


Abstract
Dopamine neurons from various animal species differ in sensitivity to the neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 1-methyl-4-phenylpyridinium (MPP+). Compared with striatal vesicles isolated from mice, those from rats have a higher density of the brain vesicular monoamine transporter (VMAT2) and a greater ability to sequester MPP+, suggesting a larger storage capacity for MPP+ in rat vesicles. In the present study, we examined whether striatal VMAT2-containing vesicles might provide protection against the neurotoxic effects of MPP+in vivo. Dose-response curves for striatally infused MPP+were determined in animals pretreated with or without a VMAT2 inhibitor. Ro 4-1284 administration (10 mg/kg i.p.; VMAT2 inhibitor) produced a 5-fold leftward shift in the MPP+ dose-response curve and a significant lowering of the EC50 concentration for MPP+-induced damage. These findings provide evidence for a substantial accumulation of MPP+ in VMAT2-containing vesicles in vivo in the rat striatum and support the hypothesis that MPP+ sequestration in vesicles can provide protection against its toxic actions. In mice, VMAT2 inhibition did not reliably enhance toxicity produced by a striatal infusion of MPP+or by systemic administration of MPTP. These data suggest that vesicular sequestration of MPP+ may be of less importance in mice than in rats as relates to protection from the toxin. The present results also reveal that although VMAT2 inhibition enhanced striatal MPP+ toxicity in the rat, the potency of MPP+ in the rat striatum was less than that in mouse striatum. This implies that there are other factors that either exacerbate MPP+ toxicity in the mouse or attenuate MPP+ toxicity in rats.
The exposure of humans or animals to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or its neurotoxic metabolite, 1-methyl-4-phenylpyridinium (MPP+), produces parkinsonism and a loss of nigrostriatal dopamine (DA) neurons. Although the mechanisms of action of MPTP and MPP+ have been well characterized (reviewed inSonsalla and Nicklas, 1992), reasons for the marked differences among animal species to susceptibility to MPTP or MPP+remain unclear (Chiueh et al., 1984; Sonsalla and Nicklas, 1992;Giovanni et al., 1994a,b; Zuddas et al., 1994). In a comparison of mice and rats, DA neurons in rats are unaffected by MPP+ concentrations that cause profound toxicity in mice, indicating that inadequate exposure to MPP+ does not account for the lack of vulnerability of rat DA neurons (Giovanni et al., 1994a,b; Zuddas et al., 1994). Furthermore, DA transporter density and MPP+ uptake into striatal synaptosomes are similar in the two species (Giovanni et al., 1994a; Staal et al., 2000), suggesting that MPP+ uptake into rat DA neurons is not a limiting factor for achieving high intraneuronal concentrations of MPP+. A possible explanation for the species difference is that the intracellular compartmentalization of MPP+ in the two species is dissimilar, such that in the rat DA neurons, there is less cytosolic MPP+ available to inhibit mitochondrial function and cause damage.
Possible sites for intracellular storage of MPP+within DA neurons are synaptic vesicles. MPP+ is accumulated into striatal vesicles via VMAT2 (Del Zompo et al., 1993;Moriyama et al., 1993; Staal et al., 2000) or into granules of chromaffin cells of the adrenal gland by VMAT1 (Reinhard et al., 1987,1989, 1990; Daniel and Reinhard, 1988; Liu et al., 1992). These observations led to the hypothesis that MPP+sequestration into vesicles may provide protection against toxicity (Reinhard et al., 1987, 1988; Liu et al., 1992; Takahashi et al., 1997;Gainetdinov et al., 1998; Speciale et al., 1998). We have found that synaptic vesicles isolated from rat striata have a greater maximum rate of [3H]MPP+ uptake (Vmax) and a greater number of [3H]dihydrotetrabenazine (ligand for VMAT2) binding sites (Bmax) than mouse striatal vesicles (Staal et al., 2000). These observations suggest that VMAT2-containing striatal vesicles in rats may have a greater storage capacity for MPP+ and that as such they may provide a mechanism that contributes to the greater resistance to MPP+ toxicity of rats versus mice. Russo et al. (1994) provided indirect evidence that MPP+ is accumulated into striatal vesicles in vivo. In MPTP-treated rats, pretreatment with reserpine to block VMAT2 resulted in lower striatal concentrations of MPP+ as would be expected if there were substantial sequestration of MPP+ into vesicles and this uptake is prevented. However, striatal MPP+ levels were not significantly reduced in reserpinized mice, although MPP+ content in adrenal glands from these animals was reduced to a similar extent as in rats. These observations, together with our in vitro findings, suggest that in vivo, rats may sequester MPP+ into striatal vesicles to a greater extent than do mice. However, whether vesicular sequestration of MPP+ in vivo in rats provides protection against the toxin has not been examined. One purpose of the present study was to determine whether the toxicity of a striatal infusion of MPP+ was enhanced in rats treated with a VMAT2 inhibitor. A second goal was to determine whether the greater accumulation of MPP+ into vesicles from rats versus mice observed in vitro might explain the lessened vulnerability of rat DA neurons to MPP+-induced toxicity in vivo.
It is hypothesized that the uptake of MPP+ into vesicles provides protection against its neurotoxicity. In support of this hypothesis, cells transfected with VMAT become resistant to MPP+ toxicity, indicating the neuroprotective value of intracellular storage of MPP+ (Liu et al., 1992, 1994). However, in vivo studies in mice have provided equivocal results. In heterozygous VMAT2 (+/−) knockout mice, MPTP-induced toxicity is enhanced compared with wild-type (WT) littermates (Takahashi et al., 1997; Gainetdinov et al., 1998) as would be expected if VMAT2 function is compromised. But in other studies in which VMAT2 was blocked by pharmacological agents, the neurotoxic effects of MPTP treatment have been reported to be unchanged (Fuller and Hemrick-Luecke, 1985; Reinhard et al., 1988) or only slightly enhanced (Melamed et al., 1985; Reinhard et al., 1988). Reasons for these differences in the effect of MPTP in mice in which VMAT2 function is reduced by pharmacological means or genetic manipulation are not readily apparent. To further examine the possible protective role of vesicles in mice, we investigated the effects of various VMAT2 inhibitors as well as dosing paradigms on toxicity produced by systemically administered MPTP.
Materials and Methods
Animals.
Male Swiss-Webster mice (30–35 g) and male Sprague-Dawley rats (250–300 g; Taconic Farms, Germantown, NY) were used in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and by the local animal care committee. The strains of mouse and rat used in this study are those in which we have previously characterized the species differences to MPTP and MPP+ (Giovanni et al., 1994a,b; Staal et al., 2000). Mice and rats were maintained at 20–22°C on a 12-h light/dark cycle with food and water available ad libitum.
Drugs and Reagents.
The reversible VMAT2 inhibitor 2-hydroxy-2-ethyl-3-isobutyl-9,10-dimethoxy-1,2,3,4,6,7-hexahydro-benzo[α]chinolizin hydrochloride (Ro 4-1284) was a gift from Hoffman-La Roche (Nutley, NJ). MPP+ iodide was purchased from Research Biochemicals Inc. (Natick, MA). MPTP was synthesized in our laboratory. All other compounds were obtained from Fisher Scientific (Springfield, NJ) or Sigma Chemical Co. (St. Louis, MO).
Microdialysis.
In vivo microdialysis was performed in freely moving animals as described by Giovanni et al. (1994a) with slight modifications. Instead of dental cement, TAK PAK (Loctite Corp., Newington, CT) was used to secure cannulas to the skull. Operations on animals were performed at least 3 days before microdialysis experiments. On the first day of the experiment (day 1), animals were anesthetized with ether and implanted with a probe a minimum of 4 h before the collection of baseline dialysate samples. Artificial cerebrospinal fluid was perfused at a flow rate of 1 μl/min, and samples were collected at 15-min intervals. After stabilization of basal DA levels, Ro 4-1284 or saline was administered. One hour later, one of several concentrations of MPP+ was infused through the dialysis probe for 15 min at a rate of 1 μl/min. On day 2, a challenge dose of MPP+ (10 mM, 15 min, 1 μl/min) was infused into the striatum, and DA overflow was measured. Probe recovery of DA was approximately 10%. Reported values are uncorrected for recovery of DA, and MPP+concentrations are uncorrected for extraction efficiency. Dialysate was analyzed for DA and metabolites by HPLC with electrochemical detection (model 420; ESA Inc., Bedford, MA; BAS 200; Bioanalytical Systems Inc., West Lafayette, IN; or model Decade; ANTEC LEYDEN BV, Leiden, the Netherlands). After completion of the experiments, probe placement was verified, and data from animals with improper placement were eliminated from the study.
Experimental Design.
On day 1, animals received saline or Ro 4-1284 (10 mg/kg i.p.), a tetrabenazine (2-oxo-3-isobutyl-9,10-dimethoxy-1,2,3,4,6,7-hexahydro-benzo[α]chinolizin hydrochloride) analog that is a reversible inhibitor of VMAT2, 1 h before the intrastriatal (i.s.) infusion of MPP+via the dialysis probe. The extent of the lesion caused by the day 1 i.s. infusion of MPP+ was assessed on day 2. This paradigm allowed VMAT2 function and DA levels to recover from the effects of the Ro 4-1284 treatment as demonstrated by the time course of Ro 4-1284 (see Figs. 1B and2B). The extent of the lesion caused by the day 1 infusion of MPP+ was assessed by infusing a challenge dose of MPP+ (10 mM) that releases all DA from remaining nerve terminals within the vicinity of the probe (Rollema et al., 1990; Giovanni et al., 1994b). Therefore, one would expect to see maximum release on day 2 if artificial cerebrospinal fluid was infused on day 1 and correspondingly lower levels of DA release on day 2 if toxic doses of MPP+ were infused on day 1 (Giovanni et al., 1994b).

Dose-response effect of Ro 4-1284 in mice and rats. Animals received Ro 4-1284 (0.5–10 mg/kg i.p.) and were sacrificed 1 h later. Striatal DA, DOPAC, and HVA were measured as described in Materials and Methods. Values are presented as percentage of control (mean ± S.E., three or four animals). DA levels were significantly (P < .05) decreased at all doses in rats and mice. DOPAC levels were significantly (P < .05) elevated at all doses tested in mice and at 0.3, 1, 3, 5, and 10 mg/kg in rats. HVA levels were significantly (P < .05) elevated at 5 and 10 mg/kg in mice and at 0.3, 1, 3, 5, and 10 mg/kg in rats. A, effects in mice. B, effects in rats.

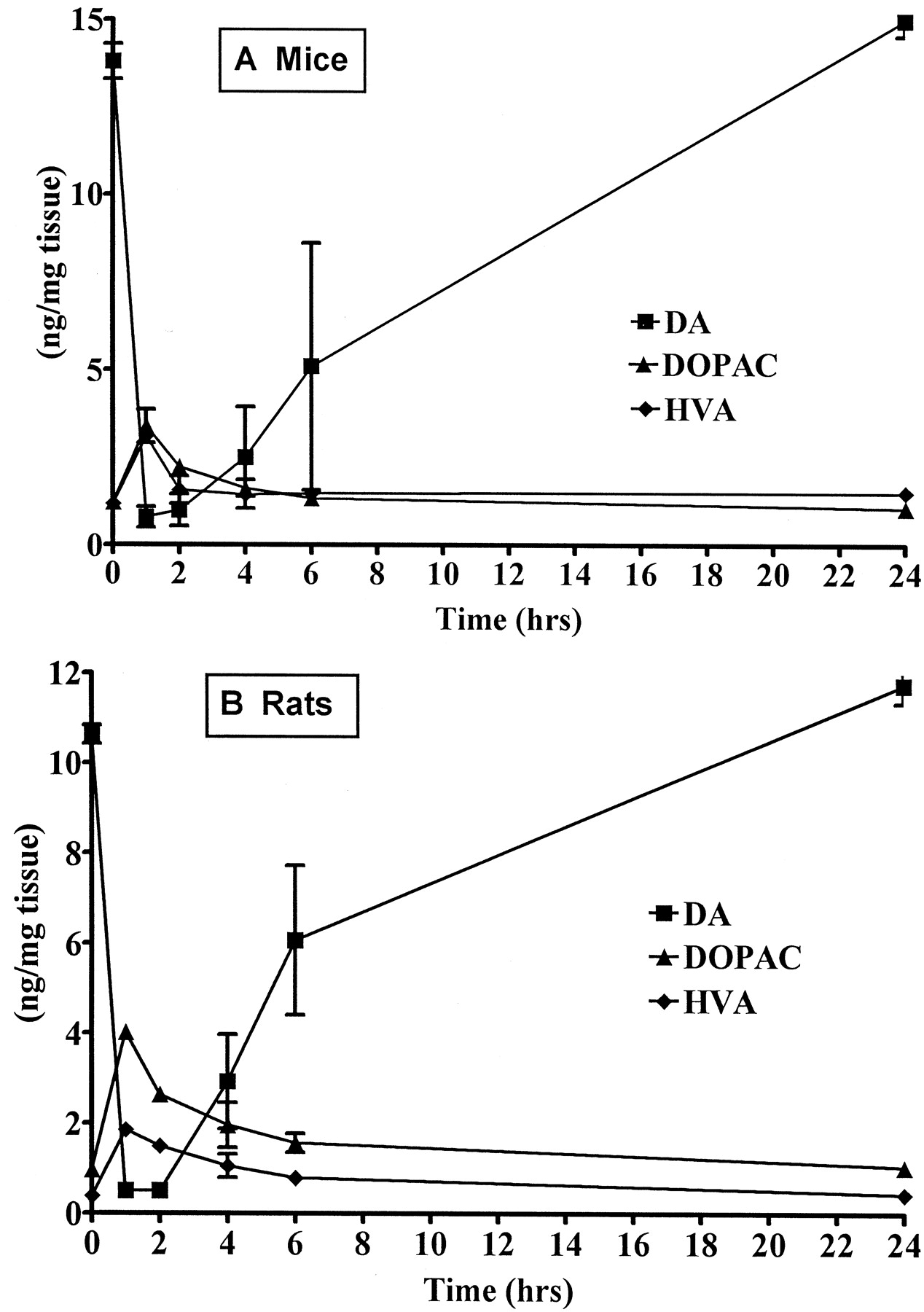
Time course for DA depletion produced by Ro 4-1284 in mice and rats. Animals were sacrificed 1, 2, 4, 6, and 24 h after administration of Ro 4-1284 (10 mg/kg i.p.). Results are the mean striatal DA content (nanogram per milligram of tissue) ± S.E. from three or four animals. DA levels were significantly (P < .05) decreased at 1, 2, 4, and 6 h after Ro 4-1284 administration in mice and rats. DOPAC levels were significantly (P < .05) elevated at 1 and 2 h after Ro 4-1284 administration in mice and at 1, 2, and 4 h after Ro 4-1284 administration in rats. HVA levels were significantly (P < .05) elevated at 1 h in mice and at 1 and 2 h after Ro 4-1284 administration in rats. A, effects in mice. B, effects in rats.
Ro 4-1284 Dose-Response and Time Course Studies.
For dose-response studies, mice and rats were treated with Ro 4-1284 (i.p.) at doses indicated and were sacrificed 1 h later. For the time course studies, mice and rats were given 10 mg/kg i.p. and sacrificed at various times after treatment. The striata were rapidly removed and homogenized; DA and its metabolites were measured.
MPTP Studies.
MPTP toxicity in mice was assessed using an acute or a subacute treatment paradigm. In the acute paradigm, reserpine (2.5 mg/kg i.p.) was administered 24 h before MPTP; Ro 4-1284 (10 mg/kg i.p.) was administered three times every 3 h starting 15 min before MPTP treatment. Mice received two to four injections of MPTP at 2-h intervals (see Table1 for total doses administered). In the subacute paradigm, mice received once daily for 5 or 9 days an injection of Ro 4-1284 (10 mg/kg i.p.) followed 20 min later by a single injection of MPTP (30 mg/kg i.p.). Alternatively, a single injection of reserpine was injected 24 h before initiation of daily injections of MPTP. Mice were sacrificed 7 or 28 days later. Striata were rapidly removed and homogenized or frozen until analyzed.
Effect of VMAT2 inhibition during acute MPTP treatment
Neurochemical Measurements.
DA and metabolite levels in striatal homogenates were measured by HPLC with electrochemical detection as described by Sonsalla et al. (1987). Striatal tyrosine hydroxylase (TH) activity was measured according to a radioenzymatic technique as described previously (Sonsalla et al., 1991).
Data Analysis.
Results were compared by Student'st test or one-way ANOVA with Tukey's post hoc evaluation (Instat; GraphPad Software, San Diego, CA). Dose-response curves were generated using Inplot (GraphPad Software) with maximum and minimum values held constant but calculations of EC50 and determination of the curve allowed. Results are expressed as mean ± S.E. and were considered significant at P < .05.
Results
Dose-Response and Time Course for Effects of Ro 4-1284 in Mice and Rats.
Dose-response and time course studies were conducted in mice and rats to determine the dose of Ro 4-1284 that would provide effective blockade of VMAT2. A dose-dependent depletion of striatal DA was observed 1 h after the treatment of mice or rats with Ro 4-1284 (Fig. 1). DA levels were maximally reduced to 3 and 5% of control in mice and rats, respectively, at 1 h after administration of the highest dose (10 mg/kg). The time courses for Ro 4-1284 effects on striatal DA content at 10 mg/kg in mice and rats were nearly identical (Fig. 2). DA depletion remained significantly reduced for at least 6 h (P< .05) but was restored by 24 h after the administration of Ro 4-1284. The DA metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were significantly increased (P< .05) by 2-fold at 1 h after the administration of Ro 4-1284 (Fig. 1) but returned to normal as DA levels increased (Fig. 2). These effects of Ro 4-1284 are similar to those produced by tetrabenazine in rats (Pettibone et al., 1984). The 10-mg/kg dose was selected for subsequent studies because a single injection of this dose provided sufficient blockade of VMAT2 for the duration of exposure to MPP+ in the microdialysis studies or to MPTP/MPP+ in the systemic toxicity studies.
Effect of VMAT2 Inhibition on Neurotoxicity of i.s. Infusions of MPP+ in Mice and Rats.
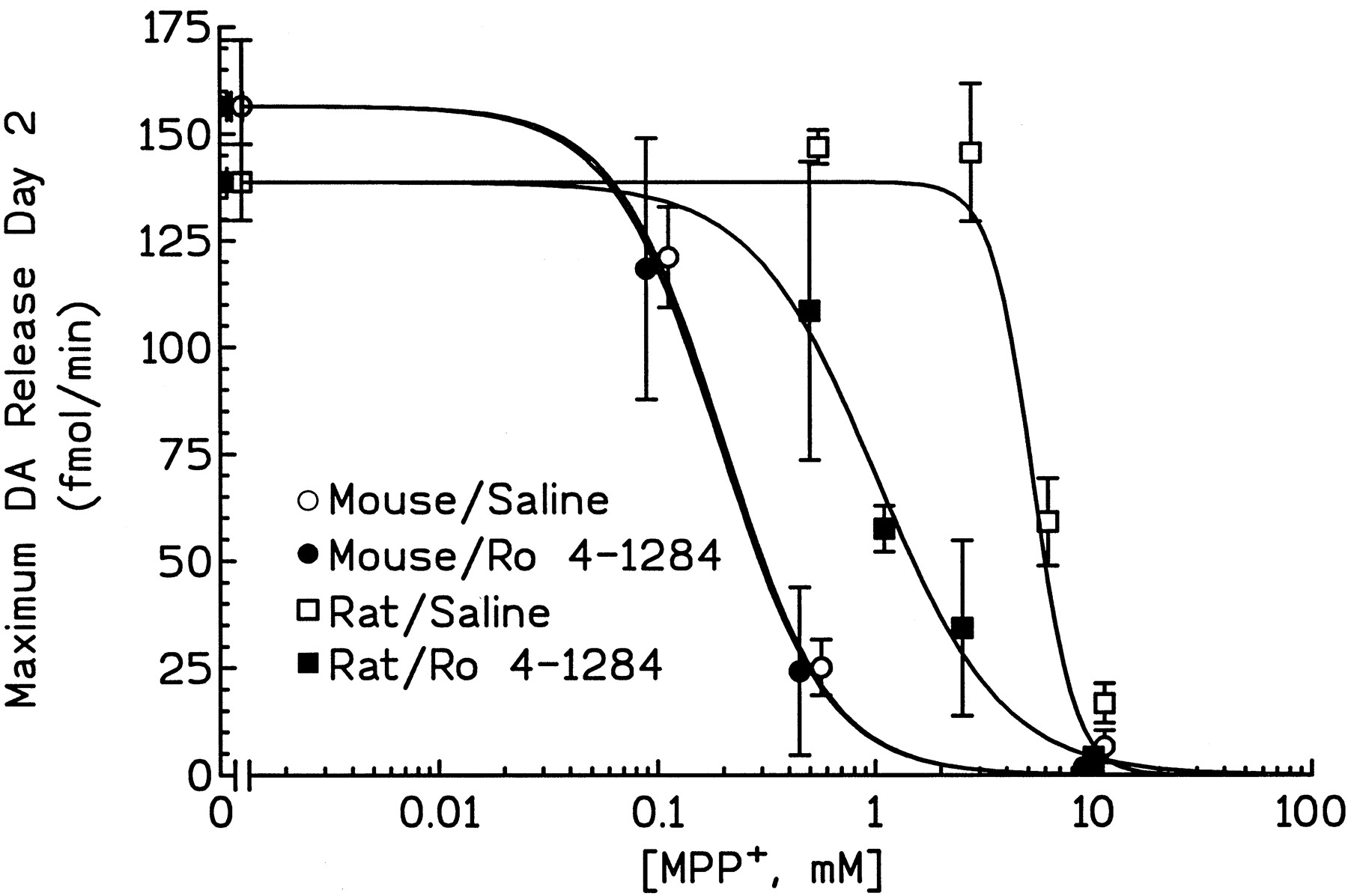
To determine the neuroprotective role of vesicular sequestration of MPP+ in mice and rats, the effect of the neurotoxin in animals in which VMAT2 was inhibited was investigated. On day 1, after probe placement and stabilization of baseline DA overflow (1–2 fmol/min), animals were treated with either saline or Ro 4-1284 (10 mg/kg i.p.) followed 1 h later by an i.s. infusion of MPP+ (0.1–10 mM; 15 μl over 15 min). On day 2, animals were challenged with a 10-mM infusion of MPP+ (15 μl over 15 min); this challenge dose releases all remaining DA within the vicinity of the probe. Dialysate samples were collected, and DA content was measured. The peak DA overflow produced by the day 2 MPP+ challenge was calculated (femtomoles per minute) and is plotted as a function of the MPP+ concentration infused on day 1 (Fig.3). The amount of DA measured at this time (day 2) reflects the content of DA remaining after day 1 MPP+ infusion and has been demonstrated by postmortem measurements to reflect toxicity to the DA nerve terminals within the vicinity of the probe (Giovanni et al., 1994b). EC50 values, calculated from the dose-response curves in mice and rats, were 0.2 and 5.1 mM, respectively. These findings demonstrate that an approximately 25-fold higher concentration of MPP+ is required to cause the same degree of toxicity in the striata of rats as in mice and are in agreement with previous reports (Giovanni et al., 1994a,b; Zuddas et al., 1994).

Dose-response curves of intrastriatally infused MPP+ in the presence or absence of VMAT2 inhibitors. Animals were treated as described in Materials and Methods. The peak DA overflow produced by the day 2 MPP+ challenge was calculated (femtomoles per minute) and is plotted as a function of the MPP+ concentration infused on day 1. Results are the mean DA overflow (femtomoles per minute) ± S.E. from three to six animals. Note that some of the data points have been slightly offset for clarity and to avoid overlap of S.E. bars.
Inhibition of VMAT2 in Rat by Systemic Ro 4-1284 Pretreatment Resulted in a 5-Fold Leftward Shift of Dose-Response Curve.
The EC50 value for day 1 MPP+in rats treated with Ro 4-1284 was 0.9 mM (compared with 5.1 mM in saline-pretreated rats). However, inhibition of VMAT2 in the mouse had no effect on the MPP+ dose-response curve; EC50 values in mice pretreated with saline or Ro 4-1284 were the same (0.2 mM). Also important to note is that although VMAT2 inhibition in rats produced a striking potentiation of MPP+ toxicity, the dose-response curve remained substantially shifted to the right of that observed in mice.
Effect of VMAT2 Inhibition on MPTP-Induced Toxicity in Mice.
The effects of VMAT2 inhibition on toxicity produced by the systemic administration of MPTP were evaluated in mice pretreated with reserpine or Ro 4-1284. Different dosing paradigms were used in an attempt to reveal a protective role for vesicular sequestration of MPP+ in mice. In one set of experiments, mice were injected with the irreversible VMAT inhibitor reserpine (2.5 mg/kg i.p.) 24 h before receiving MPTP (two i.p. injections, 15 mg/kg/injection, 2 h apart). This dose of reserpine produces an approximately 85% reduction in striatal DA at the time of MPTP treatment (data not shown). In this experiment, MPTP treatment produced a moderate reduction in striatal DA content (55%; Table 1) in mice sacrificed 28 days later. Reserpine pretreatment did not further decrease the MPTP-induced reduction in DA. Likewise, the modest reduction in TH activity (29%) produced by MPTP was not significantly enhanced by reserpine pretreatment (31% decrease). In a similar experiment in which mice received a total dose of 40 mg/kg MPTP but the dose was administered over three injections, reserpine treatment also failed to enhance the MPTP-induced reductions in DA content or TH activity (Table 1). In this study, mice were sacrificed only 8 days after reserpine treatment, thus accounting for the low DA content in the reserpinized animals. However, no significant differences in DA content were observed between the groups treated with reserpine, MPTP, or reserpine plus MPTP, although all three groups differed significantly from control. Moreover, no significant differences in TH activity were observed among any of the groups. In a third experiment, a larger dose of MPTP was administered to produce a greater lesion (total dose of 80 mg/kg; four injections of 20 mg/kg/injection at 2-h intervals). Ro 4-1284 (10 mg/kg/injection) was administered three times at 3-h intervals, starting 15 min before the first injection of MPTP. This dose and frequency of administration for Ro 4-1284 were chosen to provide sufficient inhibition of VMAT2 for the duration of the MPTP exposure (Fig. 2). A 76% reduction in DA content was observed in mice treated with only MPTP (Table 1). Concurrent treatment with Ro 4-1284 did not enhance this effect of MPTP (DA depletion was 75%).
In the subacute treatment paradigms, mice received Ro 4-1284 (single injection of 10 mg/kg i.p.) 15 min before MPTP (single injection of 30 mg/kg i.p.) for 5 or 9 days. In mice treated with only MPTP, regardless of the treatment duration, DA was reduced by approximately 70% (Table2). The concurrent treatment of mice with Ro 4-1284 and MPTP for 5 days produced a small but significantly greater DA depletion. However, in mice treated for 9 days, neither reserpine nor Ro 4-1284 enhanced the DA-depleting effects of MPTP, although there was a trend for lower DA concentrations in mice treated with Ro 4-1284 and MPTP (Table 2). Overall, these studies fail to identify any prominent effect of VMAT2 inhibition on MPTP-induced toxicity in mice regardless of whether the animals were treated acutely or subacutely with MPTP, whether VMAT2 was inhibited reversibly or irreversibly, or whether the MPTP lesion produced was moderate or extensive.
Effect of VMAT2 inhibition during subacute MPTP treatment
Discussion
The purpose of the present study was to examine the importance of vesicular sequestration of MPP+ in vivo in providing protection against its neurotoxic actions to striatal DA nerve terminals. In these experiments, the toxicity of striatally infused MPP+ was determined in animals that were pretreated with or without the VMAT2 inhibitor Ro 4-1284. The novel finding from these studies is that the pharmacological blockade of VMAT2 enhanced the neurotoxicity of MPP+ in the striata of rats and reveals that vesicular sequestration of MPP+ is a relevant form of protection in DA neurons in this species. However, VMAT2 inhibition failed to enhance MPP+ toxicity in the mouse striata, suggesting that vesicular sequestration may not be as significant in providing protection in this species as in rats. The results also demonstrated that although VMAT2 inhibition increased MPP+toxicity to striatal DA nerve terminals in the rat, the potency of MPP+ toxicity in the striatum of rats in which VMAT2 is inhibited is still less than that in mouse striata exposed to MPP+. This implies that mechanisms other than just MPP+ sequestration provide protection against MPP+ toxicity to DA neurons in rats.
A protective effect of MPP+ sequestration was first demonstrated by Reinhard and colleagues in adrenal chromaffin cells (Reinhard et al., 1987, 1988, 1990; Daniel and Reinhard, 1988). These findings led to the hypothesis that a similar mechanism may be involved in MPTP-induced neurotoxicity in the brain. The idea that the vesicular monoamine transporter renders cells more resistant to MPP+-induced toxicity was used in the cloning of VMAT1 by using MPP+ as the selecting factor (Liu et al., 1992). Since then, it has been demonstrated that striatal synaptic vesicles can sequester MPP+ and that the transport of this toxin is both proton gradient-dependent and subject to inhibition by reserpine and tetrabenazine (Del Zompo et al., 1993;Moriyama et al., 1993; Staal et al., 2000).
Several lines of evidence indicate a substantial sequestration of MPP+ into striatal vesicles of rats. In vitro, striatal vesicles isolated from rats have a large capacity to accumulate MPP+, a process that could remove significant amounts of the toxin from the cytosol of DA neurons (Staal et al., 2000). In vivo, blockade of VMAT2 significantly reduces MPP+ levels in the striatum of rats (Russo et al., 1994). However, whether the vesicular sequestration of MPP+ in vivo in rats provides protection against the toxin has not been previously demonstrated. Attempts to demonstrate this phenomenon have used pharmacological blockade of VMAT2 coupled with peripheral injections of MPTP. Unfortunately, these studies have been severely hampered by the high mortality rate seen in rats treated with both MPTP and a VMAT2 inhibitor (J. F. Reinhard, Jr., personal communication). It was the unacceptably high rate of mortality in MPTP-treated rats that led us to use the microdialysis technique for direct delivery of MPP+ into the striatum. The 2-day test-challenge microdialysis method used in these studies was developed by Rollema and colleagues in the late 1980s (reviewed inRollema et al., 1990). The technique has been used extensively to characterize the neurotoxicity of a variety of compounds in the striata of mice, monkeys, and rats (Rollema et al., 1988, 1990, 1994; Giovanni et al., 1994b). Furthermore, Giovanni et al. (1994b) demonstrated that the decreases in DA overflow observed on day 2 after the challenge dose reflect DA nerve terminal degeneration as determined by reductions in striatal tissue concentrations of DA when measured 1 week after infusion of MPP+ into the striatum.
Using this technique, we were able to directly examine the effect of VMAT2 inhibition on MPP+-induced toxicity to striatal DA nerve terminals in vivo in rats. The study demonstrates that pharmacological inhibition of VMAT2 potentiated MPP+ neurotoxicity in rats pretreated with the tetrabenazine analog Ro 4-1284 as demonstrated by the 5-fold leftward shift in the dose-response curve for MPP+toxicity. These data provide direct evidence that loss of vesicular sequestration of MPP+ due to VMAT2 inhibition increases MPP+ toxicity to DA neurons in the rat. Noteworthy, however, is that despite the leftward shift of the MPP+ dose-response curve by VMAT2 inhibition, the DA nerve terminals in the rats remained 5-fold more resistant to MPP+ than those in mice whose vesicular function was not compromised. We suggest that there are other factors that either exacerbate MPP+ toxicity in the mouse or attenuate MPP+ toxicity in rats. Studies are in progress to identify potential factors because they could provide important insight into processes that factor into the degeneration of DA neurons.
In parallel studies in mice, pharmacological VMAT2 inhibition did not enhance the toxicity of striatally infused MPP+. If vesicular sequestration of MPP+ provides significant protection against toxicity in this species, it would have been expected that its potency would have been increased in mice in which VMAT2 was blocked. Inadequate inhibition of VMAT2 in mice during the period of MPP+ infusion seems unlikely because the administration of Ro 4-1284 (10 mg/kg) produced a 95% decrease in striatal DA content at the time of MPP+ administration and had a similar time course and dose-response effect as in rats in which this dose of Ro 4-1284 was effective for enhancing MPP+ toxicity. Noteworthy is that VMAT2 density and maximal rate of MPP+transport are significantly less in striatal vesicles isolated from mice than in those from rats. This indicates a lower capacity for MPP+ storage in DA vesicles in mice. Thus, it may be that even though MPP+ is transported into vesicles of DA neurons in mice, this process is too slow or inadequate to sufficiently remove the toxin from the cytosol. The failure to enhance striatally infused MPP+ by VMAT2 blockade in mice is consistent with other studies in which pharmacological VMAT2 inhibitors have failed or only marginally enhanced toxicity produced by the systemic administration of MPTP (Fuller and Hemrick-Luecke, 1985;Melamed et al., 1985; Reinhard et al., 1988). Our experiments, in which various dosing paradigms for MPTP were used in an attempt to reveal a neuroprotective role for vesicles in mice, also demonstrated equivocal effects of pharmacological VMAT2 inhibition on MPTP toxicity. Taken together, these observations suggest that although MPP+ can be accumulated into striatal VMAT2-containing vesicles in mice, this process does not provide the extent of protection as it does in rats. It should be added, however, that in heterozygous VMAT2 (+/−) knockout mice, MPTP-induced toxicity is enhanced compared with WT littermates (Takahashi et al., 1997;Gainetdinov et al., 1998), which would be expected if VMAT2 function is compromised and vesicular storage of MPP+ is important for protection. Reasons for these differences in the effect of MPTP in mice in which VMAT2 function is reduced by pharmacological means versus genetic manipulation are not readily apparent. Acute pharmacological VMAT2 inhibition may produce responses that differ from those seen in transgenic mice in which lifetime compensatory mechanisms may have developed. In other studies of the dopamine system, experimental outcomes based on genetic models have not always agreed with those that used pharmacological approaches (Sibley, 1999).
In summary, our in vivo studies indicate that the sequestration of MPP+ into vesicles of DA neurons of rats provides substantial protection against the deleterious effects of this neurotoxin in this species. Although MPP+infusion studies in mice failed to reveal a significant neuroprotective role for vesicles in DA neurons in this species, these findings are in agreement with studies in which pharmacological VMAT2 blockade exerted little or no effect on neurotoxicity produced by the systemic administration of MPTP in mice. Our findings also reveal the importance of other as-yet-unidentified factors within the DA neurons that contribute to the species differences in vulnerability to MPP+ neurotoxicity.
Acknowledgments
We acknowledge L. Manzino for expert technical assistance.
Footnotes
-
Send reprint requests to: Dr. Patricia K. Sonsalla, Department of Neurology, University of Medicine and Dentistry of New Jersey, Robert Wood Johnson Medical School, 675 Hoes Lane, Piscataway, NJ 08854-4535. E-mail: sonsalla{at}umdnj.edu
-
↵1 This work was supported by National Institutes of Health Grant AG08479.
- Abbreviations:
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- VMAT1
- vesicular monoamine transporter 1
- VMAT2
- vesicular monoamine transporter 2
- DA
- dopamine
- DOPAC
- 3,4-dihydroxyphenylacetic acid
- HVA
- homovanillic acid
- TH
- tyrosine hydroxylase
- MPP+
- 1-methyl-4-phenylpyridinium
- i.s.
- intrastriatal
- WT
- wild-type
- Received April 6, 1999.
- Accepted February 14, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}