


Abstract
Recombinant human dopamine D4.4 receptor-mediated G protein activation was characterized in membranes of transfected mammalian (Chinese hamster ovary) cells by the use of [35S]guanosine-5′-O-(3-thio)triphosphate ([35S]GTPγS) binding. An initial series of experiments defined the conditions (3 μM GDP, 100 mM NaCl, 3 mM MgCl2) under which optimal stimulation (2.2-fold increase in specific [35S]GTPγS binding) was achieved with the endogenous agonist dopamine. The number of dopamine-activated G proteins in Chinese hamster ovary-D4.4 membranes was determined through [35S]GTPγS isotopic dilution saturation binding, yielding a B max value of 2.29 pmol/mg. This compared with a D4.4 receptorB max value of 1.40 pmol/mg determined by [3H]spiperone saturation binding, indicating that 1 or 2 G proteins were activated per D4.4 receptor and that there were few or no “spare receptors” in this cell line. Under these conditions, the efficacy for stimulation of [35S]GTPγS binding at D4.4 receptors of 12 dopaminergic agonists was determined. Several antiparkinsonian drugs, including ropinirole, quinerolane and lisuride, exhibited agonist activity at D4.4 receptors (E max = 74.3%, 72.4% and 32.2%, respectively, compared with dopamine = 100%). The EC50 values for agonist stimulation of [35S]GTPγS binding correlated well with the inhibition constants derived from competition binding with [3H]spiperone (r = +.99). However, other antiparkinsonian drugs (bromocriptine, L-DOPA and terguride) showed low affinity and/or were devoid of agonist activity at D4.4receptors. The potency at D4.4 receptors of the novel, selective D4.4 receptor antagonist L 745,870 was determined, indicating that it has high affinity (K i = 1.99 nM) without detectable agonist activity. Furthermore, L 745,870 completely inhibited dopamine-stimulated [35S]GTPγS binding with aK b value of 1.07 nM. The action of an additional 20 chemically diverse dopaminergic ligands, including clozapine, ziprasidone, sertindole, olanzapine and several other “atypical” antipsychotics, in advanced development was investigated. Each of these ligands shifted the dopamine stimulation curve to the right in a parallel manner consistent with competitive antagonism at this site and yieldingK b values (32.6, 22.4, 17.2 and 26.5 nM, respectively) that agreed closely with theirK i values (38.0, 14.9, 18.5 and 26.1 nM). In contrast, raclopride and seroquel exhibited low affinity at D4.4 receptors (K i> 1000 nM). Other compounds that showed antagonist activity at D4.4 receptors included the 5-hydroxytryptamine2A receptor antagonist fananserin (RP 62203), the sigma ligand BMY 14,802 and the D3 receptor antagonist GR 103,691. In conclusion, dopamine D4.4 receptor activity is unlikely to be an important factor in the clinical effectiveness of antiparkinsonian drugs, although low agonist efficacy at D4.4 receptors might be associated with a lesser incidence of side effects. Furthermore, antagonist activity at D4.4 receptors is a common property of many typical and atypical antipsychotic agents.
Molecular biology techniques have enabled the cloning and pharmacological characterization of multiple dopamine receptor subtypes belonging to two families. D1and D5 receptors exhibit high structural homology and similar pharmacological profiles (Sunahara et al., 1991). Likewise, D2, D3 and D4 receptors exhibit similarities in both their pharmacological profiles and their coupling to G proteins and signal transduction pathways (Levesqueet al., 1992; O’Hara et al., 1996; Tang et al., 1994; Werner et al., 1996). The D4receptor is of particular interest for several reasons. First, in situ hybridization, autoradiographic and immunohistochemical studies indicate the existence of D4-like receptor sites in limbic structures, such as cerebral cortex and hippocampus, associated with regulation of mood and cognition (Lahti et al., 1995;Matsumoto et al., 1996; Meador-Woodruff et al., 1996; Mrzljak et al., 1996). In contrast, only low levels of D4 receptors are detected in regions associated with control of locomotor activity, such as the striatum (Meador-Woodruffet al., 1996; Seeman et al., 1993b). Furthermore, the atypical antipsychotic clozapine, which is known to act as an antagonist at dopamine D2 and 5-HT2A receptors (Canton et al., 1994; Meltzer, 1996), an inverse agonist at 5-HT2C receptors (Labrecque et al., 1995) and a partial agonist at 5-HT1A receptors (Newman-Tancrediet al., 1996a), also has significant affinity at dopamine D4 receptors (Van Tol et al., 1991). This suggests that these sites may mediate some of the therapeutic actions of atypical antipsychotics. In fact, D4-like receptor up-regulation in postmortem schizophrenic brain has been observed using indirect binding techniques (Murray et al., 1995a; Seemanet al., 1993a). Second, D4 receptors have recently been discovered to display a “promiscuous” pharmacological profile, binding epinephrine and norepinephrine with high affinity, similar to that of dopamine (Lanau et al., 1997;Newman-Tancredi et al., 1997a). Hence, D4receptors may play a role in integrating dopaminergic and adrenergic transmission. Furthermore, D4 receptor activation by norepinephrine is blocked by clozapine (Lanau et al., 1997;Newman-Tancredi et al., 1997a), suggesting that some of its clinical effects may be mediated by the antagonism of noradrenergic activity at D4 receptors. Indeed, it has been suggested that noradrenergic overactivity may contribute to acute exacerbation of psychosis (Hornykiewicz, 1982; Van Kammen et al., 1990). Third, D4 receptors may be implicated in the secondary action of antiparkinsonian drugs because clozapine, which has high affinity at D4 receptors, is effective in the treatment of L-DOPA-induced psychoses (Factor et al., 1995; Meltzeret al., 1995). In fact, although dopaminergic agonists such as bromocriptine are known to be effective in alleviating the symptoms of PD (Weddell and Weiser, 1995), the dopaminergic receptor subtypes involved remain to be further defined (De Keyser et al., 1995; Jenner, 1995).
In view of the above considerations, the efficacy and potency of a range of agonists and antagonists at dopamine D4 receptors were investigated. Previous studies have revealed the existence of D4 receptor alleles, differing in the number of a 16-amino acid repeat sequence found in the putative third intracellular loop of the receptor (Van Tol et al., 1992). However, all of these alleles are negatively coupled to adenylyl cyclase activity (Asghariet al., 1995; McHale et al., 1994) and have similar binding and G protein interaction profiles (Asghari et al., 1994), although they may differ in their sensitivity to monovalent cations (Van Tol et al., 1992). Previous studies have determined the affinities of some dopaminergic ligands at D4 receptors by radioligand competition binding (Lawsonet al., 1994; Roth et al., 1995). This technique has yielded estimates of agonist efficacies at D4.4receptors by comparing their affinities at different receptor states (Lahti et al., 1996). Other studies have investigated the agonist/antagonist activity at D4 receptors of a limited number of compounds (e.g., dopamine and quinpirole as agonists and/or clozapine and haloperidol as antagonists) by adenylyl cyclase determinations (Asghari et al., 1995; Bouvieret al., 1995; Tang et al., 1994). Hence, to date, no study of a broad range of agonist efficacies and antagonist potencies in a functional test has been conducted. The present study addressed this issue by evaluation of [35S]GTPγS binding (Chabert et al., 1994) to membranes of mammalian (CHO) cells transfected with the D4.4 receptor (four repeat sequence), the most common allele in humans (Chang et al., 1996; Lichter et al., 1993). Agonist stimulation of [35S]GTPγS binding, a nonhydrolyzable analog of GTP, provides a measure of receptor-mediated G protein activation (Hilfet al., 1989; Lazareno et al., 1993). An initial characterization defined the experimental conditions under which optimum agonist stimulation of [35S]GTPγS binding was observed. Previous studies in other receptor systems (Gierschiket al., 1991; Hilf et al., 1989; Lazarenoet al., 1993; Lorenzen et al., 1993) have highlighted the importance of monovalent and divalent cations (particularly Na+ and Mg++) and of GDP as being critical for modulation of agonist activation of [35S]GTPγS binding. Furthermore, given the importance of receptor density and/or receptor reserve on functional responses, the number of receptor-coupled G proteins activated by the endogenous agonist dopamine was determined in relation to the density of receptors present in the cell line. Indeed, the stoichimetric relationship between receptors and G proteins can significantly affect the definition of agonist efficacies (Adham et al., 1993;Kenakin, 1996; Newman-Tancredi et al., 1997c). The potency and efficacy for stimulation of [35S]GTPγS binding of 12 dopaminergic agonists, including several antiparkinsonian drugs currently in development, were determined. Finally, in view of the potential use of D4 receptors as a target for antipsychotic activity, the potency of a large series of antipsychotics for blocking dopamine-induced [35S]GTPγS binding was determined and compared with the action of reference dopaminergic antagonists. In addition to the neuroleptic haloperidol and the “atypical” antipsychotic clozapine, we examined the action of ziprasidone, olanzapine, sertindole, seroquel and other putatively atypical antipsychotics in late-stage development (Goldstein, 1995). Furthermore, the action of the novel selective D4 receptor antagonist L 745,870 (Kulagowski et al., 1996), was investigated.
Methods
[3H]Spiperone binding to CHO-D4.4 cell membranes.
Saturation binding at D4.4 receptors was carried out with 8 concentrations of [3H]spiperone (100 Ci/mmol; Amersham, Les Ulis, France) from 0.02 to 2.5 nM. For competition binding experiments, the concentration of [3H]spiperone was 0.5 nM. Membranes (10–20 μg of protein) from transfected CHO cells stably expressing the human dopamine D4.4 receptor (Receptor Biology, Baltimore, MD) were incubated with [3H]spiperone at 25°C for 60 min in a buffer containing 50 mM Tris, pH 7.4, 120 mM NaCl, 5 mM KCl, 1 mM EDTA and 5 mM MgCl2. Nonspecific binding was defined with haloperidol (10 μM). Affinity (inhibition constants,Ki ) at hD4.4 receptors was determined in [3H]spiperone competition binding experiments. Isotherms were analyzed by nonlinear regression using the program Prism (GraphPAD Software, San Diego, CA) to yield IC50 values. Inhibition constants (Ki ) were derived from IC50 values according to the Cheng-Prusoff equation:Ki = IC50/(1 + L/Kd ), where L is the concentration of radioligand and Kd is the dissociation constant of [3H]spiperone at D4.4 receptors (0.37 nM).
Isotopic dilution [35S]GTPγS saturation binding to CHO-D4.4 cell membranes.
Receptor-linked G protein activation at D4.4 receptors was determined by measuring the stimulation of [35S]GTPγS (1332 Ci/mmol; New England Nuclear, Les Ulis, France) binding. Except where stated otherwise, CHO-D4.4 membranes (50 μg of protein) were incubated (20 min, 22°C) with agonists and/or antagonists in a buffer containing 20 mM HEPES, pH 7.4, 3 μM GDP, 3 mM MgCl2, 100 mM NaCl and 0.1 nM [35S]GTPγS. Nonspecific binding was defined with GTPγS (10 μM). In isotopic dilution experiments, the basal and dopamine (10 μM)-stimulated binding of radiolabeled [35S]GTPγS was inhibited with unlabeled GTPγS. Two concentration ranges of GTPγS were tested: 0 to 10 μM and 0 to 45 nM. For the former, IC50 values were derived by nonlinear regression. For the latter, saturation binding curves were derived to estimate the number of G proteins activated by dopamine. The total amount of ligand bound to G protein (BOUNDTOT) was calculated by equation 1: BOUNDTOT = [35S]GTPγSBOUND × GTPγSTOT/[35S]GTPγSCONC, where [35S]GTPγSBOUND is observed dopamine-dependent binding in the tubes (fmol/mg), [35S]GTPγSCONC is [35S]GTPγS concentration in the tubes (0.1 mM) and GTPγSTOT is [35S]GTPγSCONCplus GTPγS concentration.
Measurement of agonist efficacy and antagonist potency at D4.4 receptors.
Agonist efficacy is expressed relative to that of DA (100%), which was tested at a maximally effective concentration (10 μM) in each experiment. For antagonist tests, membranes were preincubated with dopamine and a single concentration of antagonist for 30 min before the addition of [35S]GTPγS. For concentration-response curves of the inhibition of dopamine-stimulated [35S]GTPγS binding,Kb values were calculated by equation 2: Kb = IC50/{([agonist]/EC50) + 1}, where [agonist] is agonist concentration.
For the dopamine concentration-response curves determined in the presence of a fixed concentration of antagonist, antagonist potency values (Kb ) were calculated by equation 3: Kb = [antagonist]/[(EC50′/EC50) −1], where [antagonist] is antagonist concentration, EC50′ was determined in the presence of antagonist and EC50 was determined in the absence of antagonist (dopamine alone).
Experiments were terminated by rapid filtration through Whatman GF/B filters (pretreated with 0.1% polyethyleneimine in the case of [3H]spiperone binding) using a Brandel cell harvester. Radioactivity retained on the filters was determined by liquid scintillation counting. Protein concentration was determined colorimetrically using a bicinchonic acid assay kit (Sigma, S. Quentin Fallavier, France). All results are expressed as mean ± S.E.M. of three or more independent determinations.
Compounds.
Fananserin (RP 62203) was obtained from Rhone-Poulenc Rorer (Vitry-sur-Seine, France). Lisuride and terguride were from Schering (Berlin, Germany). Ocaperidone was from Janssen (Beerse, Belgium). ORG 5222 (trans-5-chloro-2-methyl-2,3,3a,12b-tetrahydro-1H-dibenz[2,3:6,7]oxepino-[4,5-c]pyrrole) was from Organon (Oss, Netherlands). Olanzapine and quinerolane were from Eli Lilly (Indianapolis, IN). Raclopride was from Astra (Sodertalje, Sweden). Seroquel was from Zeneca (Macclesfield, UK). Sertindole was from Lundbeck (Copenhagen, Denmark). Tiaspirone and BMY 14802 (1-[4-(4-fluorophenyl)-4-hydroxybutyl]-4-(5-fluoropyrimidin-2-yl)-piperazine) was from Bristol-Myers (Wallingford, CT). (+)-7-OH-DPAT [7-hydroxy-2-(di-n-propylamino)tetralin] was from CNRS (Paris, France). dp-ADTN (5,6-dihydroxy-2-di-n-propylamino-1,2,3,4-tetrahydronaphthalene) was kindly donated by Dr. Ann Mills-Duggan (Glaxo-Wellcome, Stevenage, UK). Ropinirole, piribedil, FG 5893 (2-[4-[4,4-bis(4-fluorophenyl)butyl]-1-piperazinyl]pyridine-3-carboxylic acid), GR 103,691 (4′-acetyl-N-{4-[(2-methoxyphenyl)-piperazin-1-yl] butyl-biphenyl-4-carboxamide), risperidone, ziprasidone and L 745,870 (3-(4-[4-chlorophenyl]piperazin-1-yl)methyl-1H-pyrrolo-[2,3b]pyridine) were synthesized by J.-L. Peglion and G. Lavielle (Servier). Clozapine, bromocriptine, (−)-quinpirole and spiperone were purchased from RBI (Natick, MA). Haloperidol, (−)-apomorphine and L-DOPA were purchased from Sigma.
Results
[3H]Spiperone competition binding at D4.4 receptors.
D4.4 receptor density in CHO-D4.4 membranes was determined by [3H]spiperone saturation binding. The isotherms were monophasic, with a dissociation constant (Kd ) of 0.37 ± 0.05 nM (n = 4) and a B max value of 1.40 ± 0.12 pmol/mg protein (4) (fig. 1). In [3H]spiperone competition binding experiments, agonist isotherms were shallow, with pseudo-Hill coefficients of <0.8 (table1). In contrast, the antagonist competition isotherms were steeper and exhibited pseudo-Hill coefficients close to unity.

Saturation binding of [3H]spiperone to CHO-D4.4 cell membranes. Saturation binding was carried out by incubating [3H]spiperone (0.02–2.5 nM) with CHO-D4.4 membranes. Analysis was by nonlinear regression using the program Prism. A, Representative saturation binding isotherm. Points shown are mean of triplicate determinations from an experiment repeated on at least three occasions. B, Scatchard transformation of specific binding data from A.
Action of dopaminergic agonists at cloned human dopamine D4.4receptors
Definition of [35S]GTPγS binding conditions.
Dopamine (10 μM) stimulated [35S]GTPγS binding to CHO-D4.4 membranes in a linear manner over the first 20 min (3) of time course experiments, and a standard incubation time of 20 min was therefore used. In contrast, no stimulation of [35S]GTPγS binding was observed in membranes of untransfected CHO cells (results not shown). Basal (nonagonist-stimulated) binding of [35S]GTPγS to CHO-D4.4 membranes was dependent on the concentration of GDP present in the buffer (fig. 2B) and was reduced from ∼90,000 dpm in the absence of GDP to ∼4000 dpm at a GDP concentration of 3 μM. In contrast, agonist-dependent [35S]GTPγS binding (i.e., the difference between agonist-stimulated and basal binding) amounted to ∼5000 dpm (see legend to table 1) and was not decreased by GDP concentrations of ≤3 μM. The decrease in basal binding augmented the ratio of agonist-stimulated to basal [35S]GTPγS binding to 2.2-fold at GDP concentrations of 3 μM (fig. 2B, inset). Like GDP, NaCl reduced basal [35S]GTPγS binding, from 13,000 dpm in the absence of NaCl to 4000 dpm at a concentration of 100 mM (fig.2C). However, high concentrations of NaCl (≥200 mM) also decreased agonist-dependent [35S]GTPγS binding. Although the latter was observed in both the presence and absence of GDP and NaCl, it exhibited an absolute dependence on the presence of magnesium in the incubation medium (fig. 2D). Agonist stimulation of [35S]GTPγS binding was observed over a wide range of MgCl2 concentrations, from 0.1 to 30 mM. The effect of MgCl2 on both basal and agonist-dependent [35S]GTPγS binding was biphasic, increasing to a first maximum at ∼0.1 mM and then to a second, higher, maximum at 3 to 10 mM. A set of standard experimental conditions was defined (3 μM GDP, 3 mM MgCl2, 100 mM NaCl, 20-min incubation) that yielded the highest agonist stimulation of [35S]GTPγS binding and was used in all subsequent experiments.

Effect of (A) time, (B) GDP, (C) NaCl and (D) MgCl2 on [35S]GTPγS binding to membranes of CHO cells stably expressing cloned human D4.4 receptors. Except where shown, experiments were carried out in a buffer containing HEPES (20 mM, pH 7.4), GDP (3 μM), MgCl2 (3 mM) and [35S]GTPγS (0.1 nM) for 20 min at 22°C. Nonspecific binding was defined with GTPγS (10 μM). Points shown are mean of triplicate determinations from representative experiments repeated on at least three independent occasions. DA, dopamine. □, Basal [35S]GTPγS binding (no dopamine). ▪, Agonist-stimulated [35S]GTPγS binding (with 10 μM dopamine). A, Specific basal and dopamine-stimulated [35S]GTPγS binding determined over time points ranging from 1 to 60 min. B, Specific basal and dopamine-stimulated [35S]GTPγS binding determined in the presence of GDP concentrations between 0 and 100 μM. Inset, effect of GDP concentration on agonist stimulation ratio. The stimulation ratio was calculated as specific dopamine-stimulated [35S]GTPγS binding divided by specific basal [35S]GTPγS binding. C, Specific basal and dopamine-stimulated [35S]GTPγS binding determined in the presence of concentrations of NaCl between 0 and 200 mM. D, Specific basal and dopamine-stimulated [35S]GTPγS binding determined in the presence of concentrations of MgCl2 between 0 and 100 mM. Inset, effect of MgCl2 concentration on agonist stimulation ratio.
Isotopic dilution [35S]GTPγS saturation binding.
Inhibition of basal [35S]GTPγS binding to CHO-D4.4 membranes with GTPγS (0.1 nM to 10 μM) exhibited a low affinity component (IC50 = 110 ± 18 nM). In contrast, inhibition of dopamine (10 μM)-stimulated [35S]GTPγS binding produced biphasic isotherms with IC50(high) = 4.4 ± 1.9 nM (4) and IC50(low) = 257 ± 67 nM (4) (fig. 3A). [35S]GTPγS saturation binding isotherms were derived for the high-affinity binding component by isotopic dilution with GTPγS (0–45 nM; fig. 3B). These yielded an apparentKd for [35S]GTPγS binding to the high-affinity (agonist-dependent) binding site of 15.0 ± 4.2 nM and a B max of 2.29 ± 0.44 pmol/mg (4) (fig. 3B). The Kd value did not differ significantly from the IC50(high)value above (P > .05, two-tailed t test).

Effect of GTPγS on [35S]GTPγS binding to membranes of CHO stably expressing cloned human D4.4 receptors. Experiments were carried out in a buffer containing HEPES (20 mM, pH 7.4), GDP (3 μM), MgCl2 (3 mM) and [35S]GTPγS (0.1 nM) for 20 min at 22°C. Representative curves are shown in which each point is the mean of duplicate determinations. Similar results were obtained in at least three independent experiments. A, Basal and dopamine (10 μM)-stimulated [35S]GTPγS binding determined in the presence of concentrations of GTPγS between 0 and 10 μM. B, Basal and dopamine (10 μM)-stimulated [35S]GTPγS binding determined in the presence of concentrations of GTPγS between 0 and 45 nM. These data were transformed as described in the text to generate a saturation binding isotherm for agonist-dependent [35S]GTPγS binding. Inset, Scatchard plot of the saturation binding isotherm.
Agonist and antagonist action at D4.4receptors.
The EC50 values for stimulation of [35S]GTPγS binding by agonists, including the antiparkinsonian drugs quinerolane, (−)-apomorphine, (+)7-OH-DPAT and lisuride, correlated (r = .99, P < .01) with their binding affinity (Ki ; table 1; see fig. 5B). In contrast, other clinically used antiparkinsonian drugs (e.g., bromocriptine, piribedil) had low affinity and/or efficacy at D4.4 receptors. None of the compounds, other than dopamine, acted as full agonists for stimulation of [35S]GTPγS binding.

Agonism and antagonism at cloned human D4.4 receptors defined by [35S]GTPγS binding. Experiments were carried out in a buffer containing HEPES (20 mM, pH 7.4), GDP (3 μM), MgCl2 (3 mM) and [35S]GTPγS (0.1 nM) for 20 min at 22°C. Nonspecific binding was defined with GTPγS (10 μM). Points shown are mean of triplicate determinations from representative experiments repeated on at least three independent occasions. [35S]GTPγS binding is expressed as a percentage of the maximal stimulation given by dopamine. A, Agonist concentration-response curves. B, Correlation of minus log EC50 values (pEC50) for agonist stimulation of [35S]GTPγS binding with minus logK i values (pK i; table 1) for inhibition of [3H]spiperone binding. The correlation coefficient was .99 (P < .001). C, Shift of dopamine concentration-response [35S]GTPγS binding curve in the presence of fixed concentrations of antagonists (table 2). D, Correlation of minus logK b values (pK b) for antagonist potency with minus log K i values (pK i, table 2) for inhibition of [3H]spiperone binding. The correlation coefficient was .99 (P < .001). The point corresponding to FG 5893 was not included in the calculation of the correlation.
The antagonist activity of a range of dopaminergic ligands was tested, including the novel, selective D4 receptor ligand L 745,870 (Ki = 1.99 nM). Like spiperone, haloperidol and clozapine, L 745,870 concentration-dependently and completely blocked the stimulation of [35S]GTPγS binding induced by 1 μM dopamine (Kb = 1.07 nM; fig. 4and table 2). L 745,870 (100 nM) also shifted the dopamine concentration-response curve to the right, with an 89-fold increase in EC50 (8910 ± 1250 nM), yielding aKb value of 1.19 nM (table3 and fig. 5C). Other compounds that showed antagonist activity were the dopamine D3 receptor ligand GR 103,691, the serotonin 5-HT2A receptor antagonist fananserin (RP 62,203), the sigma ligand BMY 14,802 and the antiparkinsonian drug terguride.
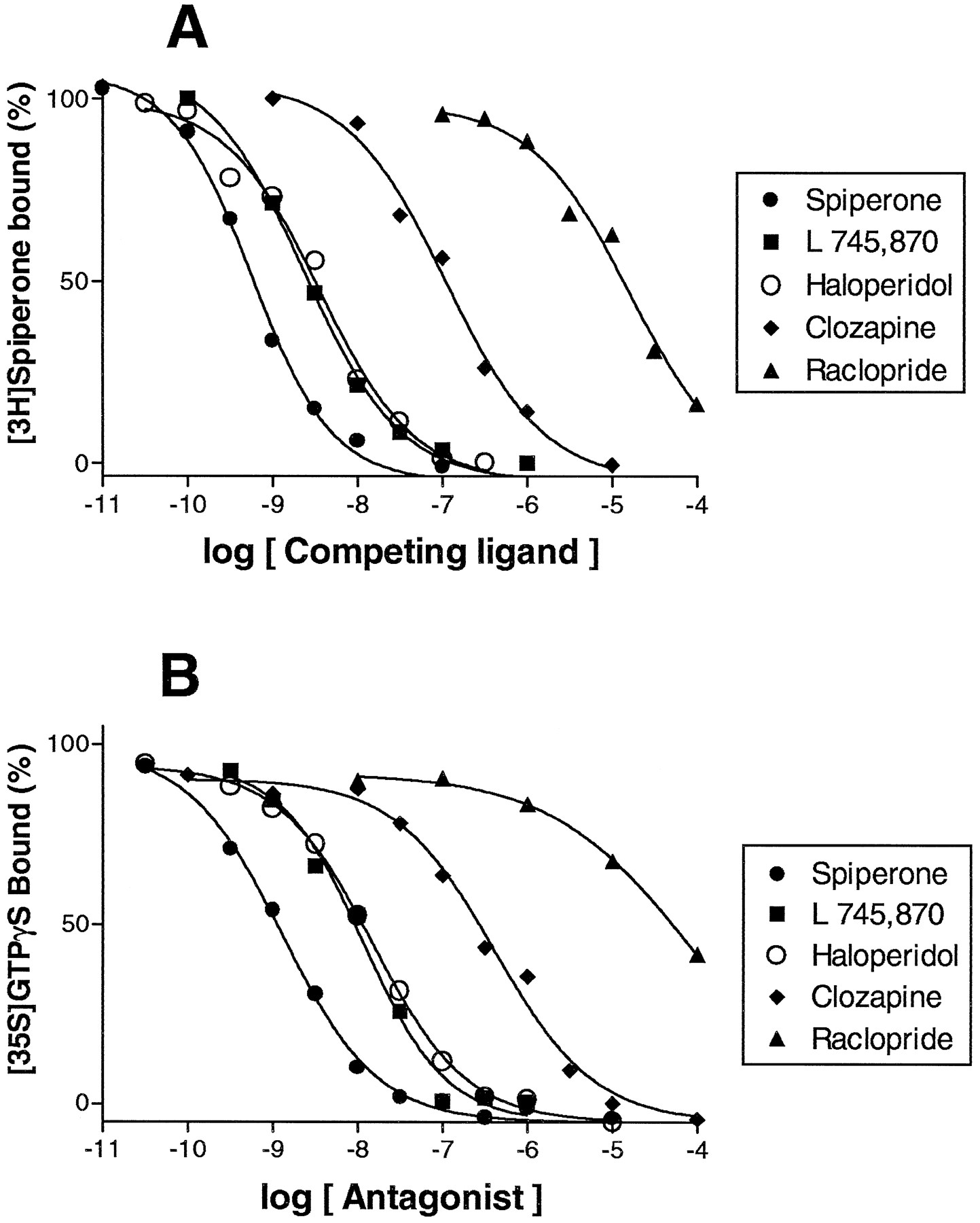
Competition binding and antagonism at cloned human D4.4 receptors by dopaminergic antagonists. A, Representative [3H]spiperone competition binding isotherms. B, Antagonism of dopamine (1 μM)-stimulated [35S]GTPγS binding. Points shown are mean of triplicate determinations from representative experiments repeated on at least three occasions.
Antagonism of dopamine-stimulated [35S]GTPγS binding to CHO-D4.4 membranes
Action of dopaminergic antagonists at cloned human D4.4receptors
The effect on [35S]GTPγS binding of a range of antipsychotics was tested, including clozapine, olanzapine, risperidone and ziprasidone, none of which altered [35S]GTPγS binding from basal levels when tested alone. However, fixed concentrations of antagonist shifted the dopamine concentration-response curve to the right in a parallel manner, consistent with competitive antagonism at D4.4 receptors (table 3). For all the compounds tested, theKb values calculated from these shifts agreed closely with their respectiveKi values (r = .99, P < .01; fig. 5D), except FG 5893, which showed a 6-fold lowerKb value thanKi value (P < .05, table 3) and was not included in the calculation of correlation coefficient.
Discussion
Effects of GDP, NaCl and MgCl2 on [35S]GTPγS binding to CHO-D4.4 membranes.
[35S]GTPγS binding affords a measure of receptor-mediated G protein activation (the first step of the signal transduction pathway) and is applicable regardless of the second-messenger system(s) involved. In CHO-D4.4 cell membranes, [35S]GTPγS binding was modulated by buffer concentrations of GDP. The latter reduced the level of basal (non-agonist-stimulated) binding, without affecting agonist-dependent binding. Hence, as GDP concentration increased, the ratio of agonist-stimulated to basal [35S]GTPγS binding increased to 2.2-fold at a GDP concentration of 3 μM (fig. 2B, inset). This compares with stimulation ratios of 1.4-, 2.2-, 2.5- and 3-fold for 5-HT1D α, 5-HT1D β, muscarinic and 5-HT1Areceptors, respectively (Hilf et al., 1989; Newman-Tancrediet al., 1996b; Thomas et al., 1995). Like GDP, NaCl reduced basal binding of [35S]GTPγS but reduced dopamine-stimulated binding only at concentrations of >100 mM. Similar data have been reported for agonist stimulation of [35S]GTPγS binding at alpha-2 adrenoceptors (Tian et al., 1994).
[35S]GTPγS binding to CHO-D4.4 membranes has an absolute requirement for magnesium, similar to that observed for A1 adenosine receptors (Lorenzen et al., 1993). In the present study, the modulatory effects of magnesium were complex, exhibiting a biphasic action on agonist-dependent [35S]GTPγS binding (fig. 2D). The [35S]GTPγS binding peak at a MgCl2concentration of 3 to 10 mM probably reflects conditions that favor the formation of a ternary complex of agonist/receptor/G protein. In contrast, agonist stimulation of [35S]GTPγS binding at a MgCl2 concentration of 0.1 mM is low, because these conditions may be less favorable for the formation of the ternary complex. However, MgCl2 also had a biphasic effect onbasal [35S]GTPγS binding, suggesting that Mg++ ions may have modulatory effects on G proteins themselves. These may reflect altered levels of G protein attachment to cell membranes. For example, transducin solubility increases at Mg++ concentrations of <0.1 mM (Bornancin et al., 1989), suggesting that the association of G proteins to membrane-bound receptors would be favored by higher Mg++concentrations. Although the exact mechanistic basis for the biphasic action of magnesium is unclear, the present observations agree with similar biphasic effects reported for muscarinic acetylcholine receptors (Hilf et al., 1989) and fMet-Leu-Phe (fMet) chemotactic receptors (Gierschik et al., 1991). For subsequent experiments, a magnesium concentration of 3 mM was selected, providing the highest ratio of dopamine-stimulated over basal [35S]GTPγS binding (fig. 2D).
The buffer composition chosen was similar to that of Chabert et al. (1994) for [35S]GTPγS binding to D4.4 -transfected Sf9 insect cells and for the other receptors mentioned above. This suggests that buffer conditions that yield optimal agonist stimulation of [35S]GTPγS binding may be similar for many receptor and cell types. Furthermore, the present data show that there is no necessity for agonist to be present for G protein activation to occur. Rather, G protein activation can be induced, in the absence of agonist, by selecting conditions that favor coupling of G protein to receptor (millimolar magnesium, low sodium and low GDP). These factors do not, however, appear to influence the ability of dopamine to further stimulate [35S]GTPγS binding, suggesting that an active receptor conformation is induced by agonists that is not achieved by merely manipulating buffer conditions. Thus, basal and agonist-stimulated [35S]GTPγS binding may reflect different activation states of the D4.4receptor. In addition, the presence of a basal level of receptor-mediated G protein activation enables, in principle, the identification of compounds that inhibit G protein activation (inverse agonists). Xanthine amine congener, for example, lowers basal G protein activation at A1 adenosine receptors (Freissmuth et al., 1991), whereas methiothepin and ketanserin inhibit [35S]GTPγS binding to membranes of CHO cells stably expressing 5-HT1D α and 5-HT1D β receptors (Thomas et al., 1995). In the present system, however, the level of basal [35S]GTPγS binding was minimized to facilitate the definition of agonist effects. Hence, reduction of basal binding by inverse agonists, may be relatively small. Furthermore, the ability to detect inverse agonist activity may depend on the presence of a high receptor to G protein ratio. Indeed, in a CHO cell line manipulated to express a high level of 5-HT1A receptors (without a change in G protein number), the inverse agonist spiperone exhibited increased negative efficacy (Newman-Tancredi et al., 1997b, 1997c).
Determination of G protein number in CHO-D4.4 membranes by [35S]GTPγS isotopic dilution.
Unlabeled GTPγS inhibited basal [35S]GTPγS binding to CHO-D4.4 monophasically and with low affinity [IC50(low) = 110 nM]. In contrast, GTPγS inhibited agonist-stimulated [35S]GTPγS binding biphasically, with an additional high-affinity site [IC50(high) = 4.4 nM] as well as a low-affinity binding component. Two points should be made regarding these data. First, the IC50 values are a function of the GDP concentration in the assays, because GTPγS in effect competes with GDP for binding to G proteins. Second, whereas the low-affinity binding component reflects inhibition of the endogenous GDP/GTP exchange rate of all CHO-D4.4 G proteins, the high-affinity binding component reflects only inhibition of agonist-stimulated GDP/GTP exchange at D4.4 receptor-linked G proteins (fig. 3A; Tian et al., 1994). TheKd value for this high-affinity component (15 ± 4.2 nM) was not significantly different from the IC50(high) above, but serotonin-activated recombinant human 5-HT1A receptors, also expressed in CHO cells, display aKd value for [35S]-GTPγS of only 1.29 ± 0.13 nM (Newman-Tancredi et al., 1977c P < .01, two-tailedt test, compared with theKd value for D4.4receptors). This suggests that D4.4 and 5-HT1Areceptors may differently activate G proteins in the same host cell line. In fact, CHO-K1 cells express Gi α 2 and Gi α 3, both of which can couple to 5-HT1A receptors (Raymond et al., 1993), whereas D4.4 receptor coupling to Gi α 2 may be a cell-dependent property because it is observed in mouse fibroblast CCL1.3 cells but not MN9D mesencephalic cells (Tang et al., 1994). Additional studies are therefore required to determine which G protein subtype or subtypes are activated by D4 receptors in CHO cells.
Information regarding the receptor/G protein stoichiometry in CHO-D4.4 cells can be obtained from theB max values for dopamine-stimulated [35S]GTPγS binding in CHO-D4.4 cells (2.29 pmol/mg) and the B max value for D4.4receptor expression (1.40 pmol/mg). These data indicate that 1 or 2 dopamine-activated G proteins are labeled in CHO-D4.4membranes per D4.4 receptor. This is similar to that seen for atrial natriuretic factor receptors (1 G protein/receptor; Khurana and Pandey, 1995) and for mu opioid receptors (2 G proteins/receptor; Traynor and Nahorski, 1995). However, much higher degrees of amplification have been observed for fMet chemotactic receptors (20 G proteins/receptor; Gierschik et al., 1991) and for human cardiac muscarinic acetylcholine receptors (50–80 G proteins/receptor; Böhm et al., 1994). Further investigation is required to elucidate the origin of these widely varying ratios, but they may be a function of the rapidity of cycling between different receptor subtypes and their respective G proteins. Indeed, in a comparative study in rat striatal membranes, muand sigma opioid receptors activated 20 G proteins/receptor, whereas cannabinoid receptors only activated 3 G proteins/receptor (Simet al., 1996).
Agonist stimulation of [35S]GTPγS binding to CHO-D4.4 membranes.
The action at D4.4 receptors of 12 dopaminergic agonists was characterized. Their EC50 values for activation of [35S]GTPγS binding correlated closely with theKi values obtained for inhibition of [3H]spiperone binding, and their order of potency agrees with that previously reported for D4 receptors (Chabertet al., 1994; Tang et al., 1994; Van Tol et al., 1991). In the case of agonist competition binding isotherms, the presence of more than one receptor affinity state was suggested by the low (<.8) pseudo-Hill coefficients (table 1), probably reflecting ligand binding to G protein-coupled and -uncoupled forms of the receptor.
In measurements of [35S]GTPγS binding, all the agonists exhibited efficacies (relative to dopamine) of markedly <100%, with the highest (∼74%) observed with ropinirole and quinerolane. In contrast to the present data, Lahti et al. (1996) reported that the efficacy of (−)-apomorphine at D4.4 receptors, estimated by the ratio of ligand affinities for G protein-coupled and -uncoupled receptor states, was about double (80%) that seen here (≈40%; table 1). Similarly, Chabert et al. (1994) found that (−)-apomorphine exhibited an efficacy of 85%, whereas two further partial agonists tested here (dp-ADTN and (−)-quinpirole) were full agonists at D4.4 receptors expressed in insect Sf9 cells. At least two possibilities may account for these differences. First, although buffer conditions were similar, the incubation temperature in the present study was lower (22°C) than that used by Chabert et al. (1994) (30°C). A temperature rise may augment the apparent efficacy of partial agonists through thermodynamic facilitation of GDP release from G proteins (Lorenzen et al., 1996). However, in control experiments incubated at 30°C, the efficacy of (−)-apomorphine was increased by only 5% to 10%,1 which is insufficient to account for the 45% difference in efficacies. Second, the D4.4 receptor B max value in the CHO cells used here (1.40 pmol/mg) was 4-fold lower than that in theSf9 cells used by Mills et al. (1993) (5–6 pmol/mg). This suggests that apparent agonist efficacies in Sf9 cells may be higher due to the presence of “spare” receptors. Indeed, a high ratio of receptor to G protein in Sf9 cells would be expected to increase agonist efficacy, as has been shown for weak partial agonists such as pindolol at 5-HT1B receptors and eltoprazine at 5-HT1A receptors (Adham et al., 1993; Newman-Tancredi et al., 1997c).
The agonists tested in the present study include several compounds that are in development for the treatment of PD, such as quinerolane, bromocriptine and ropinirole. Dopaminergic agonists are known to alleviate the symptoms of PD (De Keyser et al., 1995;Wolters et al., 1995), but their exact mechanism of action is unclear, and may involve multiple dopamine receptor subtypes (Jenner, 1995). Quinerolane, (−)-apomorphine and lisuride displayed high affinity for D4.4 receptors, but bromocriptine and piribedil displayed low or negligible affinity at this site. Furthermore, although quinerolane was an efficacious agonist (Emax = 72.4%), lisuride had only weak partial agonist activity (Emax = 32.2%), and terguride is an antagonist at D4.4 receptors (see tables 1 and 2). It is concluded that no correlation exists between the antiparkinsonian effects of these drugs and their activity at D4.4 receptors. However, given that dopaminergic agonists can have propsychotic actions (Factoret al., 1995) and the possible importance of D4.4 receptors in mediating mood dysfunctions, an antiparkinsonian drug with antagonist activity at D4.4 , such as the ergot terguride (trans-dihydrolisuride), may present a therapeutic advantage. Indeed, terguride attenuates parkinsonian symptoms in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned monkeys and suppresses hyperactivity induced by apomorphine treatment (Akaiet al., 1993), whereas in clinical trials, terguride was effective against PD with only a low incidence of psychotic side effects (Filipova et al., 1988). Thus, it may be hypothesized that antiparkinsonian drugs with low efficacy at D4.4 receptors may induce less-pronounced psychotic side effects. This is consistent with the observed effectiveness of clozapine, which has significant affinity and antagonist activity at D4 receptors, in countering L-DOPA-induced psychosis in PD patients (Factor et al., 1995; Meltzer et al., 1995).
Antagonism of dopamine-stimulated [35S]GTPγS binding to CHO-D4.4 membranes.
In agreement with previous reports, clozapine showed marked affinity (Ki = 38 nM) at D4.4receptors (table 3). This indicates that in our hands, clozapine is ∼2-fold selective for D4.4 compared with D2receptors (Ki = 76 nM) (Millanet al., 1995), although using different radioligands and experimental conditions, other authors have reported selectivities of ≤17-fold (Durcan et al., 1995; Lawson et al., 1994; Van Tol et al., 1991). In contrast, the novel D4 receptor antagonist L 745,870 has negligible affinity at D2 receptors (Ki = 890 nM)2 but high affinity at D4.4receptors. Its Ki value (1.99 nM) is in the same range as that (0.4 nM) reported by Kulagowski et al. (1996). Although it has no effect alone, L 745,870 completely antagonized dopamine-stimulated [35S]GTPγS binding (fig. 4B), with a Kb value of 1.07 nM, and shifted the dopamine concentration-response curve to the right with a Kb value of 1.19 nM (table 2), confirming its high potency at D4.4 receptors.
The present study tested the antagonist activity and/or affinity of 19 other dopaminergic ligands at D4.4 receptors, including several antipsychotics in late-stage development. Olanzapine, which was designed to exhibit a similar receptorial profile to that of clozapine (which is also a dibenzazepine derivative), displayed a similar affinity at D4.4 receptors (Ki = 26.1 nM). However, other dibenzazepine derivatives, ORG 5222 and seroquel, displayed widely differing affinities at this site (Ki = 0.78 and 2290 nM, respectively). In fact, seroquel has generally low affinity at a range of receptors, including dopamine D1, D2 and D3(pKi = 5.4, 6.2 and 6.5, respectively; Schotte et al., 1996). All the butyrophenone compounds tested (spiperone, haloperidol, ocaperidone, risperidone and ziprasidone) showed marked affinity at D4.4 receptors, as did the arylpiperazines tiaspirone and FG 5893. Interestingly, BMY 14,802 (another arylpiperazine), originally described as a selectivesigma-site ligand (Taylor and Dekleva, 1987), had significant affinity at D4.4 receptors (Ki = 24.5 nM), whereas othersigma ligands, DUP 734 and rimcazole, did not (Ki > 1000 nM). Similarly, fananserin (RP 62203), originally presented as a selective 5-HT2A antagonist (Doble et al., 1992), has high affinity at D4.4 receptors (Ki = 4.99 nM). This result agrees with Heuillet et al. (1996), who reported aKi value of 2.9 nM at CHO-D4.2 receptors. Ligands selective for D1(SCH 23,390) and D2/D3 receptors (raclopride) showed low affinity, but the D3 receptor antagonist GR 103,691 exhibited moderate affinity at D4.4 receptors (Ki = 83.6 nM), which is in agreement with previous reports (63 nM) (Murray et al., 1995b).
Previous studies have shown the antagonist activity of clozapine and haloperidol at D4 receptors (Asghari et al., 1995; Chabert et al., 1994). The present study confirms these findings and characterized 19 other compounds, showing thatKb values corresponded closely to respective Ki values (r = .99, fig. 4D). In contrast, Asghari et al. (1995) found, surprisingly, that clozapine and haloperidol were equipotent for antagonism of dopamine-inhibited adenylyl cyclase activity, and Chabertet al. (1994) reported pA 2 values for clozapine and haloperidol 1 order of magnitude lower than theKb values here. These discrepancies may be due to the different cell lines used, the different receptor expression levels or the different functional test adopted ([35S]GTPγS binding or adenylyl cyclase). Nevertheless, the present data demonstrate the ability of all the antipsychotics tested to shift the dopamine stimulation curve to the right in a parallel manner, which is consistent with competitive antagonism at D4 receptors. The Kb values of spiperone, L 745,870, haloperidol and clozapine calculated for inhibition of dopamine-stimulated [35S]GTPγS binding agreed closely with those calculated for the shift in the dopamine concentration-response curves (tables 2 and 3). None of the drugs tested, including L 745,870, terguride and clozapine, exhibited any intrinsic agonist activity. This indicates that they act as “neutral” or “silent” antagonists in this system, although experiments in a cell line with a high receptor expression level might reveal weak agonist activity (Newman-Tancredi et al., 1997c). Taken together, the present data found no distinction in (lack of) intrinsic activity at D4.4 receptors between typical antipsychotics such as haloperidol and atypical antipsychotics such as clozapine, risperidone and olanzapine.
The physiological significance of D4 receptors is unclear, but the controversial up-regulation of D4-like receptors in postmortem schizophrenic brain tissue (Murray et al., 1995;//Reynolds, 1996; Seeman et al., 1993a) suggests that an interaction at these sites may be involved in the etiology of the disease. However, the benzamide antipsychotic raclopride, which is effective in countering productive schizophrenic symptoms, has low affinity at D4.4 receptors (table 3), whereas the potent and selective D4 receptor antagonist L 745,870 is ineffective in treating acutely psychotic patients (Kramer et al., 1996). Nevertheless, raclopride induces a high incidence of extrapyramidal symptoms and poorly treats negative schizophrenic symptoms, whereas L 745,870 did not induce extrapyramidal symptoms, and its therapeutic interest in treating negative and cognitive symptoms is as yet unknown. These observations, together with the known distribution of D4-like receptors in frontal cortex and hippocampus and on GABAergic neurons (Matsumoto et al., 1996; Meador-Woodruff et al., 1996; Mrzljak et al., 1996), suggest that the functional significance of D4 receptors may be related to deficit symptoms, cognitive dysfunction or anxiodepressive states. Indeed, the D4receptor antagonist (and antiparkinsonian drug) terguride significantly attenuated negative, but not positive, schizophrenic symptoms in clinical trials (Olbrich and Schanz, 1988, 1991).
Conclusions.
[35S]GTPγS binding methodology has been applied to recombinant human dopamine D4.4receptors expressed in a mammalian (CHO) cell line. In this system, high experimental concentrations of GDP (3 μM), NaCl (100 mM) and MgCl2 (3 mM) are necessary to obtain optimum ratios of agonist-induced over basal [35S]GTPγS binding. CHO-D4.4 cells display a high ratio of dopamine-activated G proteins to receptors, indicating the absence of spare receptors and enabling partial agonist activity to be defined. A range of antiparkinsonian drugs exhibited widely varying affinities and efficacies at D4.4 receptors, suggesting that activity at this site is not an important factor in their clinical effectiveness, although D4.4 receptor agonism may be associated with side effects on mood. In contrast, a range of neuroleptic and atypical antipsychotics antagonized the dopamine-induced stimulation of [35S]GTPγS binding, suggesting that D4receptor antagonism may be a potentially clinically important feature of many antipsychotic drugs.
Acknowledgments
We thank Paul Chazot, Chris Breivogel, Frederic Bornancin and Ann Mills-Duggan for helpful discussions.
Footnotes
-
Send reprint requests to: Dr. Adrian Newman-Tancredi, Department of Psychopharmacology, Institut de Recherches Servier, 125, Chemin de Ronde, 78290 Croissy-sur-Seine (Paris), France. E-mail: 101511,274{at}compuserve.com
-
↵1 A. Newman-Tancredi and C. Chaput, unpublished observations.
-
↵2 A. Newman-Tancredi and C. Chaput, unpublished observations.
- Abbreviations:
- 5-HT
- 5-hydroxytryptamine
- CHO
- Chinese hamster ovary
- CHO-D4.4
- Chinese hamster ovary cells expressing dopamine D4.4 receptors, GTPγS, guanosine-5′-O-(3-thio)triphosphate
- PD
- Parkinson’s disease
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- Received September 23, 1996.
- Accepted March 17, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}