


Abstract
Although certain antiparkinson agents interact with serotonin (5-HT) receptors, little information is available concerning functional actions. Herein, we characterized efficacies of apomorphine, bromocriptine, cabergoline, lisuride, piribedil, pergolide, roxindole, and terguride at human (h)5-HT1A, h5-HT1B, and h5-HT1D receptors [guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding], and at h5-HT2A, h5-HT2B, and h5-HT2C receptors (depletion of membrane-bound [3H]phosphatydilinositol). All drugs stimulated h5-HT1A receptors with efficacies (compared with 5-HT, 100%) ranging from modest (apomorphine, 35%) to high (cabergoline, 93%). At h5-HT1B receptors, efficacies varied from mild (terguride, 37%) to marked (cabergoline, 102%) and potencies were modest (pEC50 values of 5.8–7.6): h5-HT1D sites were activated with a similar range of efficacies and greater potency (7.1–8.5). Piribedil and apomorphine were inactive at h5-HT1B and h5-HT1D receptors. At h5-HT2A receptors, terguride, lisuride, bromocriptine, cabergoline, and pergolide displayed potent (7.6–8.8) agonist properties (49–103%), whereas apomorphine and roxindole were antagonists and piribedil was inactive. Only pergolide (113%/8.2) and cabergoline (123%/8.6) displayed pronounced agonist properties at h5-HT2B receptors. At 5-HT2C receptors, lisuride, bromocriptine, pergolide, and cabergoline were efficacious (75–96%) agonists, apomorphine and terguride were antagonists, and piribedil was inactive. MDL100,907 and SB242,084, selective antagonists at 5-HT2A and 5-HT2C receptors, respectively, abolished these actions of pergolide, cabergoline, and bromocriptine. In conclusion, antiparkinson agents display markedly different patterns of agonist and antagonist properties at multiple 5-HT receptor subtypes. Although all show modest (agonist) activity at 5-HT1A sites, their contrasting actions at 5-HT2A and 5-HT2C sites may be of particular significance to their functional profiles in vivo.
Several lines of evidence implicate serotonergic pathways in the etiology and treatment of Parkinson's disease. First, patients exhibit decreased levels of 5-HT reuptake sites and of the 5-HT metabolite 5-hydroxyindoleacetic acid, observations possibly related to depressive symptoms comorbid with motor dysfunction (Mayeux, 1990). Second, the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, which depletes nigrostriatal pools of dopamine (DA) and induces a Parkinson's disease-like syndrome, reduces striatal levels of 5-HT (Pérez-Otaño et al., 1991). Third, the antiparkinson agent and DA precursor l-dihydroxyphenylacetic acid (l-DOPA) displaces 5-HT from serotonergic neurons innervating the striatum, wherein it is transformed into DA (Arai et al., 1996; Kannari et al., 2001). Fourth, actions of antiparkinson agents at 5-HT receptors (Newman-Tancredi et al., 2002) may participate in their influence upon the motor, mood, and cognitive symptoms of Parkinson's disease, although serotonergic properties do not underlie their ability to restore motor function per se (see Discussion). The significance of individual subtypes of 5-HT receptor to Parkinson's disease and its management may be outlined as follows.
5-HT1A receptors are enriched in regions controlling motor function, such as the striatum, nucleus accumbens, and frontal cortex (Barnes and Sharp, 1999; Millan et al., 2000): in the striatum, the density of 5-HT1A receptors is elevated after damage to nigrostriatal dopaminergic pathways (Frechilla et al., 2001). Both presynaptic 5-HT1Aautoreceptors and their postsynaptic counterparts exert a modulatory influence upon dopaminergic transmission, motor function, mood, and cognition (Wadenberg, 1996; Barnes and Sharp, 1999; Meneses, 1999;Millan, 2000). Notably, 5-HT1A agonists reverse reserpine-induced hypoactivity (Ahlenius and Salmi, 1995) and abrogate the motor disruption elicited by agonists (dyskinesias) and antagonists (catalepsy) at dopaminergic receptors (Wadenberg, 1996; Bibbiani et al., 2001). 5-HT1B receptors are highly expressed in the substantia nigra, striatum, and corticolimbic structures wherein they modify the activity of serotonergic, cholinergic, and glutamatergic pathways (Bruinvels et al., 1993; Barnes and Sharp, 1999). Although 5-HT1B receptors inhibit DA release in the striatum (Sarhan et al., 2000), they are facilitatory to DA release in the nucleus accumbens wherein, in interaction with dopaminergic terminals, they influence motor function (Przegalinski et al., 2001; Yan and Yan, 2001). However, activation of mesolimbic DA release is implicated in reward mechanisms as well as disturbances of cognition and mood associated with schizophrenia: this is of note because psychosis is a serious problem in Parkinson's disease (Friedman and Factor, 2000; Audinot et al., 2001). Inasmuch as dendritic 5-HT1D receptors complement the inhibitory influence of (terminal) 5-HT1Breceptors upon serotonergic transmission, they may mimic certain of their functional roles. Furthermore, their high concentration in the basal ganglia is of potential pertinence to antiparkinson agents (Bruinvels et al., 1993, Barnes and Sharp, 1999).
There is a high concentration of 5-HT2A receptors in corticolimbic structures controlling motor function and mood as well as in the striatum, wherein they are up-regulated upon elimination of nigrostriatal dopaminergic input (Numan et al., 1995; Barnes and Sharp, 1999; Gresch and Walker, 1999). Some 5-HT2Areceptors are located on substance P-containing output neurons, but the majority are localized on corticostriatal and pallidostriatal afferents: antagonism of the latter sites may reduce the induction of dyskinesias by antiparkinson agents (Gresch and Walker, 1999; Bubser et al., 2001; Naidu and Kulkarni, 2001). Activation of 5-HT2A receptors enhances DA release in the striatum (Ng et al., 1999; De Deurwaerdère and Spampinato, 2001), frontal cortex (Millan, 2000; Millan et al., 2000), and nucleus accumbens (Bowers et al., 2000; Yan et al., 2000), actions underlying their complex, facilitatory influence upon motor function (Millan et al., 1999; McMahon and Cunningham, 2001). However, 5-HT2A receptors in the nucleus accumbens are implicated in the pathogenesis of schizophrenia, raising the possibility that their stimulation may contribute to psychiatric symptoms in Parkinson's disease (Roth and Meltzer, 1995; Friedman and Factor, 2000). Indeed, clozapine, an antipsychotic agent displaying potent antagonist properties at 5-HT2A receptors, attenuates psychotic symptoms in Parkinsonian patients (Roth and Meltzer, 1995; Friedman and Factor, 2000). Although actions at 5-HT2B sites controlling cardiovascular, respiratory, and gastrointestinal function may be relevant to side effects of antiparkinson agents, their functional significance in the central nervous system remains unclear (Barnes and Sharp, 1999). In contrast, 5-HT2C receptors, which are enriched in the substantia nigra and basal ganglia (Wolf and Schutz, 1997; Barnes and Sharp, 1999; Fox and Brotchie, 1999), are of particular pertinence to Parkinson's disease. 5-HT2C agonists exert an inhibitory influence upon striatal, mesolimbic, and frontocortical DA release (Millan et al., 2000; De Deurwaerdère and Spampinato, 2001; Di Matteo et al., 2001) and, correspondingly, suppress motor behavior (Kennett et al., 1996). Accordingly, 5-HT2C receptor antagonists attenuate catalepsy induced by blockade of striatal D2 receptors (Reavill et al., 1999) and potentiate actions of dopaminergic agonists in models of Parkinson's disease (Fox and Brotchie, 1999). Activation of 5-HT2C receptors is also implicated in processes underlying the induction of tremor (Sarkar et al., 2000) and (at subthalamic loci) dyskinesias (Eberle-Wang et al., 1996; Barwick et al., 2000). Finally, 5-HT2C antagonist properties alleviate depressive and anxious states (Kennett et al., 1996; Millan et al., 2000).
Because a detailed characterization of the serotonergic properties of antiparkinson agents has yet to be undertaken, we investigated the efficacies of diverse antiparkinson agents at h5-HT1A, h5-HT1B, h5-HT1D, h5-HT2A, h5-HT2B, and h5-HT2Creceptors Efficacy at “5-HT1 ” receptor subtypes was determined by measuring G protein activation with established [35S]GTPγS binding methodologies (Newman-Tancredi et al., 1999). Efficacy at “5-HT2 ” receptor subtypes was determined using a measure of phospholipase C activation, depletion of membrane-bound [3H]phosphatidylinositols (PI) (Cussac et al., 2002b).
Materials and Methods
Determination of Agonist Efficacy at Recombinant h5-HT1A, h5-HT1B, and h5-HT1DReceptors by [35S]GTPγS Binding.
Efficacy at cloned h5-HT1A, h5-HT1B, and h5-HT1D receptors was determined by measuring stimulation of [35S]GTPγS binding, as described previously (Newman-Tancredi et al., 1999). Briefly, membranes prepared from Chinese hamster ovary (CHO) cells stably expressing h5-HT1A, h5-HT1B, or h5-HT1D receptors were incubated at 22°C for 20 min (h5-HT1A) or 30 min (h5-HT1B and h5-HT1D) with drugs or 5-HT in the following buffer: 20 mM HEPES pH 7.4, 10 mM NaCl, 3 μM GDP, 3 mM MgCl2, and 0.1 nM [35S]GTPγS. Agonist efficacy is expressed relative to that of 5-HT (defined as 100%), which was tested at a maximally effective concentration (10 μM) in each experiment. Experiments were terminated by rapid filtration through GF/B filters (Whatman, Maidstone, UK) using a Packard Instrument Company, Inc. (Downers Grove, IL) 96-well cell harvester and radioactivity determined by liquid scintillation counting. Binding densities (Bmax values) at h5-HT1A, h5-HT1B, and h5-HT1D receptors were 3.6, 8.5, and 1.6 pmol/mg, respectively.
Determination of Agonist Efficacy at h5-HT2A, h5-HT2B, and h5-HT2C Receptors by [3H]PI Depletion.
The functional activity of antiparkinson compounds at h5-HT2A, h5-HT2B, and h5-HT2Creceptors (VSV isoform) was determined as described previously (Cussac et al., 2002). Briefly, cells were labeled with 2 μCi/ml of [3H]myoinositol (10–20 Ci/mmol) for 24 h. Cells were washed and then incubated at 37°C for 30 min with drugs in Krebs-LiCl buffer: 15.6 mM NaH2PO4 pH 7, 120 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 1.2 mM CaCl2, 0.6% (w/v) glucose, 0.04% (w/v) bovine serum albumin, and 10 mM LiCl. At h5-HT2A, h5-HT2B, and h5-HT2Creceptors in each case, in the absence of agonists, ∼40,000 dpm was typically detected, compared with ∼25,000 dpm in the presence of a maximally effective concentration of 5-HT (10 μM). Agonist efficacy is expressed relative to that of 5-HT (defined as 100%), which was tested at a maximally effective concentration (10 μM) in each experiment. For antagonist studies, cells were preincubated for 15 min with drug before the addition of 5-HT: 1 μM for h5-HT2A receptors and 0.03 μM for h5-HT2B and h5-HT2Creceptors. In additional studies, the influence of the selective 5-HT2A receptor antagonist MDL100,907 and of the selective 5-HT2C antagonist SB242,084 against actions of pergolide, cabergoline, and bromocriptine were determined. Membranes were recovered by rapid filtration, and [3H]PI content was determined by scintillation counting. Bmax values at h5-HT2A, h5-HT2B, and h5-HT2C receptors were 2.0, 3.0, and 18 pmol/mg, respectively.
Data Analyses.
Binding isotherms were analyzed by nonlinear regression using the program PRISM (GraphPad Software, San Diego, CA). In antagonist studies, KB values were calculated, as described previously (Newman-Tancredi et al., 1999), according to the equation KB = IC50/{[(2 + (agonist/EC50)nH)nH−1] − 1}, where IC50 is the inhibitory concentration50 of the antagonist, agonist is agonist concentration, EC50 is the effective concentration50 of 5-HT alone, andnH is Hill coefficient of the agonist stimulation isotherm. The EC50 values of 5-HT at h5-HT2A, h5-HT2B, and h5-HT2C receptors used in these calculations were 32, 3.1, and 1.3 nM, respectively. Protein concentrations were determined by use of a bicinchoninic acid kit (Sigma, St. Quentin Fallavier, France).
Drugs.
Lisuride maleate and terguride were donated by Schering (Berlin, Germany). Bromocriptine and pergolide methanesulfonate were purchased from Sigma/RBI (Natick, MA). Apomorphine hydrochloride was purchased from Sigma. Roxindole methanesulfonate was donated by Merck (Darmstadt, Germany), and talipexole was provided by Boehringer Ingelheim GmbH (Ingelheim, Germany). Cabergoline was obtained from Farmitalia Carlo Erba (Rueil-Malmaison, France). Pramipexole dihydrochloride, ropinirole, piribedil hydrochloride,R-(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidine-methanol base [(+)-MDL100,907], ondansetron hydrochloride, and (6-chloro-5-methyl-1-[6-(2-methylpyridin-3-yloxy) pyridin-3-yl carbamoyl] indoline) hydrochloride (SB242,084) were synthesized by Institut de Recherches Servier (Paris, France) chemists.
Results
Drugs Evaluated.
The antiparkinson agents examined herein were those demonstrated to possess significant affinity (pKi ≥ 6.0) at sites studied in competition binding assays documented in the accompanying article (Millan et al., 2002). Thus, quinpirole, quinelorane, talipexole, and TL99 were not included in the present article and both pramipexole and ropinirole were evaluated only at h5-HT1Areceptors.
Drug Actions at h5-HT1A Receptors.
At a maximally effective concentration (10 μM), 5-HT enhanced [35S]GTPγS binding at h5-HT1A receptors by ∼1.5-fold relative to basal values; it displayed a pEC50 value of 7.7 (Fig. 1; Table1). All ligands stimulated [35S]GTPγS binding at h5-HT1A receptors, with efficacies ranging from partial for apomorphine (Emax = 35%) to full for cabergoline (93%) and lisuride (98%). Potencies for stimulation of [35S]GTPγS binding varied considerably from low (piribedil, pEC50 = 5.2) to pronounced (lisuride, 8.90). Pramipexole exhibited partial agonist properties (62.6 ± 8.5%) at high concentrations (pEC50, 4.9). Ropinirole was also a weak (pEC50, 5.3) partial agonist (73.0 ± 4.0%). The correlation coefficient, r (Pearson product-moment), between pEC50 values and pKi values determined in competition experiments (Millan et al., 2002) was 0.91 (P < 0.05).

Actions of antiparkinson agents at h5-HT1A receptors expressed in CHO cells. [35S]GTPγS binding was carried out as described underMaterials and Methods. Binding is expressed as a percentage of that observed with a maximally effective concentration (10 μM) of 5-HT (defined as 100%). Values shown are from representative experiments performed in triplicate and repeated on at least three occasions.
Efficacies (Emax values) and potencies (pEC50 values) of antiparkinson agents at recombinant h5-HT1A, h5-HT1B, and h5-HT1D receptors
Drug Actions at h5-HT1B Receptors.
At a maximally effective concentration (10 μM), 5-HT stimulated [35S]GTPγS binding at h5-HT1B receptors by ∼1.5-fold relative to basal values; it displayed a pEC50 value of 8.1 (Fig. 2; Table 1). All ligands displayed agonist properties except for apomorphine (no stimulation, 10 μM) and piribedil (not tested, pKi < 5.0;Millan et al., 2002). Certain drugs showed low intrinsic activity (such as terguride, 37%), whereas others were highly efficacious (such as pergolide, 90%, and cabergoline, 102%). Drug potencies (pEC50) were modest, ranging from 5.7 (cabergoline) to lisuride (7.6).

Actions of antiparkinson agents at h5-HT1B receptors expressed in CHO cells. [35S]GTPγS binding was carried out as described underMaterials and Methods. Binding is expressed as a percentage of that observed with a maximally effective concentration (10 μM) of 5-HT (defined as 100%). Values shown are from representative experiments performed in triplicate and repeated on at least three occasions.
Drug Actions at h5-HT1D Receptors.
At a maximally effective concentration (10 μM), 5-HT increased [35S]GTPγS binding at h5-HT1D receptors by ∼1.5-fold relative to basal values; it displayed a pEC50 value of 8.9 (Fig. 3; Table 1). Apomorphine (no stimulation at 10 μM) was inactive and piribedil (pKi < 5.0; Millan et al., 2002) was not evaluated. Potencies of other drugs for stimulation of [35S]GTPγS binding were greater (pEC50 values 1 to 2 log units higher) than at h5-HT1B receptors. Relative efficacies also differed between h5-HT1D versus h5-HT1B receptors. For example, bromocriptine was more efficacious, whereas cabergoline was less efficacious.

Actions of antiparkinson agents at h5-HT1D receptors expressed in CHO cells. [35S]GTPγS binding was carried out as described under under Materials and Methods. Binding is expressed as a percentage of that observed with a maximally effective concentration (10 μM) of 5-HT (defined as 100%). Values shown are from representative experiments performed in triplicate and repeated on at least three occasions.
Drug Actions at h5-HT2A Receptors.
5-HT depleted [3H]PI with a pEC50 value of 7.5 (Fig. 4; Table2). Terguride, lisuride, bromocriptine, cabergoline, and pergolide also exhibited, in order of increasing efficacy, agonist properties. These actions were expressed with high potencies (pEC50 values) ranging from 7.6 (terguride) to 8.8 (pergolide). In contrast, apomorphine and roxindole blocked stimulation of [3H]PI depletion by 5-HT (10 μM) with pKbvalues of 6.4 and 7.7, respectively. Piribedil was inactive. The agonist properties of pergolide, bromocriptine, and cabergoline (1.0 μM) were concentration-dependently abolished by the selective 5-HT2A receptor antagonist MDL100,907 with pKb values close to its pKb for blockade of [3H]PI depletion elicited by 5-HT, that is, pKbvalues were 10.09 ± 0.13, 10.01 ± 0.09, and 10.15 ± 0.27 compared with 9.97 ± 0.11 for 5-HT.
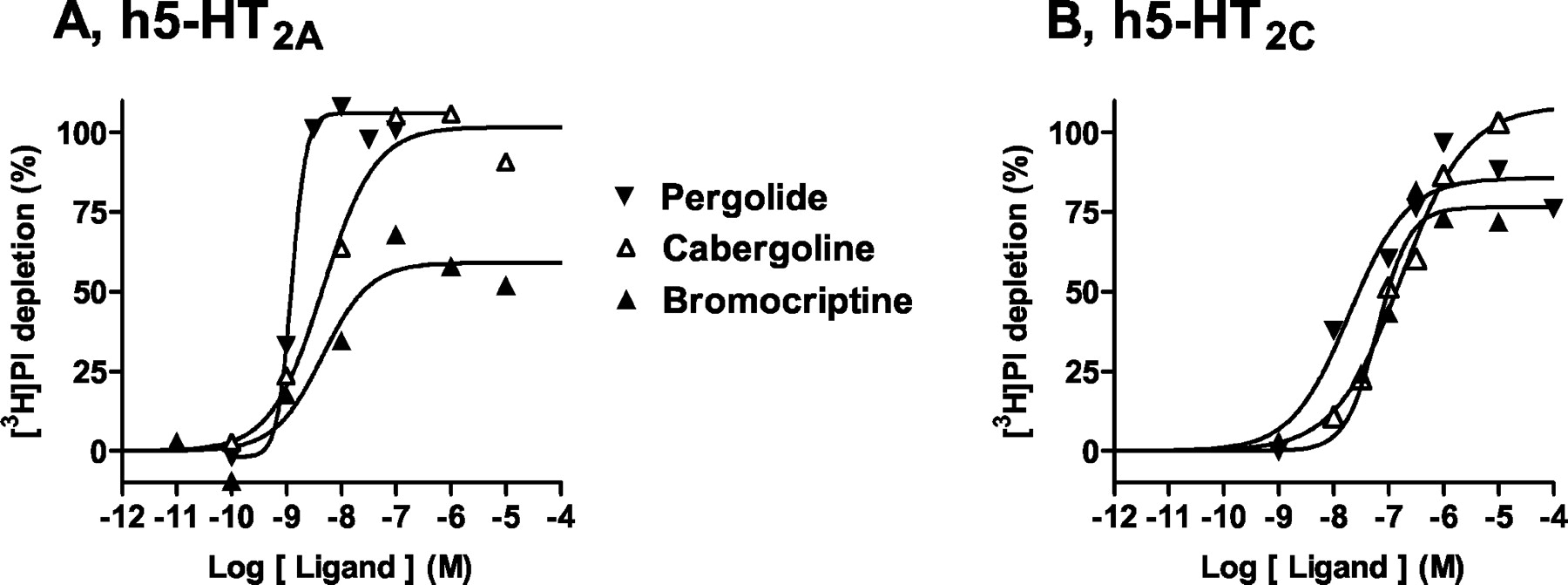
Actions of pergolide, cabergoline, and bromocriptine at h5-HT2A and h5-HT2C receptors. Phospholipase C activity was assessed by determination of [3H]phosphatidylinositol depletion in membranes of CHO cells expressing h5-HT2A (A) or h5-HT2Creceptors (B). Depletion is expressed as a percentage of that observed with a maximally effective concentration (10 μM) of 5-HT (defined as 100%). Values shown are from representative experiments performed in triplicate and repeated on at least three occasions.
Efficacies (Emax values) and potencies (pEC50 or pKb values) of antiparkinson agents at recombinant h5-HT2A, h5-HT2B, and h5-HT2C receptors
Drug Actions at h5-HT2B Receptors.
5-HT depleted [3H]PI with a pEC50 value of 8.52 (Table 2). Only cabergoline and pergolide acted as agonists in potently (pEC50 values of 8.59 and 8.22, respectively) stimulating [3H]PI depletion with high efficacy (123 and 113%, respectively). All other ligands acted as antagonists with potencies ranging from weak (piribedil, 5.99) to pronounced (bromocriptine, 8.89).
Drug Actions at h5-HT2C Receptors.
5-HT depleted [3H]PI with a pEC50 value of 8.9 (Fig. 4; Table 2). Cabergoline and pergolide behaved as efficacious (96 and 87%) agonists, albeit with potencies lower than those at h5-HT2A and h5-HT2B receptors. Both bromocriptine and lisuride also revealed high efficacy (79 and 75%, respectively) in enhancing [3H]PI depletion. On the other hand, terguride, apomorphine, and roxindole manifested antagonist properties. Finally, in line with its low affinity (pKi < 5.0; Millan et al., 2002), piribedil was inactive. The agonist properties of pergolide, bromocriptine, and cabergoline (1.0 μM) were concentration-dependently blocked by the selective 5-HT2C receptor antagonist SB242,084, with pKb values close to its pKb for blockade of [3H]PI depletion by 5-HT, that is, pKb values were 9.73 ± 0.04, 9.81 ± 0.06, and 9.35 ± 0.13 compared with 9.53 ± 0.14 for 5-HT.
Discussion
The present study demonstrates that antiparkinson agents display contrasting profiles of agonist and antagonist activity at multiple subtypes of 5-HT receptor implicated in the etiology and management of Parkinson's disease.
h5-HT1A Receptors.
Using [35S]GTPγS binding, a measure of coupling to G proteins, the clinically active antiparkinson agent lisuride displayed pronounced potency and efficacy at h5-HT1A receptors (Newman-Tancredi et al., 1999). These observations are consistent with agonist properties at 1) 5-HT1A receptors coupled to adenylyl cyclase in rat hippocampus, 2) postsynaptic 5-HT1A receptors controlling behavioral parameters, and 3) 5-HT autoreceptors inhibitory to serotonergic neurons (Barnes and Sharp, 1999; Millan et al., 2000). A further ergot, terguride, likewise stimulated h5-HT1A receptors, although data from in vivo models are lacking. Jackson et al. (1995) reported that bromocriptine possesses high affinity for native 5-HT1A sites and assumed that its increase of 5-HT turnover reflected antagonist properties at 5-HT1A autoreceptors. This explanation seems unlikely in light of its marked efficacy at h5-HT1A receptors. Thus, a more likely interpretation for the enhancement of serotonergic transmission by bromocriptine is its antagonist actions at inhibitory α2-AR heteroceptors (Millan et al., 2002;Newman-Tancredi et al., 2002). Cabergoline and pergolide, which display modest affinity for h5-HT1A receptors (Millan et al., 2002), markedly enhanced [35S]GTPγS binding. Although in vivo correlates of their actions remain to be documented, the potent agonist properties of roxindole at h5-HT1A sites coincide well with its suppressive influence upon central serotonergic transmission (Newman-Tancredi et al., 1999).
Actions of antiparkinsonian agents at 5-HT1Areceptors are of interest because their engagement abrogates the induction of dyskinesias by l-DOPA in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates (Bibbiani et al., 2001). Furthermore, in pilot studies with the 5-HT1A partial agonist, buspirone, an attenuation of spontaneous and l-DOPA-induced dyskinesias was seen in Parkinson's disease patients (Bonifati et al., 1994). However, there is no compelling evidence from experimental models (Ahlenius and Salmi 1995) that selective engagement of 5-HT1Areceptors exerts clinically meaningful antiparkinson actions or that 5-HT1A receptors are involved in the antiparkinson profiles of drugs evaluated herein. Furthermore, 5-HT1A agonists exert a complex influence upon locomotor behavior and blunt DA release in the striatum (Barnes and Sharp, 1999; De La Garza and Cunningham, 2000; Millan, 2000): they also attenuate l-DOPA-induced DA release in the striatum from serotonergic neurons bearing 5-HT1A autoreceptors (Arai et al., 1996; Kannari et al., 2001). These observations, together with the low activity at 5-HT1A receptors of clinically effective antiparkinson agents, such as pramipexole, indicate that their stimulation is neither necessary nor sufficient for treatment of Parkinson's disease. Nevertheless, in association with agonist actions at dopaminergic receptors, modest agonist properties at 5-HT1A receptors may improve motor dyskinesias (vide supra), mood, and cognitive performance (Barnes and Sharp, 1999;Meneses, 1999). This hypothesis justifies clinical evaluation.
h5-HT1B and h5-HT1D Receptors.
Lisuride and bromocriptine interact with native 5-HT1B sites, and notwithstanding species differences (Barnes and Sharp, 1999; Audinot et al., 2001), they exerted agonist actions at h5-HT1B receptors herein with high and modest potency, respectively. Furthermore, the structurally related ergot derivatives terguride, pergolide, and cabergoline likewise revealed high efficacies, whereas the chemically distinct roxindole is a weak partial agonist (Newman-Tancredi et al., 1999). Except for modest affinities of bromocriptine and lisuride at native 5-HT1D receptors (Barnes and Sharp, 1999), and the low efficacy of roxindole at h5-HT1Dsites (Newman-Tancredi et al., 1999), actions of antiparkinson drugs at h5-HT1D sites have not been reported. The demonstration then that bromocriptine, lisuride, and the other ergots terguride, cabergolide, and pergolide (but neither piribedil nor apomorphine) are agonists at h5-HT1D receptors was unanticipated. Moreover, drug potencies were ∼1 to 2 log units higher than at h5-HT1B sites. In view of the implication of 5-HT1B receptors in the control of dopaminergic transmission, motor behavior, and mood, and of the high concentration of 5-HT1D receptors in human basal ganglia (see Introduction), evaluation of their potential significance in the beneficial and deleterious actions of antiparkinson agents would be justified. However, the lack of activity of apomorphine, pramipexole, and other agents at these sites indicates that their stimulation is not required for therapeutic activity.
h5-HT2A Receptors.
A preliminary report documented partial agonist actions of bromocriptine at SH-SY5Y cells expressing h5-HT2A receptors coupled to cytosolic inositol phosphates (Mitchell et al., 1998). Using the complementary approach of depletion of membrane-bound [3H]PI (Cussac et al., 2002b), bromocriptine similarly activated phospholipase C at h5-HT2A receptors, consistent with its high affinity for h5-HT2A (Millan et al., 2002). Lisuride likewise behaved as a potent partial agonist, corroborating its modest efficacies at h5-HT2A receptors in human embryonic kidney-293 cells (inositol phosphates), NIH-3T3 cells (agonist/antagonist binding ratios), and CHO and SH-SY5Y cells (intracellular Ca2+ levels) (Porter et al., 1999;Egan et al., 2000; Jerman et al., 2001). Like lisuride, a further ergot possessing high affinity for 5-HT2A sites, pergolide (Hagen et al., 1994; Millan et al., 2002), was an efficacious agonist at h5-HT2A receptors, actions mimicked by terguride and cabergoline. In contrast, the structurally distinct roxindole, a potent ligand of h5-HT2A sites (Millan et al., 2002), and apomorphine behaved as antagonists at h5-HT2A receptors, whereas piribedil was inactive. The pronounced agonist properties of pergolide at h5-HT2A sites coincide with the finding that 5-HT2A receptors participate in its induction of hyperlocomotion in rats (Moore et al., 1999).
Although the latter observation is consistent with the facilitatory influence of 5-HT2A sites upon locomotor behavior in general (Millan et al., 1999), its relevance to the motor dysfunction of Parkinson's disease is unclear, and (selective) activation of 5-HT2A sites is not known to be associated with antiparkinson properties. Moreover, antagonist actions at 5-HT2A receptors, probably on corticostriatal-pallidostriatal afferents, improve extrapyramidal motor dysfunction (Gresch and Walker, 1999; Bubser et al., 2001; Oh et al., 2001). Furthermore, stimulation of 5-HT2Areceptors and DA release in limbic structures may be related to the psychiatric (hallucinogenesis) side effects of antiparkinson agents (Roth and Meltzer, 1995; Millan et al., 1999; see Introduction). Nevertheless, antiparkinson agents devoid of affinity for 5-HT2A sites (such as pramipexole) also display psychiatric effects as well as therapeutic efficacy. Thus, experimental and clinical studies comparing antiparkinson drugs with differing efficacies/potencies at h5-HT2A receptors would help elucidate their significance to beneficial and undesirable actions of antiparkinson agents.
h5-HT2B Receptors.
In contrast to h5-HT2A receptors, lisuride is a potent antagonist at h5-HT2B receptors (Porter et al., 1999; Jerman et al., 2001; Cussac et al., 2002), and all other antiparkinson agents weakly (piribedil) to potently (bromocriptine) blocked h5-HT2B receptors except cabergolide and pergolide, which were potent agonists. The significance of central h5-HT2B receptors to antiparkinson agents remains unclear because the only functional role ascribed to their activation is a reduction in anxiety, an observation awaiting confirmation (Duxon et al., 1997). On the other hand, activation of peripheral populations may influence respiratory and gastrointestinal function (Barnes and Sharp, 1999).
h5-HT2C Receptors.
Herein, lisuride displayed significant efficacy at h5-HT2C receptors (VSV isoform), in analogy to its partial agonist properties at the VNV isoform, whereas it exhibited low efficacy at SH-SY5Y cells expressing the wild-type (INI) isoform (Egan et al., 2000; Jerman et al., 2001). The contrasting lack of agonist activity of lisuride in a previous study of CHO cells expressing the VSV isoform (Porter et al., 1999) presumably relates to the low receptor density (0.2 pmol/mg) compared with this study (18 pmol/mg). Indeed, the high receptor reserve for induction of [3H]PI depletion in our cellular model favors low levels of drug efficacy (Cussac et al., 2002a,b). Thus, the antagonist properties of apomorphine, roxindole, and terguride at h5-HT2C receptors herein are of special note. This high sensitivity should also be borne in mind as regards the marked efficacy of cabergoline and pergolide, the only drugs showing potent and high efficacy at all 5-HT2 receptor subtypes. Bromocriptine has high affinity for h5-HT2C receptors (Millan et al., 2002) and mimicked the high efficacy of lisuride at h5-HT2C sites herein. Although 5-HT2C antagonist properties may improve the influence of antiparkinson agents upon mood and motor function (see Introduction), the risk of weight gain and proepileptic actions should not be neglected (Barnes and Sharp, 1999; Fox and Brotchie, 1999). On the other hand, 5-HT2C agonist properties may oppose the favorable influence of antiparkinson agents upon motor function and mood.
General Discussion.
Together with the two accompanying articles (Millan et al., 2002; Newman-Tancredi et al., 2002), the present observations evoke several general comments. First, the most striking conclusion of this comprehensive evaluation of antiparkinson agents is that they cannot be regarded as a homogeneous group of “dopaminergic agonists”. Like other classes of drug, such as antipsychotics, they present contrasting patterns of interactions with multiple subtypes of monoaminergic receptor. Imaging studies in Parkinson's disease patients would be instructive in identifying the receptors with which they interact at clinically used doses. Second, the use of cloned, heterologously expressed populations of human receptors under uniform conditions offered important advantages for our comparative studies. However, drug potencies, efficacies, and in vivo actions depend upon a multitude of factors, including colocalization of different receptor types permitting intracellular interactions and formation of heterodimers, coupling to various subtypes of G protein that can be differentially recruited by specific agonists, receptor density, and both isoform and species differences (Backstrom et al., 1999; Barnes and Sharp, 1999; Devi, 2001; Jerman et al., 2001; Cussac et al., 2002a,b). Moreover, although comprehensive, the present studies could be extended to other receptor types, such as 5-HT3, 5-HT4 and 5-HT6 receptors, which are also of potential relevance to the actions of antiparkinson agents (Barnes and Sharp, 1999). Third, as emphasized throughout these articles, the relevance of monoaminergic properties of antiparkinson agents is not restricted to their influence upon motor performance but is equally pertinent to mood and cognitive function. Furthermore, sensory disturbances, including pain, are an important feature of Parkinson's disease (Chulder and Dong, 1995), and monoaminergic mechanisms exert a pronounced influence upon the perceptive and affective-cognitive dimensions of pain via actions at central and peripheral loci (Millan, 2002). Fifth, the involvement of monoaminergic receptors in potentially deleterious actions of antiparkinson agents should not be neglected. Finally, the present observations provide a springboard for additional studies of the role of individual monoaminergic receptors in the pathogenesis and management of Parkinson's disease. They suggest, further, the need for a reexamination of previous experimental and clinical findings of differences and similarities in the functional profiles of antiparkinson agents that have not integrated the notion of contrasting actions at diverse monoaminergic receptors. Additional comparative studies of the actions of antiparkinson drugs in animal models of Parkinson's disease and in Parkinsonian patients would be instructive. Such functional studies, complementary to the present cellular approach, should permit reasonable predictions concerning the therapeutic potential of future antiparkinson agents.
Conclusions
Antiparkinson drugs display a diverse pattern of agonist and antagonist actions at multiple classes of 5-HT receptor. These observations complement those of the accompanying articles (Millan et al., 2002; Newman-Tancredi et al., 2002) in underlining the heterogeneity of antiparkinson agents. Although modest agonist activity at 5-HT1A sites may be favorable, marked stimulation of 5-HT2 receptor subtypes may well be disadvantageous. Thus, although serotonergic mechanisms are clearly not essential for therapeutic activity in Parkinson's disease, further elucidation of the functional significance of the contrasting actions of antiparkinson agents at multiple subtypes of 5-HT receptor would be of considerable interest.
Acknowledgments
We thank Marianne Soubeyran for secretarial assistance and Valérie Pasteau, Christine Chaput, and Laetitia Marini for technical assistance.
Footnotes
-
DOI: 10.1124/jpet.102.039883
- Abbreviations:
- 5-HT
- serotonin
- DA
- dopamine
- l-DOPA
- l-dihydroxyphenylacetic acid
- [35S]GTPγS
- guanosine 5′-O-(3-[35S]thio)triphosphate
- h
- human
- PI
- phosphatidylinositol
- CHO
- Chinese hamster ovary
- Received June 14, 2002.
- Accepted July 22, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}