


Abstract
The neuronal transporters for the monoamines dopamine, serotonin, and norepinephrine are plasma membrane proteins that serve vital functions in the reuptake and control of synaptic neurotransmitter levels. They are also targets for abused and therapeutic drugs and play pivotal roles in neurological disorders such as depression, schizophrenia, and Parkinson's disease. There is increasing evidence that some activities of these carriers are subject to acute control by treatments that affect phosphorylation pathways, but the molecular basis for this is not understood. Recent work suggests that these regulatory processes may involve phosphorylation of the transporters by protein kinase C and other kinases, and may occur by affecting intrinsic transport activity or by controlling transporter cell surface expression. Phosphorylation-mediated regulation of monoamine transporters provides the potential for acute presynaptic control of neurotransmitter levels during normal neurophysiologic events, and dysregulation of these processes may lead to inappropriate transmitter clearance that contributes to the etiology of neurological disorders.
The plasma membrane transporters for the monoamines dopamine (DA), norepinephrine (NE) and serotonin (5-HT) are part of a family of Na+/Cl--dependent symporters that rapidly clear neurotransmitter from the synaptic space. This activity is essential for appropriate neural function as it terminates synaptic transmission and allows for receipt of subsequent incoming signals. Because the dopamine, norepinephrine, and serotonin transporters (DAT, NET, and SERT) are the primary means of clearance for their cognate transmitters, increases or decreases in their activities may provide mechanisms for fine-tuning the spatial and temporal dynamics of neurotransmission, and dysregulation of their activities may underlie disorders of transmitter imbalance such as depression, attention deficit hyperactivity disorder, and schizophrenia.
In addition to their roles in neurotransmitter homeostasis, DAT, NET, and SERT are major targets for psychostimulants such as cocaine, which blocks transmitter reuptake, and amphetamine (AMPH) and methamphetamine (METH), which are carried by the proteins and induce efflux of endogenous substrate through transport reversal. The net effect of these and related drugs is to increase synaptic neurotransmitter concentrations, which leads to immediate and long-term changes associated with addiction (Torres et al., 2003). Enhancement of monoamine levels by transporter blockade also underlies the action of therapeutic compounds such as fluoxetine (Prozac), bupropion (Wellbutrin), and methylphenidate (Ritalin), used to treat depression and attention deficit disorder. Monoamine carriers function as gateways for cell-specific entry of neurotoxins including 1-methyl 4-phenylpyridinium, oxidized dopamine metabolites, and environmental chemicals such as pesticides that may induce the selective neuronal death found in Parkinson's disease and other neuropathologies (Miller et al., 1999). Radioactive analogs of monoamines and cocaine are also widely used as in vivo imaging agents for monoaminergic neurons in clinical and experimental applications.
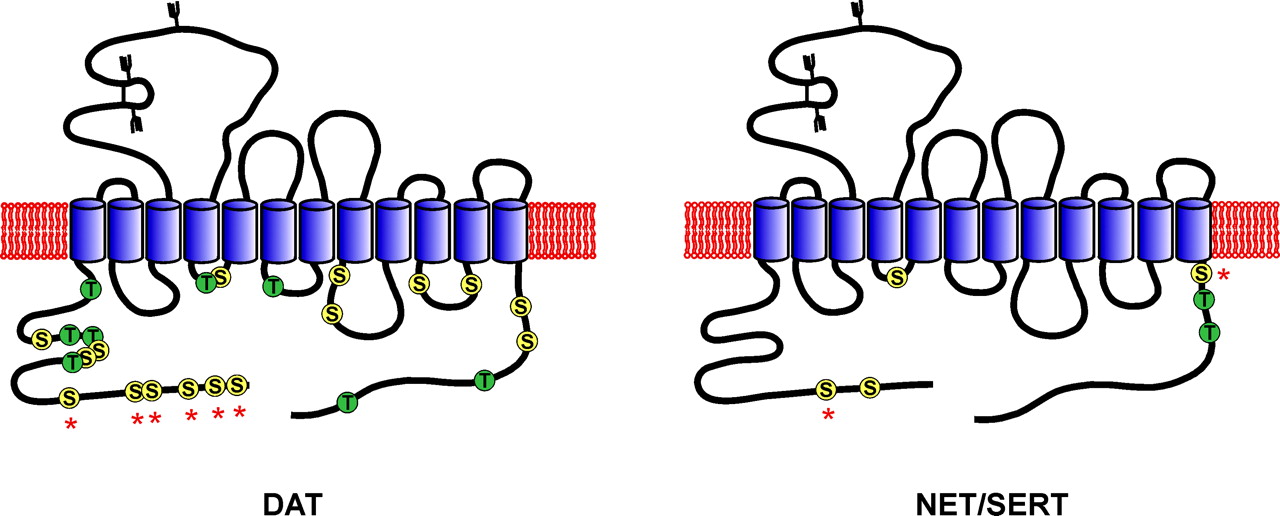
Since the cloning of DAT, NET, and SERT over a decade ago, there has been widespread interest in the possibility that their activities undergo regulation by phosphorylation-mediated processes, which would provide a mechanism for rapid modulation of transmitter clearance in response to specific physiological demands. The proteins share a similar structure consisting of twelve transmembrane-spanning (TM) domains with intracellularly oriented N and C termini (Fig. 1). Within the TM domains, the proteins show considerable sequence homology presumed to reflect a common mechanism for substrate translocation, whereas the connecting loops and cytoplasmic tails are more divergent and may represent sites for unique protein functions (Torres et al., 2003). The N- and C-terminal tails and intracellular loops (ILs) of these proteins contain numerous serine (S), threonine (T), and tyrosine (Y) residues that could serve as phosphorylation sites, and many of these residues are present within consensus sequences for protein kinase C (PKC), protein kinase A (PKA), and calcium/calmodulin-dependent protein kinase (CaMK). Many studies have now demonstrated that various transporter properties including net transmitter uptake, reverse transport, subcellular distribution, and interaction with binding partners are affected by a wide variety of second messengers and phosphorylation conditions (reviewed in Zahniser and Doolen, 2001). In some of these studies, concomitant phosphorylation of the transporters has been examined, but it is not known if this is a requisite step in these regulatory processes or if some events are mediated by phosphorylation of associated proteins. This review describes the currently known characteristics of monoamine transporter phosphorylation (summarized in Table 1) and the most closely associated regulatory processes. Other aspects of regulation of these and related neurotransmitter transporters can be found in several excellent recent reviews (Zahniser and Doolen, 2001; Robinson, 2002; Torres et al., 2003). The data emerging from these studies demonstrate both common and unique phosphorylation properties for the monoamine transporters and suggest that regulatory activities may occur by multiple molecular mechanisms.

Schematic diagram of DAT, NET, and SERT structures and potential phosphorylation sites. Left, composite diagram showing Ser and Thr residues rat and human DAT that have been mutated to nonphosphate acceptors (Chang et al., 2001; Granas et al., 2003; Foster et al., 2003a; Lin et al., 2003). The asterisks indicate the cluster of serines that contain basal and OA/PKC-stimulated phosphorylation sites (Foster et al., 2002; Granas et al., 2003). Right, composite diagram showing Ser and Thr residues in NET and SERT that have been mutated to nonphosphate acceptors (Sakai et al., 1997; Blakely et al., 1998; Bonisch et al., 1998). Asterisks indicate SERT residues phosphorylated in vitro by PKA (Blakely et al., 1998).
Summary of DAT, SERT, and NET phosphorylation properties
Effects of Protein Kinases
The human, rat, and mouse isoforms of the dopamine transporter have been identified as phosphoproteins by metabolic 32PO4 labeling of the protein in rat and mouse striatal tissue and in heterologously expressing cultured cells (Huff et al., 1997; Vaughan et al., 1997; Cowell et al., 2000; Granas et al., 2003; Lin et al., 2003). In the absence of exogenous treatments, DAT exhibits basal phosphorylation detectable with 32PO4 labeling times as short as 30 min, demonstrating the presence and rapid turnover of constitutive phosphate. Treatment of tissue with PKC activators, such as phorbol 12-myristate 13-acetate (PMA) or protein phosphatase inhibitors such as okadaic acid (OA), leads to increased DAT phosphorylation that occurs within 5 min and plateaus at 3- to 4-fold above basal by 20 min (Huff et al., 1997; Vaughan et al., 1997). Comparable treatments produce 15 to 50% reductions in DA transport Vmax (Huff et al., 1997; Vaughan et al., 1997; Granas et al., 2003), whereas the Km for DA and the binding affinity of cocaine analogs are not strongly affected. It is currently not known if PKC phosphorylates DAT directly or acts by a downstream kinase, and attempts in our laboratory to demonstrate in vitro phosphorylation of purified DAT with PKC, PKA, PKG, or CaMK have been negative (R. Vaughan, unpublished result). PKC-stimulated down-regulation of DA transport activity is accompanied by dynamin-dependent endocytosis of DAT (Daniels and Amara, 1999; Melikian and Buckley, 1999; Granas et al., 2003), which has been taken as evidence compatible with the reduced transport velocity as sequestered transporters would not be able to interact with extracellular dopamine. Although DA uptake is regulated by many second messengers and treatments not directly linked to PKC, effects of other kinases on DAT phosphorylation have been examined in only a few studies. PKA conditions increase DA transport velocity (Batchelor and Schenk, 1998), but have no pronounced effect on DAT phosphorylation (Vaughan et al., 1997), suggesting a control mechanism independent of transporter phosphorylation. Increased DAT activity and cell surface expression has also been found in cells treated with insulin or transfected with constitutively active PI3 kinase (PI3K) or MAP kinase (MAPK) (Carvelli et al., 2002; Moron et al., 2003). In this case, preliminary experiments demonstrated suppression of basal DAT phosphorylation by PI3K and MAPK inhibitors (Lin et al., 2003), suggesting a potential involvement of MAPK and PI3K in promoting constitutive phosphorylation and cell surface expression of DAT. Although further work is needed to verify these results, they suggest the intriguing possibility for reciprocal regulation of DAT by PKC and other kinases.
SERT phosphorylation has also been demonstrated by metabolic labeling with 32PO4 in hSERT-HEK 293 cells (Ramamoorthy et al., 1998; Ramamoorthy and Blakely, 1999), and by Western blotting of rat brain synaptosomes with anti-phosphoserine antibodies (Najib et al., 2000). These studies showed, as with DAT, that SERT undergoes basal phosphorylation that is acutely increased (5- to 10-fold) by treatments with PKC activators and protein phosphatase inhibitors. SERT subcellular distribution shifts to internal compartments in response to these treatments, and this is accompanied by 25 to 50% decreases in transport Vmax with no change in substrate or cocaine affinity (Qian et al., 1997). PMA-induced phosphorylation of SERT plateaus between 10 and 30 min, but unlike DAT, the response to OA increases for at least 2 h (Ramamoorthy et al., 1998). Since OA-induced phosphorylation produced by blockade of dephosphorylation in the absence of exogenous kinase activators reflects constitutive kinase activity, this suggests a slower catalysis of basal phosphorylation. SERT phosphorylation also shows robust stimulation by PKA and PKG activators (Ramamoorthy et al., 1998), although this is not associated with transport down-regulation. This may indicate that PKA/PKG-dependent SERT phosphorylation accompanies an as yet unknown activity or that the cellular context in which phosphorylation was obtained was insufficient to support a functional response. SERT phosphorylation levels are additive with all combinations of OA, PMA, and cholera toxin, consistent with basal, PKC-stimulated, and PKA-stimulated phosphorylation occurring on different sites, and OA-induced phosphorylation is not blocked by inhibitors or PKC, PKA, or CaMK, suggesting that none of these kinases catalyzes basal phosphorylation.
The transport velocity and surface expression of NET in heterologous and endogenous systems is also acutely decreased by PMA treatment and by activation of receptors linked to PKC and other pathways (Apparsundaram et al., 1998; Jayanthi et al., 2004). Although fewer phosphorylation studies have been done for NET, the protein expressed endogenously in rat trophoblasts undergoes basal metabolic 32PO4 labeling that is increased by PMA and OA (Jayanthi et al., 2004).
The kinetic, dose, and inhibitor characteristics of PKC-induced DAT, NET, and SERT sequestration, down-regulation, and phosphorylation show strong correlations consistent with the processes being mechanistically linked. These findings are reminiscent of the phosphorylation of G-protein-coupled receptors that serves to initiate arrestin binding and clathrin-mediated endocytosis associated with receptor desensitization and down-regulation. This originally led to the hypothesis that stimulated phosphorylation of monoamine transporters may be a signal leading to their intracellular sequestration, possibly by acting as a binding motif for endocytic proteins. Recent work suggests that this is not the case for DAT (Granas et al., 2003; Khoshbouei et al., 2004), but the experiments testing this hypothesis for NET and SERT have not yet been performed. An alternative hypothesis is that phosphorylation of transporters regulates their interactions with SNARE proteins such as syntaxin 1A, a protein best characterized as a plasma membrane docking site for synaptic vesicle proteins. Coimmunoprecipitation and coexpression assays have shown that syntaxin 1A binds to NET and SERT, which promotes their presence and regulates their activity at the neuronal surface (Quick, 2002, 2003; Sung et al., 2003). Both transporters bind to syntaxin 1A through N-terminal tail regions that contain potential phosphorylation sites, and the NET-syntaxin 1A interaction is decreased by PMA (Quick, 2003; Sung et al., 2003). SERT phosphorylation is also increased by treatment of synaptosomes with tetanus toxin, which cleaves the synaptic vesicle SNARE protein synaptobrevin (Najib et al., 2000). These results implicate a role for monoamine transporter phosphorylation with regulation of SNARE interactions that may control transmitter clearance by affecting transporter cell surface levels or intrinsic activity.
Whereas these models have interpreted kinase effects on transporters and transmitter uptake with respect to trafficking, PKC also modulates reverse substrate transport, which presumably occurs at the cell surface. AMPH-induced efflux of substrate through DAT and NET is blocked by PKC inhibitors (Kantor and Gnegy, 1998; Kantor et al., 2001), and DA efflux through DAT displays AMPH-independent stimulation by PMA (Cowell et al., 2000). Both PMA-stimulated efflux and PMA-stimulated DAT phosphorylation are blocked by cocaine (Cowell et al., 2000), and N-terminal DAT serines are required for AMPH-induced DA efflux (Khoshbouei et al., 2004). These findings are consistent with an association of the phosphorylated state of the transporters with their efflux mode and suggest that DAT phosphorylation promotes an intrinsic protein conformation favorable for reverse transport.
Effects of Psychostimulants and Substrates
Another process associated with phosphorylation of DAT and SERT is feedback regulation of uptake by substrate transport. For SERT this occurs by modulation of PKC actions on uptake and phosphorylation. Treatment of hSERT-HEK 293 cells with PMA in combination with 5-HT, AMPH, or other substrates results in pronounced attenuation of the PMA-stimulated level of SERT phosphorylation and loss of PMA-induced SERT internalization (Ramamoorthy and Blakely, 1999). The substrate effects were prevented by cocaine and were not induced by 5-HT receptor agonists, demonstrating that transport activity was required to produce the responses. Thus the ability of PKC to induce SERT endocytosis is counteracted in an actively transporting protein, providing an adaptive feedback mechanism for maintaining the transporter at the cell surface during periods of high transport demand.
Substantially different effects of substrates have been found for DAT. In the absence of PKC activators, in vitro treatments of rat striatal synaptosomes or hDAT-HEK 293 cells with AMPH, METH, or DA induce internalization and functional down-regulation of DAT that is blockable by cocaine (Saunders et al., 2000; Sandoval et al., 2001; Chi and Reith, 2003). Similar processes may occur in vivo, as rats given a single injection of METH displayed rapid and reversible reductions in striatal DA uptake (Fleckenstein et al., 1997). METH-induced transport down-regulation in synaptosomes was blocked with a PKC inhibitor (Sandoval et al., 2001), indicating a link for DAT, as with SERT, between PKC and substrate-induced processes. Comparable treatments of cells or striatal tissue with AMPH or METH also lead to increased DAT phosphorylation that can be blocked by cocaine and PKC inhibitors (Cervinski and Vaughan, 2003), consistent with involvement of DAT phosphorylation in the internalization and down-regulation mechanisms. In contrast with SERT, however, substrates increase rather than decrease DAT phosphorylation, and the transport-induced feedback regulation of DAT decreases rather than increases its cell surface availability. Thus, in the case of DAT, these processes may serve a neuroprotective function limiting the movement of high doses of potentially neurotoxic substrate into the cell. Not all cell types support DA-induced trafficking of DAT, however (Daniels and Amara, 1999), indicating the importance of the cellular context for determining associated molecular mechanisms. It is not known if the effects of substrates on DAT and SERT phosphorylation occur due to altered properties of the actively transporting protein as a substrate for kinases or phosphatases, or if transported substrates modify the activities or subcellular localization of kinases or phosphatases, which impacts their actions on transporters.
Application of cocaine alone to cells or tissue has no pronounced effect on DAT phosphorylation (Cervinski and Vaughan, 2003), although acute cocaine treatments lead to modest increases in DA transport and DAT cell surface expression (Daws et al., 2002). However, cocaine blocks PMA-induced phosphorylation of DAT (Colwell et al., 2000), but not of SERT (Ramamoorthy and Blakely, 1999). Thus substrate and inhibitor classes of psychostimulants have distinct effects on monoamine transporter phosphorylation, and the same psychostimulant classes produce different effects on SERT and DAT phosphorylation and functional responses, implicating differential molecular mechanisms of action. Despite these differences, the results found in these studies demonstrate previously unknown effects of psychostimulant drugs that are distinct from their direct actions on monoamine transport and that lead to adaptive functional responses potentially mediated by phosphorylation.
Transporter Dephosphorylation
The importance of dephosphorylation pathways for DAT, NET, and SERT is highlighted by the substantial increases in their levels of phosphorylation produced by inhibition of phosphatases with OA. For all three transporters, the level of phosphorylation produced by OA equals or exceeds those induced by PKC alone (Ramamoorthy et al., 1998; Foster et al., 2002, 2003b; Jayanthi et al., 2004), suggesting the presence in resting tissue of biochemical pressure toward maintenance of transporters in a more dephosphorylated state. Thus, if phosphorylation leads to endocytosis or loss of plasma membrane syntaxin 1A interactions, transporters in the resting neuron would be maintained at the cell surface where they will be available for activity, and if phosphorylation promotes reverse transport, transporters in the resting neuron would be maintained in an inwardly transporting conformation. For all of these scenarios, dephosphorylation would promote a situation favoring synaptic transmitter clearance, and input from PKC or other second messengers would alter the physiology toward reduced clearance/prolonged neurotransmission.
The phosphatases contributing to SERT dephosphorylation were examined in hSERT-HEK 293 cells using inhibitors specific for the major serine/threonine protein phosphatase (PP) classes, PP1, PP2A, and PP2B (Ramamoorthy et al., 1998.) The strongest phosphorylation increases were seen with OA and calyculin A, which inhibit PP1 and PP2A; a lesser response was seen with the PP2A-specific inhibitor microcystin, and no response was seen with the PP2B inhibitor cyclosporine, which together suggests that the transporter is dephosphorylated by both PP1 and PP2A. In addition, SERT from rat frontal cortex and cultured cells coimmunoprecipitated with enzymatically active PP2Ac, the catalytic subunit of PP2A (Bauman et al., 2000). This association was disrupted by OA and PMA, suggesting that bound PP2Ac maintains SERT in a dephosphorylated state and that increased SERT phosphorylation follows from disruption of the complex. Coapplication of 5-HT and PMA to cells prevented the PMA-induced disruption of the SERT-PP2Ac complex, which would be predicted to maintain SERT dephosphorylation, a mechanism consistent with the transport-dependent suppression of PMA-induced phosphorylation described above.
Stimulation of DAT phosphorylation in rat synaptosomes by phosphatase inhibitors showed a profile similar to SERT, being strongly increased by OA and calyculin, modestly increased by microcystin, and not changed by cyclosporine (Vaughan et al., 1997), consistent with dephosphorylation by both PP1 and PP2A. The effects of PP1 and PP2A were examined independently in striatal broken cell homogenates that accommodated the introduction of inhibitors highly specific for each phosphatase (Foster et al., 2003b). In this system DAT phosphorylation was strongly stimulated by PP1 inhibitor 2 and modestly stimulated by PP2A inhibitor 1. PP1 was also able to completely dephosphorylate purified 32PO4-labeled DAT in vitro at physiological substrate and enzyme concentrations, showing the potential for this enzyme to act directly on the protein in vivo. Although PP2Ac coimmunoprecipitates with DAT (Bauman et al., 2000), purified PP2A, PP2B, and protein tyrosine phosphatase did not dephosphorylate 32PO4-labeled DAT in vitro. Together these results provide strong evidence that PP1 is a major phosphatase for DAT but are also consistent with additional actions by PP2A.
Although NET dephosphorylation has not been extensively investigated, it also coimmunoprecipitates with PP2Ac (Bauman et al., 2000) and its phosphorylation is strongly stimulated by OA (Jayanthi et al., 2004), consistent with effects of PP1 and/or PP2A. Thus with respect to phosphatase action, the cocaine-sensitive neurotransmitter transporters share considerable similarities. Because PP1 and PP2A protein inhibitors can be regulated in vivo by second messengers (Greengard 2001), this presents the possibility for indirect modulation of DAT, NET, and SERT phosphorylation levels through effects on dephosphorylation.
Identification of Phosphorylation Sites
Phosphoamino acid analysis of 32PO4-labeled DAT prepared from both rat brain and cultured cells has demonstrated that basal, PMA-stimulated, and OA-stimulated phosphorylation of DAT occurs primarily on serine (Foster et al., 2002). There is a minor amount (<1%) of phosphothreonine present, and to date, there is no evidence from metabolic labeling or Western blotting for phosphotyrosine. Two lines of evidence have demonstrated that most or all phosphorylation of DAT occurs on a cluster of serines near the distal end of the N-terminal tail. Peptide mapping of 32PO4-labeled protein prepared from rat striatum identified domains from the NH2 tail that contain basal, OA-stimulated, and PKC-stimulated phosphorylation, whereas no evidence was found for 32PO4-labeling of sequences containing the C-terminal tail or interhelical loops (Foster et al., 2002). These and additional data indicate that most of the basal and OA/PKC-stimulated phosphorylation of rDAT occurs at one or more of the six serines present at the distal end of the cytoplasmic N terminus (Figs. 1 and 2). Mutants in which each of these serines are individually changed to alanine undergo easily detectable basal and stimulated 32PO4-labeling (Foster et al., 2003a), demonstrating that phosphorylation occurs at more than one residue. Only serines 7 and 21 in this cluster are found within consensus PKC sites (Fig. 2), compatible with noncanonical phosphorylation occurring at other sites by other kinase(s). Phosphorylation at the end of the long NH2 tail suggests a location ideal for flexible interactions with other proteins and/or with DAT internal loops or C-terminal tail, and the finding that phosphorylation occurs at multiple sites indicates the potential for hierarchical or graded phosphorylation that may result in fine control of associated functions.

Comparison of rat (r) and human (h) DAT, SERT, and NET N-terminal tail sequences up to approximate point of entry into TM1. Potential phosphorylation sites for serine (S) threonine (T) and tyrosine (Y) residues are shown in red, blue, and green, respectively. Numbers at top and bottom indicate residue positions for DAT and SERT, asterisks indicate consensus PKC sites, and the enclosed sequence near TM1 indicates the region of highest homology in these domains.
Consistent with the mapping results from the native protein, mutagenesis studies have shown that truncation of hDAT at valine 22, which deletes the first five serines in the human isoform (Figs. 1 and 2) results in the loss of most or all of the basal and PMA-stimulated 32PO4-labeling (Granas et al., 2003). Mutants with this truncation or with S→A conversions at all five serines (Khoshbouei et al., 2004) display wild-type levels of PMA-induced down-regulation and endocytosis, suggesting that phosphorylation is not required for these events. Mutation of multiple S/T residues in other regions of the protein also had no effects on down-regulation or internalization (Granas et al., 2003), showing that phosphate acceptor sites in other parts of the protein were not contributing to these effects. Although these studies do not support a link between DAT phosphorylation and sequestration, conditions in which these processes are dissociated in vivo have not yet been identified using wild-type transporters. Other mutagenesis studies of DAT did not identify S/T residues clearly involved in PKC-dependent phosphorylation or functional effects (Lin et al., 2003) or utilized lengthy PMA treatments that preclude comparison with results of acute studies (Chang et al., 2001). The DAT serine and threonine residues that have been examined by mutagenesis are summarized in Fig. 1.
Less is currently known about phosphorylation sites on NET and SERT. Basal and PKC-stimulated phosphorylation of SERT in brain tissue is detected with phosphoserine antibodies (Najib et al., 2000), and N- and C-terminal tail fusion proteins of SERT undergo in vitro phosphorylation by PKC on multiple serines and threonines and by PKA on serine 13 and serine 599 (Blakely et al., 1998). The relative proportions of phosphoserine and phosphothreonine in the wild-type protein are not known, and to date there is no evidence for phosphotyrosine. The in vitro studies suggest, as with DAT, that SERT phosphorylation occurs at multiple N-terminal serines or threonines, but that unlike DAT, SERT may also undergo C-terminal phosphorylation. Other potential phosphate acceptor sites that have been mutated in expressed SERT include Ser8 and Ser13 in the NH2 tail, Ser277 in IL2, and Thr603 and Thr613 in the COOH tail (Sakai et al., 1997). NET residue Ser259 in IL2, which is homologous to SERT Ser277, has also been examined (Bonisch et al., 1998). These sites are shown in Fig. 1. The PMA down-regulation response was not changed in these mutants, but the proteins were not analyzed for phosphorylation. Because the phosphate acceptor sites in NET and SERT are unknown, mutagenesis studies on phosphorylation-dependent function have not yet been possible.
Comparison of potential phosphate acceptor sites and their adjacent sequences in the NH2 tail domains in DAT, NET, and SERT reveals some regions with little obvious similarity but also homologous regions surrounding the conserved threonines proximal to TM1 and the serine/threonine clusters that include PKC consensus motifs at Ser7 and Ser8 near the distal ends of DAT and SERT (Fig. 2). This presents the potential for phosphorylation of these transporters to occur on both similar and distinct sites, which may relate to the similarities and differences between the proteins in phosphorylation characteristics and regulated processes. Although SERT and DAT have the most potential for phosphorylation site similarities, there are no serines in the NH2 tail of NET. Thus PKC/OA-dependent NET phosphorylation must be occurring on NH2 tail threonines and/or in a different domain of the protein, a situation that is significantly different from DAT, its closest homolog.
Another issue is the relationship between basal and stimulated phosphorylation sites. Basal and PKC/OA-stimulated phosphorylation of DAT occur on the N-terminal serine cluster, but the specific sites within that cluster have not been identified, and it is currently not known if the sites phosphorylated in each condition are the same or different. For SERT, the additivity of phosphorylation induced by OA, PKC, and PKA suggests that basal and stimulated phosphorylation occurs on distinct residues. Basal and stimulated phosphorylation on the same sites suggests control of processes by increasing the number of phosphorylated transporters that undergo a constitutive activity such as trafficking (Loder and Melikian 2003) in which the equilibrium may be determined by phosphorylation, whereas phosphorylation of distinct basal and stimulated sites is compatible with induction of a nonconstitutive function.
Summary
The finding that the psychostimulant-sensitive monoamine transporters undergo phosphorylation that may be related to complex regulation of their activities leads to a previously unappreciated level of understanding of their normal physiological functions and is crucial to interpreting the actions of the ever-increasing body of therapeutic and diagnostic agents that act on these proteins. We are at an early stage of understanding these processes and many phosphorylation characteristics remain to be elucidated, including identification of the receptors and enzymes that control these processes in the brain, determination of phosphorylation sites and stoichiometry, identification of the subcellular compartments in which phosphorylation and dephosphorylation occur, and correlation of the subcellular distribution and functional activity of transporters with their phosphorylation state. Clarification of these properties will enhance our understanding of the role of transporter phosphorylation and indicate the potential for related processes to be susceptible to dysregulation in psychiatric disorders and drug abuse.
Acknowledgments
Thanks are given to Dr. James Foster, Mark Cervinski, Steven Adkins, Benchaporn Pananusorn, and Heather Holden, who contributed to these studies.
Footnotes
-
The Vaughan laboratory is supported by grants RO1 DA13147 and RO1 DA15175 from the National Institute on Drug Abuse and from the National Science Foundation (ND EPSCoR).
-
DOI: 10.1124/jpet.103.052423.
-
ABBREVIATIONS: DA, dopamine; DAT, dopamine transporter; NE, norepinephrine; NET, norepinephrine transporter; SERT, serotonin transporter; 5-HT, serotonin; TM, transmembrane domain; IL, intracellular loop; PMA, phorbol 12-myristate 13 acetate; OA, okadaic acid; AMPH amphetamine; METH, methamphetamine; PKC, protein kinase C; PKA, protein kinase A; PKG, protein kinase G, CaMK, Ca2+-calmodulin-dependent kinase; PP, protein phosphatase; PP2Ac, catalytic subunit of PP2A; PP2B protein phosphatase 2B; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein kinase; HEK, human embryonic kidney.
- Received February 19, 2004.
- Accepted March 23, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}