in the absence of nigericin was only 7.6 μmol/mg protein/h. Thus, >90% of the K+-stimulated ATPase activity was dependent on the addition of the K+ ionophore, nigericin, showing that this fraction of the vesicles was K+-impermeant. Pi released was measured by the method of Yoda and Hokin (1970) and protein concentration was determined by a modified Lowry method (Lowry et al., 1951) with 0.1% SDS.
H,K-ATPase Activity in Ion-Leaky Gastric Vesicles. One microgram of protein was incubated in the presence of 1 mM Mg-ATP in 100 mM Pipes-200 mM Tris-HCl buffer, pH 7.4, 250 mM sucrose, and 0.5 to 5.0 mM KCl. Soraprazan was used in nanomolar concentrations for Ki calculation and at 10–4 to 10–9 M for IC50 determination. The reaction was terminated by addition of a 2:1 mixture of 4.5% (w/v) malachite green and 42 g/liter ammonium molybdate. The phosphomolybdate complex was measured at 690 nm in a multiwell spectrophotometer, as described previously (Vagin et al., 2002). Graphic determination of Ki was performed according to Dixon (1953). IC50 values were calculated with the help of GraphPad Prism (version 4.02; GraphPad Software Inc., San Diego, CA).
Proton Transport Activity. Acidification of the gastric vesicles was measured by the quenching of acridine orange. The vesicles at 10 μg/ml were suspended in a medium containing 250 mM sucrose, 150 mM KCl, 3 mM MgCl2, 1 μM acridine orange, 4 mM PIPES-8 mM Tris buffer. pH 7.4, and 10 μg of valinomycin to allow K+ access to the interior of the vesicles. Transport was initiated by the addition of 2 mM ATP (pH 7.4), and the fluorescence of acridine orange was measured over time at an excitation wavelength of 480 nm and emission at 530 nm (Rabon et al., 1978; Wolosin and Forte, 1981) in a VICTOR3 multilabel counter from Perkin Elmer. The inhibitors were added at the indicated concentrations after maximal acidification of the vesicles 30 min after start of the reaction by ATP.
Reversibility. Reversibility experiments were carried out with ion-leaky gastric vesicles as described above. To measure the reversibility of inhibition by soraprazan in the presence of 1 mM KCl, recovery of H,K-ATPase activity after dilution of the incubation mixture was measured. The starting concentration was 2.5 μM soraprazan in two independent experiments conducted in triplicate, and the dilutions were 2-, 5-, 10-, 20-, 50- and 100-fold at constant KCl. H,K-ATPase activity was set to 100% at any dilution without inhibitor.
[14C]Aminopyrine Accumulation in Intact Gastric Glands. Gastric acid secretion is stimulated by gastrin, histamine, and acetylcholine via the receptors on the parietal or the enterochromaffin-like cell. These physiologic stimuli influence the intracellular cyclic AMP and Ca2+ levels, thus leading to relocation and activation of H+,K+-ATPase. Instead of the physiologic agonists, the membrane-permeant dibutyryl cAMP was used to stimulate receptor-independent acid secretion in isolated gastric glands. Accumulation of the weak base [dimethyl-amine-14C]aminopyrine ([14C]AP) in the acidic compartment of the canaliculi serves as an indirect measure of acid secretion and forms the basis of measurement of acid secretion in this in vitro model of the mammalian stomach. Intact gastric glands were prepared from anesthetized New Zealand rabbits (weight 2–3 kg) by high-pressure perfusion of the stomach, separation of the fundic mucosa, and subsequent collagenase digestion of fragments of the mucosa (Berglindh et al., 1976; Berglindh and Obrink, 1976). After the gastric glands were washed several times, they were suspended in Krebs-Henseleit solution containing 2 mg/ml rabbit serum albumin and 2 mg/ml glucose. Glands were incubated for 30 min at 37°C in a shaker bath (200 oscillations/min) in the presence of 0.125 μM [14C]AP (113 μCi/μmol) at pH 7.4. Glands were stimulated with 1 mM dibutyryl cAMP in absence or presence of soraprazan (concentration range 3 nM –100 μM). The reaction was stopped by centrifugation (10 s at 20,000g). After centrifugation, the accumulation of [14C]AP in the glands was calculated as follows: radioactivity was measured in an aliquot of the supernatant (200 μl) and in the precipitate after dissolution in 1 ml of 1 N NaOH. To calculate the amount of protein, the Eppendorf tubes were weighed empty, with protein (wet weight) and with freeze-dried protein (dry weight). This ratio of supernatant and pellet protein radioactivity was used to calculate the accumulation of [14C]AP in the glands. The inhibitor concentration required to achieve 50% inhibition (IC50) of [14C]AP accumulation was determined by fitting the equation for the expected inhibition pattern to the data points.
Soraprazan Binding to Ion-Leaky Gastric H,K-ATPase. [3H]Soraprazan binding studies were carried out at 20°C. In saturation experiments to determine the Kd value, ion-leaky gastric vesicles (0.01–0.02 mg/ml) were resuspended in a buffer composed of 20 mM Tris-HCl, pH 7.0, 2 mM MgCl2, and 2 mM ATP (pH 7.0 by Tris) in the presence of increasing concentrations of [3H]soraprazan (0.1 nM –1 μM). Nonspecific binding was determined in the presence of a 100 fold excess of unlabeled soraprazan over the concentration range of [3H]soraprazan used. The enzyme suspension (1 ml) was incubated at 20°C for 30 min and rapidly filtered through a nitrocellulose membrane filter (HAWP Millipore filter, 0.45 μm) prewet with a solution composed of 20 mM Tris-HCl, pH 7.0, 10% polyethylene glycol 3350 that was placed on top of a glass fiber filter. The membrane was washed five times with 2.5 ml of a buffer composed of 20 mM Tris-HCl, pH 7.0, and 10% polyethylene glycol 3350 to remove unbound inhibitor. The membrane was put into a 20-ml scintillation vial, dimethylacetamide (0.5 ml) was added to dissolve the membrane, and 14 ml of scintillation solvent was added and counted. Binding of [3H]soraprazan was determined by subtracting the non-specific binding of [3H]soraprazan, obtained in the presence of the 100-fold excess of nonradioactive soraprazan, from the amounts of [3H]soraprazan bound to the membrane in the absence of the cold inhibitor.
In KCl competition experiments, a fixed concentration of [3H]soraprazan (18 nM) was incubated in the presence of varying concentrations of KCl (0.001–300 mM) at 20°C for 30 min. An aliquot at a given concentration of KCl was taken out, and the radioactivity bound to the enzyme was measured as described above. All experiments were performed in triplicate or more, and the average of the results was used for analysis.
Na,K-ATPase Measurement. The Na,K-ATPase purified from rabbit kidney was purchased from Professor H. J. Apell (University of Konstanz, Konstanz, Germany). Inhibitor activity on the Na,K-ATPase was measured in the same way as the H,K-ATPase activity in ion-leaky gastric vesicles with the exception that NaCl was added at a 12 mM final concentration.

Chemical structure of SCH28080 and of soraprazan [(7R,8R,9R)-2,3-dimethyl-8-hydroxy-7(2-methoxyethoxy)-9-phenyl-7,8,9,10-tetrahydro-imidazo-[1,2-h][1,7]-naphthyridine].
pH-Metry in the Gastric Fistula Dog. Male Beagle dogs (Boehringer Ingelheim, Biberach/Riβ, Germany and Harlan, Borchen, Germany) were used. At an age of 1 to 2 years, a metallic cannula (V4a or titan) was placed in an artificial fistula at the lowest part of the gastric corpus near the greater curvature. At the beginning of the present study, the animals were aged 2 to 4 years. Their body weight was between 12 and 19 kg (mean ± S.E.M., 14.3 ± 2.3 kg). They were kept in groups of two to four animals and housed at 20 to 23°C, in 55 to 65% relative humidity under a seasonally varying light/dark rhythm. They received water-presoaked standard dog diet (Provimi Kliba, Kaiseraugst, Switzerland) once daily at 10 AM with tap water ad libitum. For 20 to 22 h before and during the day of the experiment, the animals were fasted.
The experimental procedure has been described in detail elsewhere (Postius et al., 1991). Briefly, on the experimental day, the animals were supplied with an ambulatory pH meter containing a solid-state storage unit (Digitrapper pH100; Medtronic, Düsseldorf, Germany) and a programmable infusion pump (Panomat P; Disetronic, Burgdorf, Switzerland). Intragastric pH was measured by means of a combined pH-glass electrode (type 440-M3; Ingold, Urdorf, Switzerland) inserted into the gastric cannula. Gastric acid secretion was stimulated by continuous subcutaneous infusion of pentagastrin (6 μg/kg/h). Recording of intragastric pH started at 8 AM, and pentagastrin infusion began at 9 AM. At 10:30 AM, the animals received the test substance orally. The experiment was terminated at 7:30 AM the next day, and pH readings were transformed by the Polygram98 program (Medtronics) to yield individual 24-h pH profiles. With application of a second program (Stat-pHac2000; Leif Fransson, Karlskrona, Sweden), the individual pH profiles of one treatment group were processed to establish median pH for intervals of 10 min each. Calculation of the significance of the pH levels achieved was performed by use of nonparametric analysis according to the Kruskal-Wallis test. The statistical tools are included in StatpHac2000.
Drugs for dog studies were soraprazan granulate (ALTANA Pharma AG, Konstanz, Germany) and esomeprazole (Nexium MUPS; AstraZeneca Pharmaceuticals LP, Wilmington, DE). Oral administration of the calculated amounts per body weight was performed comparably for both drugs in hard gelatin capsules. Encapsulation has been demonstrated not to affect the release and absorption characteristics of enteric-coated omeprazol MUPS in man (Schaltenbrand et al., 2001). With each animal, a drug-free control run was done. The comparison of both drugs was done as a randomized intraindividual dose-response study with six animals. Oral doses were 1, 3, 9, and 27 μmol/kg.
Primers and Conditions for TaqMan PCR. Human RNAs were obtained from several sources: ABS (Basel, Switzerland), Ambion (Huntingdon, UK), Ardais Corp. (Lexington, MA), Biocat (Heidelberg, Germany), Stratagene (Amsterdam, The Netherlands), and AXXAM srl (Milan, Italy). The purity and integrity of all RNAs was assessed on the Agilent 2100 bioanalyzer with the RNA 6000 NanoChip reagent set (Agilent Technologies, Böblingen, Germany). Samples were treated with DNase to remove traces of contaminating genomic DNA. RNAs were quantified with the Nanodrop ND-1000 spectrophotometer (PEQLAB Biotechnologie GmbH, Erlangen, Germany) and then stored at –80°C. Additionally, the human tissue RNA collection from AXXAM srl in combination with AXXAM srl's TaqMan analysis service was used. RNA samples were exchanged, and TaqMan PCR procedures were cross-validated between ALTANA Pharma AG and AXXAM srl. RNAs for every tissue came from several independent donors from both sexes. RNA (1 μg) was reverse-transcribed using random hexanucleotide primers (Roche Applied Science, Mannheim, Germany), dNTPs (PCR 3 Mix; Larova, Teltow, Germany), and avian myeloblastosis virus (AMV) reverse transcriptase (Roche Molecular Biochemicals, Mannheim, Germany) at 42°C for 1 h. All cDNAs were diluted with Tris-HCl buffer (1 mM Tris and 0.1 mM EDTA pH 8.0, Ambion) to a final concentration of 2 ng/μl and stored at –20°C until further use. As a control for genomic DNA contamination each RNA sample was incubated also in the absence of avian myeloblastosis virus reverse transcriptase, and this sample was run alongside in the TaqMan PCR runs. All primers and VIC-labeled probes were obtained from Applied Biosystems (Darmstadt, Germany). The assay on demand HS00167575_m1 was used for specific detection of the gastric H,K-ATPase α subunit. The following endogenous 18S rRNA control primers and probe were used: sense 5′-CGGCTACCACATCCAAGGAA-3′, antisense 5′-GCTGGAATTACCGCGGCT-3′, and probe 5′-VIC-TGCTGGCACCAGACTTGCCCTC-TAMRA-3′.
TaqMan PCRs were run on ABI 7900 HT and ABI 7700 Sequence Detection Systems (Applied Biosystems). Each PCR reaction was performed in a total volume of 25 μl, containing 2.5 μl of cDNA, 12.5 μl of qPCR Mastermix Plus (Eurogentec, Seraing, Belgium), 1.25 μl of the commercial primer/probe set, and nuclease-free water (Ambion) in 96-well plate format. 18S rRNA primers and probe were used at 50 nM each. The TaqMan PCR parameters were 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of amplification (95°C denaturation for 20 s and 60°C annealing/extension for 1 min). Each run included a water control to check for DNA contamination and probe degradation. All PCRs were performed in triplicate for each sample. Expression levels were calculated from ΔCt with expression in stomach fundus being normalized to 100%·
Immunohistochemistry. Paraffin-embedded human normal tissue slides were supplied by Biocat and DCS (Hamburg, Germany) and were stained as follows in brief. After deparaffination and rehydration to DCS LabLine buffer (AL120R500; DCS), tissue slides were treated with protease (8038; Sigma, Munich, Germany). Slides were blocked with dual endogenous enzyme block (S2003; Dako, Hamburg, Germany) followed by a biotin blocking system (X0590; Dako). Then samples were blocked with 10% normal donkey serum (017-000-001; Dianova, Hamburg, Germany) in Tris-buffered saline. Monoclonal antibody 1H9 (D031-3; MoBiTec, Göttingen, Germany) directed against gastric H,K-ATPase α subunit was used 1:2000 in common antibody diluent (HK156-5K) from DCS with 5% donkey serum. An isotype control antibody was used as negative control (N1698; Dako). Secondary biotin-SP-conjugated Affini Pure donkey anti-mouse IgG (715-065-150; Dianova) was used at 1:1000. Alkaline phosphatase was introduced by use of the StreptABC complex system from Dako (K0391). Signal was visualized with Sigma Fast Red (F4648) and samples were counterstained with ChemMate hematoxylin (S2020; Dako). Tissue slides were mounted on Kaiser glycerin gelatin (109242; Merck, Darmstadt, Germany). Microscopic pictures were taken with an Axiovert 200 microscope from Carl Zeiss (Göttingen, Germany) in combination with Zeiss AxioVision software (version 4.5).

Lineweaver-Burk plot, showing the data used for Ki determination with ion-leaky gastric vesicles. The control line without inhibitor indicates a Km for potassium of 0.57 mM, which is in accord with the literature.

Reversibility of inhibition by soraprazan. Activity of H,K-ATPase was measured in the presence and absence of 2.5 μM soraprazan as the starting concentration. Upon dilution by 2-, 5-, 10-, 20-, 50- or a 100-fold in assay buffer with constant potassium at 1 mM, H,K-ATPase activity in presence of soraprazan returned to almost 100% of control because of dissociation of soraprazan. For each dilution, activity without inhibitor was set as 100% control. Activity in the undiluted reaction was 2.8 nmol of phosphate/h · μg of protein with 2.5 μM soraprazan.
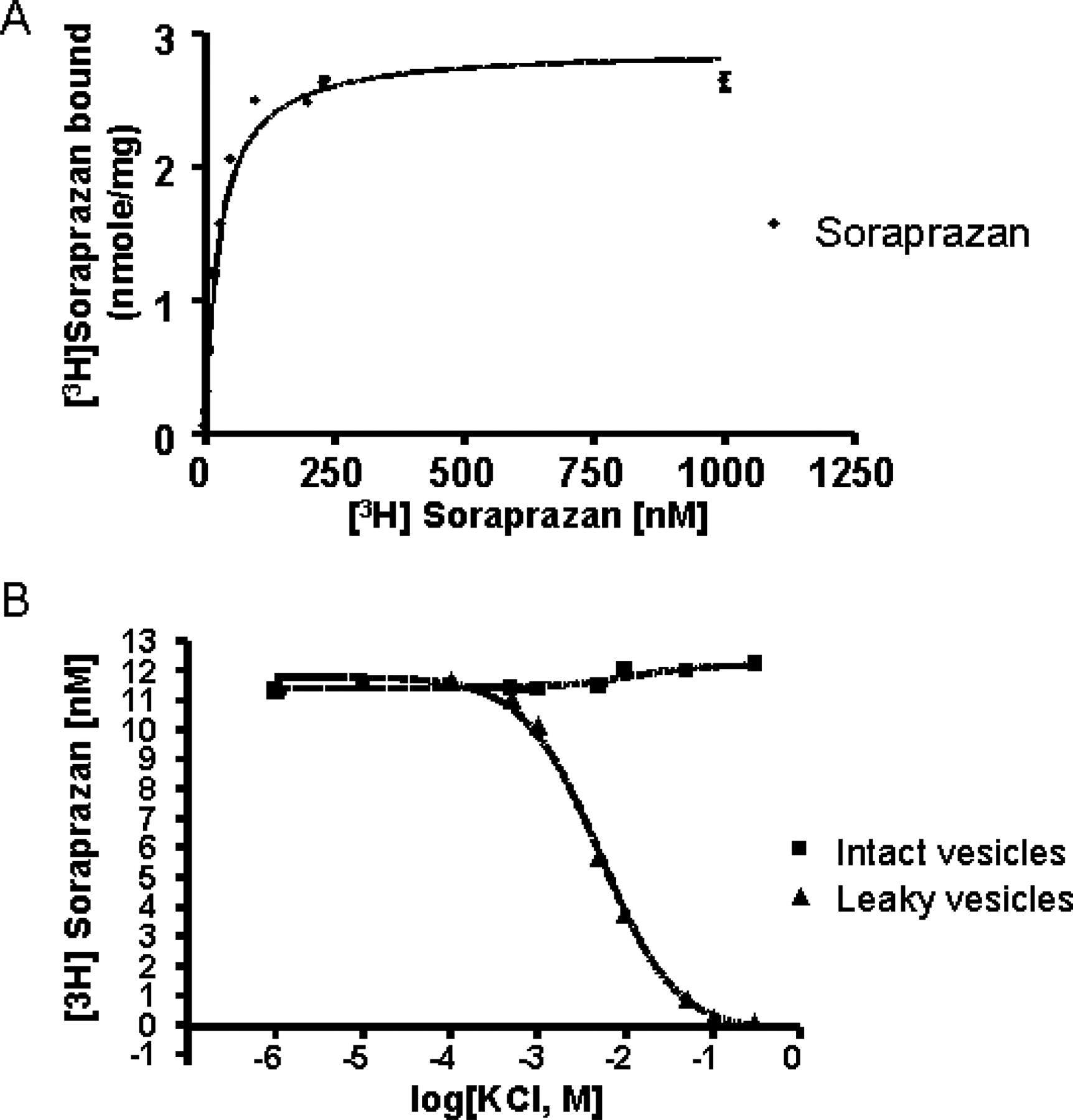
A, gastric vesicles (11 μg/ml) were incubated in a buffer composed of 20 mM Tris-HCl, pH 7.0, 2 mM MgCl2, 2 mM ATP (pH 7.0 by Tris), and nigericin (2 μg/ml), in the presence of soraprazan. Soraprazan bound to the enzyme was determined as described under Materials and Methods. A Bmax of 2.89 ± 0.1 nmol/mg protein was obtained at 20°C. B, soraprazan binding was determined at 20°C in the presence of 2 mM Mg-ATP. The intact gastric vesicles curve represents incubation in the absence of nigericin and the leaky vesicle curve represents incubation in the presence of nigericin. The enzyme (10 μg/ml) was incubated in a buffer composed of 20 mM Tris-HCl, pH 7.0, 2 mM MgCl2, 2 mM ATP, ±5 μg of nigericin/ml, 18 nM [3H]soraprazan, and KCl (0.001–300 mM). No effect of KCl addition was seen in this experiment in the absence of nigericin. In contrast, in the presence of nigericin, a Km(app) of 5.2 mM KCl in the presence of 18 nM soraprazan was observed. Each measurement was an average of three experiments.
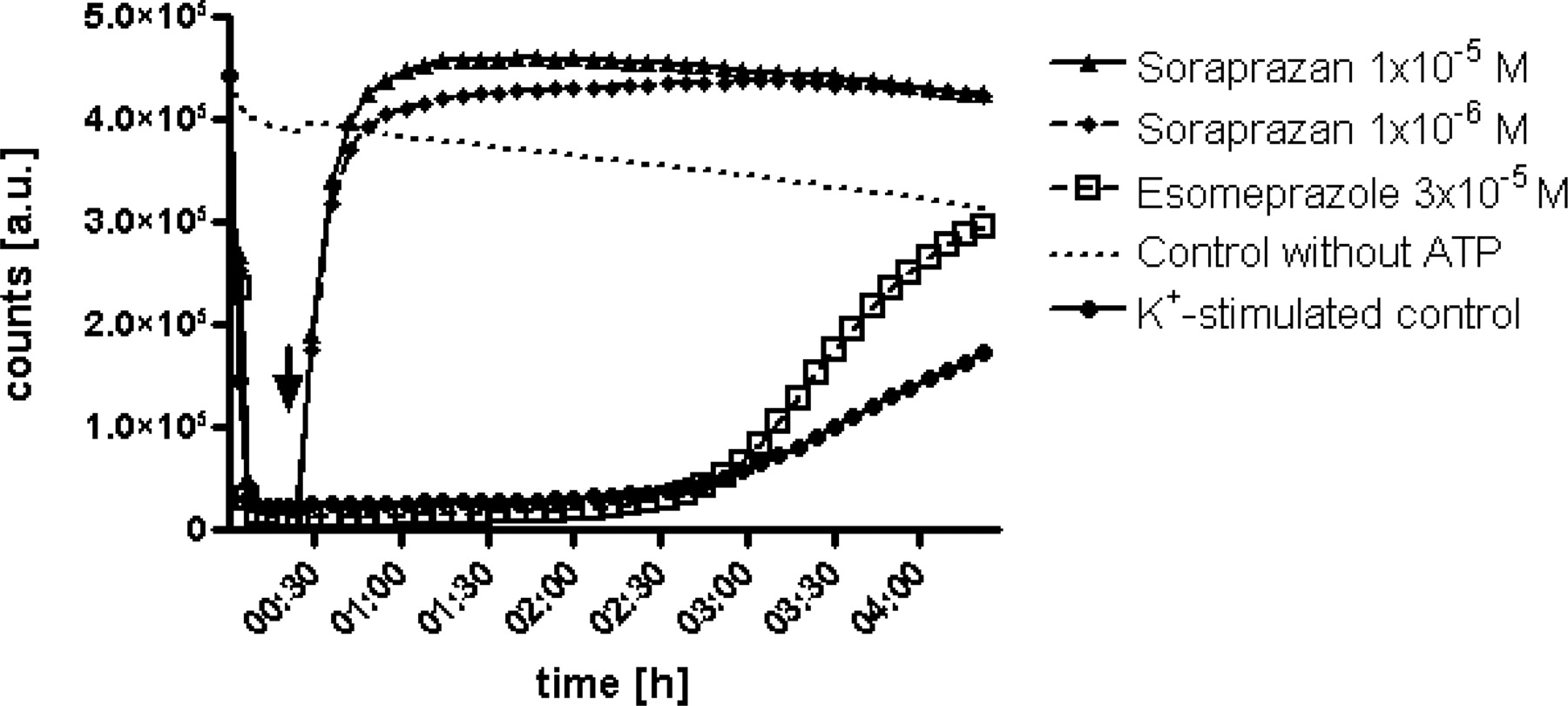
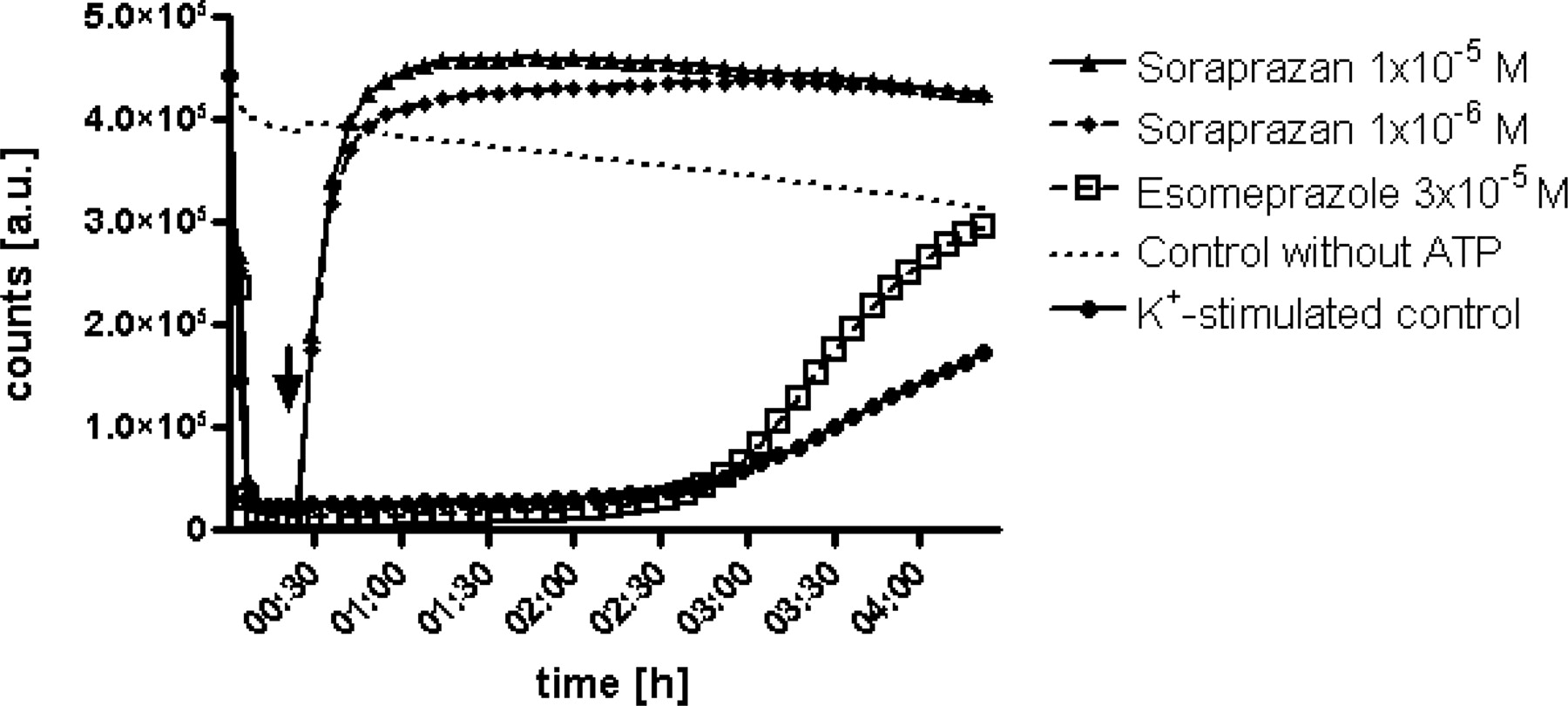
A proton gradient was created in intact vesicles by gastric H,K-ATPase as soon as the enzymatic reaction was started by ATP. Upon addition of the inhibitors (indicated by arrow), inhibition by soraprazan occurred instantaneously, whereas esomeprazole showed the typical delay of acid-dependent activation of a PPI. The inhibition by soraprazan reached its half-maximal level in <5 min after addition of the inhibitor. This immediate and acid-independent mechanism of inhibition is observed with reversible interaction of a substrate with an enzyme. Esomeprazole reached half-maximal inhibition only within hours in this leaky vesicle system. Furthermore, soraprazan displayed a higher pH elevation in this system. Diagrams show arbitrary units (a.u.) over time, which represent relative intravesicular pH values.
Protein Determination. The protein contents of the membrane vesicle preparations were determined according to Lowry et al. (1951).
Chemicals. Soraprazan and esomeprazole [6-methoxy-2-((S)-[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl)-1H-benzimidazole] were obtained from ALTANA Chemical Research. [3H]Soraprazan (specific activity 30 Ci/mmol) was obtained from Amersham. Ionophores were dissolved in methanol. Soraprazan and esomeprazole were dissolved in dimethyl sulfoxide. These solvents were shown not to affect ATPase activity in any assay at the concentrations used. All other reagents were analytical grade or better.
Results
Structure-Activity Relationship of Naphthyridines versus Imidazopyridine. Soraprazan with the benzene ring orthogonal to the imidazopyridine (Fig. 1) was synthesized as described previously (Senn-Bilfinger et al., 2006). The generation of a relatively rigid bridged structure such as a naphthyridine, promised greater efficacy and more metabolic stability than the more flexible SCH28080.
Soraprazan Is Highly Potent in Vitro. Soraprazan is a potent inhibitor of gastric H,K-ATPase. It has an IC50 of 0.1 μM when measured in ion-leaky vesicles in the presence of 1 mM potassium. Soraprazan also effectively inhibits dibutyryl cAMP-stimulated [14C]AP accumulation in isolated gastric glands with an IC50 of 0.19 μM (0.09–0.40 μM geometric mean from n = 6 with 95% confidence limits), which is similar to the IC50 (0.2 μM) found for SCH28080, the first of the reversible proton pump inhibitors with an imidazo[1,2α] pyridine structure (Wallmark et al., 1987).
Kinetics of Soraprazan on the Gastric H,K-ATPase. The Lineweaver-Burk plot shown in Fig. 2 demonstrates that, in ion-leaky vesicles, soraprazan is a potent K+-competitive inhibitor of the H,K-ATPase. The Ki of soraprazan for inhibition of the H,K ATPase is 6.4 nM (arithmetic mean from four separate experiments). From one of these experiments, the K+-competitive characteristic of inhibition was derived (Fig. 2). The validity of this experiment was demonstrated by Km determination for potassium from the same data of 0.57 mM. Soraprazan binds to the H,K-ATPase in ion-leaky vesicles with a Kd of 26.4 ± 3.4 nM and a Bmax of 2.89 nmol/mg. This result was very similar to that found for intact vesicles (Ki, 47 nM; Kd, 30.9 nM) (Shin et al., 2005). The binding of soraprazan in the presence of 2 mM Mg-ATP (Kd 28.45) was in agreement with the Ki of 6.4 nM (Ki, 47 nM; Kd, 30.9 nM) (Shin et al., 2005). Comparable data for SCH28080 have been published (Kd, 45 nM; Ki, 24 nM) (Wallmark et al., 1987; Keeling et al., 1988, 1989).

Relative expression of the gastric H,K-ATPase α subunit in human organs/tissues with significantly detectable expression. Expression was detected by TaqMan PCR, and mean values and S.D. were calculated from ΔCt. The mean of stomach fundus was set as 100%. The number of independent donors measured was stomach cardia, n = 3; fundus, n = 12; corpus, n = 4; antrum, n = 3; pyloric sphincter, n = 3; pancreas, n = 7; adrenal gland, n = 4; adrenal gland cortex, n = 2; adrenal gland medulla, n = 2; and cerebellum, n = 4.
Inhibition by Soraprazan Is Fully Reversible. Consistent with the Lineweaver-Burk plot (Fig. 2), data from dilution inhibition experiments (Fig. 3) reveal that the inhibition of H,K-ATPase by soraprazan is fully reversible. The inhibition of the H,K ATPase after 2-, 5-, 10-, 20-, 50-, and 100-fold dilution subsequently reached the activity seen in the absence of inhibitor (100%).
Additionally, the radioactive binding experiments demonstrate that potassium can displace the radiolabeled soraprazan from its binding site (Fig. 4). There was a concentration-dependent displacement of drug binding by K+ in the presence of nigericin. KCl did not displace drug binding in intact vesicles because the cation does not access the luminal drug binding site. However, in the presence of the ionophore nigericin, KCl displaced the drug, because K+ was able to reach the luminal surface of the enzyme, the location of the inhibitor binding site. There was a concentration-dependent displacement of drug binding by K+ in the presence of nigericin. The Km(app) was 5.2 mM at 18 nM soraprazan. This shows that binding of soraprazan interferes with binding of K+ to the luminal binding site as previously found for SCH28080 (Keeling et al., 1988, 1989; Shin et al., 2005).
Soraprazan Is Highly Selective for H,K-ATPase. Soraprazan has a high selectivity for the H,K-ATPase versus the Na,K-ATPase. The Ki for inhibition of the Na,K-ATPase was found to be 14.6 μM (arithmetic mean from three separate experiments). The geometric mean ± S.D –log Ki was 4.83 ± 0.06 M. This compares with a Ki of 6.4 nM for the H,K-ATPase, demonstrating a selectivity for the latter by a factor of >2000.
Rate of Inhibition by Soraprazan. As demonstrated in Fig. 5, soraprazan inhibits H,K-ATPase immediately without a lag phase. This is the expected result for inhibitors of an enzyme that do not require activation. Upon addition of the compound (arrow) soraprazan immediately raised the intravesicular pH. This rapid inhibition is independent of intravesicular pH. Esomeprazole, in contrast, showed the typical delayed inhibition of a pH-activated PPI with ∼3 h between compound addition and half-maximal inhibition in this assay system (Shin et al., 2004).
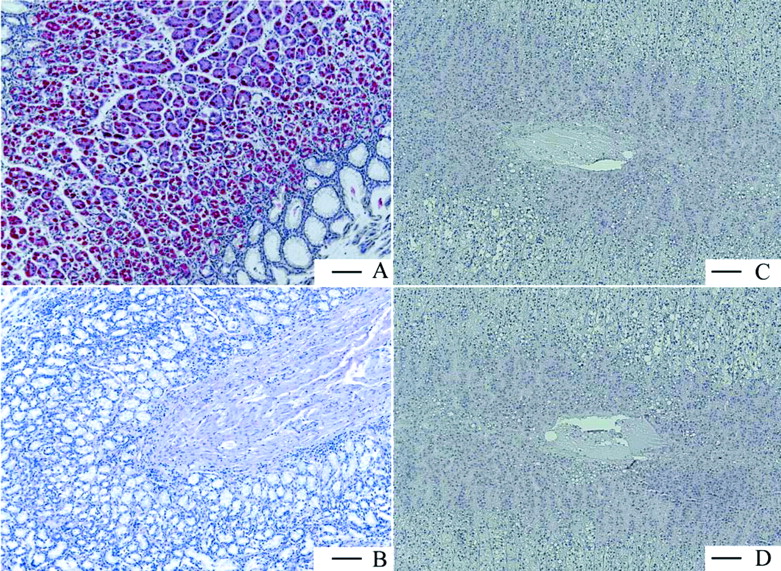
Gastric H,K-ATPase Is a Stomach-Specific Target. The following human organs, tissues, and cells were tested for expression of gastric H,K-ATPase α and β subunits, namely, parts of the gastrointestinal tract from esophagus to colon, kidney, eye, retinal pigment epithelium, pancreas, adrenal gland, skeletal muscle, breast, ovary, placenta, uterus, fallopian tube, lymph node, spleen, heart, tongue, skin, liver, bladder, trachea, bronchus, lung, different parts of the central and peripheral nervous system, testis, prostate, different blood vessels, various adipose tissues, parotid and thyroid gland, white blood cells, thymus, tonsil, bone marrow, and larynx. Figure 6 shows the results for only the organs and tissues in which the TaqMan PCR was positive for the α subunit of H,K-ATPase. Displayed are all organs/tissues with 0.2% of the H,K-ATPase α subunit expression found in stomach fundus. It can be seen that α subunit expression is maximal in stomach and marginally present in some other tissues. Not shown is the respective expression of the β subunit, but the data were negative for adrenal gland and cerebellum. Because, without the β subunit the α subunit is unstable and degraded (Geering, 2001; Vagin et al., 2005), these results show that no significant expression of functional heterodimeric H,K-ATPase is present in organs other than gastric mucosa. These findings were confirmed on the protein level by immunohistochemistry with human tissue slides (Fig. 7). Whereas stomach fundus displayed a strong and very specific staining of the H,K-ATPase α subunit in parietal cells, the adrenal gland was completely negative as was the concomitant isotype control.
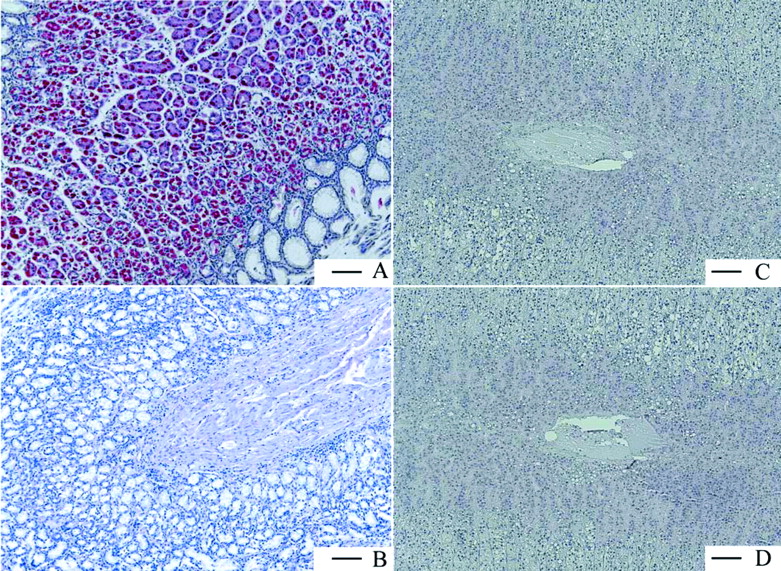
Immunohistologic staining of the gastric H,K-ATPase α subunit in human paraffin tissue slides from stomach fundus (A) and adrenal gland (C) together with the respective isotype negative controls (B and D). The pictures clearly show that no specific staining and no unspecific background was observed in B, C, and D. Scale bar = 100 μm.
Superiority of Soraprazan over Esomeprazole in the Gastric Fistula Dog. In the gastric fistula dog in vivo model, the efficacy of compounds on acid secretion and thus on intragastric pH can be determined. Figure 8 shows the intragastric 24-h pH profiles of both compounds upon oral administration of 1, 3, 9, and 27 μmol/kg. Although both compounds elevate the intragastric pH in a dose-dependent manner, the very straight and clear pH profile of soraprazan differs strikingly from the inconsistent pH-elevating effect of esomeprazole. Statistics of the 24-h profiles are compiled in Table 1. Accordingly, mean times to pH 4 and 6 are fast with all doses of soraprazan (Fig. 8, a–d) but significantly later and highly variable with all doses of esomeprazole (Fig. 8, e–h). At 1 μmol/kg, only one animal reaches pH 4 but none reaches pH 6 with esomeprazole in contrast to three animals for soraprazan for both pH levels. The lowest dose of soraprazan produces pH 4 and 6 faster than any dose of esomeprazole used. The 22-h pH median achieves values of 3.7 and 6.7 at 9 and 27 μmol/kg soraprazan, whereas the same doses of esomeprazole yielded pH medians of 1.9 and 2.2. Neutral pH was reached with 3 and 27 μmol/kg soraprazan and esomeprazole, respectively; 3 μmol/kg soraprazan produces a very consistent intragastric pH-plateau (Fig. 8, b) that is statistically significant compared with controls (p < 0.01). In contrast, even 27 μmol/kg esomeprazole do not produce consistent and reliable intragastric neutrality (Fig. 8, h). Both onset of action and maximum pH reached are much more variable with esomeprazole, which here is clearly inferior to soraprazan (Fig. 7, e–h). No statistical significance against control (Fig. 7, a) could be detected for esomeprazole, whereas 6 h of significant pH elevation was found with soraprazan. A very common measure from clinical studies, the period of time at a pH greater than 4.0 (Bell et al., 1992), is 24.4, 49.6, and 87.4% versus 0, 15, and 35.2% for 3, 9, and 27 μmol/kg soraprazan and esomeprazole, respectively. In this model, soraprazan showed clear superiority over esomeprazole with respect to the extent and duration of pH elevation, speed of rise of pH, and interindividual variation.
Statistical description of the 24-h pH-metry profiles shown in Fig. 8
Twenty-four-hour pH profiles were recorded for six gastric fistula dogs treated with soraprazan and esomeprazole, respectively. Profiles were compared with respect to speed and extent of pH rise as well as stability of the pH plateau reached. Values are calculated on the basis of six intraindividually and randomly performed experiments. t1/2 values for both compounds were determined in separate dog studies.

Dose-dependent effects of soraprazan and esomeprazole on intragastric 24-h pH profiles in the pentagastrin-stimulated gastric fistula dog. Abscissa, time of day; ordinate, intragastric pH. Intraindividual comparison with n = 6 dogs. The diagrams show medians (solid curves) and 25 and 75% quartiles (shaded areas). Continuous stimulation of acid secretion is done by s.c. infusion of pentagastrin from 9:00 AM to 7:30 AM on the next day (open frame in each diagram). Administration of drugs at 10:30 AM (▴): soraprazan (a–d) and esomeprazole (e–h). Oral doses were 1, 3, 9, and 27 μmol/kg. For statistical description of the 24-h pH profiles, see Table 1.
Discussion
Full control of gastric acid secretion has not yet been obtained in clinical practice. It is believed that full inhibition of secretion would alleviate, in particular, nocturnal acid breakthrough that may result in nocturnal symptoms or nocturnal GERD (Ang and Fock, 2006).
Although the PPIs of the substituted pyridyl methyl sulfinyl benzimidazole class treat most of the acid-related complications in the upper gastrointestinal tract, there is still a need to improve suppression of gastric acid secretion to obtain better symptom relief. The drawbacks of the PPIs relate to their mechanism of inhibition and the pharmacokinetics of this class of drugs. The mechanism of inhibition by PPIs requires food intake not less than ½ h after administration of the drug because acid secretion in the secretory canaliculus of the parietal cell has to be induced by food via cholinergic and histaminergic pathways. Acidification on the luminal side of the pump in the membrane of the secretory canaliculus is necessary for accumulation of the PPI and then for the acid-catalyzed conversion of the PPI prodrugs into the active, thiophilic sulfenic acid form that is able to bind to various luminal cysteines of the H,K-ATPase, forming a covalent disulfide and thereby inhibiting the enzyme and, in consequence, acid secretion. Food stimulation of acid secretion and a plasma half-life of ∼1 h make the window for inhibition of active proton pumps very narrow. Acid secretion during day and night therefore cannot be controlled by once daily dosage of PPI in the morning. The mechanism of acid control by the PPIs also involves the dynamics of partial inhibition of the H,K-ATPase after the first dose only, the recovery of acid secretion during 24 h due to food activation of inactive H,K-ATPase, the reversal of inactivated enzyme (Huber et al., 1995), and the daily 25% de novo biosynthesis of new enzyme (Gedda et al., 1995; Shin and Sachs, 2002).
Therefore, a drug has been developed that can immediately and more completely inhibit the H,K-ATPase independent of food and time of administration, because at an adequate dose, no pump would escape inhibition. Soraprazan showed in vitro potency with an IC50 of 0.19 μM in gastric glands and bound to the H,K-ATPase with a Kd of 28.27 nM. This value is close to the Ki for the enzyme. K+-competitive inhibition and full reversibility were shown, the latter by three independent methods (Lineweaver-Burk plot, dilution experiments, and reversal of radiolabeled compound binding). One main characteristic of these reversible inhibitors, the fast onset of action, could be shown impressively in vitro in comparison with esomeprazole (Fig. 5). Vesicles in this in vitro system do not acidify as much as do the canaliculi in vivo, and therefore esomeprazole is not activated at an adequate rate. However, even with faster activation of esomeprazole in vivo, soraprazan is expected to be faster in any system because of its mode of action independent of low pH. Because of the incomplete acidification of vesicles in this in vitro setting, this system emphasizes the advantage of acid-independent inhibition of P-CABs over PPIs. Selectivity against Na,K-ATPases and other ATPases by a factor of >2000 along with the exclusive expression of gastric H,K-ATPase demonstrated in human stomach suggests that no effects on ATPase-mediated H+ or K+ homeostasis in other organs should be found.
Soraprazan demonstrated an impressive in vivo efficacy and superiority to esomeprazole in the fistula dog model. Standard deviations for esomeprazole were always much higher, which translates into higher interindividual variations in dogs and probably also in patients. The in vivo results in the dog clearly show the dose-independent duration of action of esomeprazole. With PPIs, the time of acid inhibition is achieved by blockade of stimulated parietal cells. Because of the short half-life of 0.5 h in dog, an increase in dose does not lead to prolonged duration of action. In contrast, inhibition of acid secretion by soraprazan is not dependent on active parietal cells but rather on the availability of effective plasma concentrations. Therefore, an increasing dose of soraprazan leads to prolongation of pH elevation as seen in classic receptor pharmacology. It is of note that the relation of the half-lives of soraprazan and esomeprazole (2.7 and 1.1 h; R. Huber, unpublished data) is comparable in humans. Therefore, it is likely that the relative effects of both compounds in humans are comparable to those seen in dogs on day 1.
Soraprazan is a potent and reversible inhibitor of the H,K-ATPase and introduces a new class of drugs for therapy of acid-related diseases. There is an increasing awareness of unmet medical needs in GERD therapy today, in particular, complete relief from heartburn, day and night. P-CABs may fulfill this goal for acid-related pain in GERD by raising the intragastric pH to a reliable plateau of pH 6 to 7 and therefore quickly eliminating pain caused by acid reflux into the esophagus. In addition to bringing fast symptom relief to patients with GERD, these effects are expected to be of great value for treating gastrointestinal bleeding and producing pain relief in intensive care patients. The pharmacology of compounds in this class with a fast onset of action favors them for on-demand use in clinical practice. With soraprazan, therefore, it is possible to effectively ablate gastric acid secretion for any period of time. This ablation would eliminate the problems associated with PPIs, such as nocturnal acid breakthrough, and also allow rational on-demand therapy (Sachs et al., 2002; Ang and Fock, 2006). The immediate and profound inhibition of acid secretion, as shown for soraprazan, promises significant improvement in therapeutic efficacy over PPIs in acid-related diseases.
Footnotes
-
W.A.S. and M.H. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.120428.
-
ABBREVIATIONS: GERD, gastroesophageal reflux disease; PPI, proton pump inhibitor; soraprazan, BYK61359, (7R,8R,9R)-2,3-dimethyl-8-hydroxy-7(2-methoxyethoxy)-9-phenyl-7,8,9,10-tetrahydro-imidazo-[1,2-h][1,7]-naphthyridine; P-CAB, potassium-competitive acid blocker; PIPES, piperazine-N,N′-bis(2-ethanesulfonic acid); [14C]AP, [dimethyl-amine-14C]aminopyrine; MUPS, multiple unit pellet system; PCR, polymerase chain reaction; esomeprazole, 6-methoxy-2-((S)-[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl)-1H-benzimidazole.
- Received January 24, 2007.
- Accepted March 14, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}