


Abstract
In situ clinical measurement of receptor occupancy (RO) is challenging, particularly for solid tumors, necessitating the use of mathematical models that predict tumor receptor occupancy to guide dose decisions. A potency metric, average free tissue target to initial target ratio (AFTIR), was previously described based on a mechanistic compartmental model and is informative for near-saturating dose regimens. However, the metric fails at clinically relevant subsaturating antibody doses, as compartmental models cannot capture the spatial heterogeneity of distribution faced by some antibodies in solid tumors. Here we employ a partial differential equation (PDE) Krogh cylinder model to simulate spatiotemporal receptor occupancy and derive an analytical solution, a mechanistically weighted global AFTIR, that can better predict receptor occupancy regardless of dosing regimen. In addition to the four key parameters previously identified, a fifth key parameter, the absolute receptor density (targets/cell), is incorporated into the mechanistic AFTIR metric. Receptor density can influence equilibrium intratumoral drug concentration relative to whether the dose is saturating or not, thereby influencing the tumor penetration depth of the antibody. We derive mechanistic RO predictions based on distinct patterns of antibody tumor penetration, presented as a global AFTIR metric guided by a Thiele Modulus and a local saturation potential (drug equivalent of binding potential for positron emissions tomography imaging) and validate the results using rigorous global and local sensitivity analysis. This generalized AFTIR serves as a more accurate analytical metric to aid clinical dose decisions and rational design of antibody-based therapeutics without the need for extensive PDE simulations.
SIGNIFICANCE STATEMENT Determining antibody-receptor occupancy (RO) is critical for dosing decisions in pharmaceutical development, but direct clinical measurement of RO is often challenging and invasive, particularly for solid tumors. Significant efforts have been made to develop mathematical models and simplified analytical metrics of RO, but these often require complex computer simulations. Here we present a mathematically rigorous but simplified analytical model to accurately predict RO across a range of affinities, doses, drug, and tumor properties.
Introduction
Target engagement information is critical for guiding dose selection of monoclonal antibodies, which can often be tolerated at high doses without exhibiting dose-limiting toxicities in first-in-human trials. This makes dose optimization for antibodies challenging since doses that are too low may lack efficacy while doses that are too high increase the risk for antidrug antibody responses and treatment cost. Mathematical modeling has played a significant role in predicting first-in-human doses for small molecules (Poulin and Theil, 2002b; Poulin et al., 2015) and can also assist in the dosing of antibodies (Baxter et al., 1995; Cao and Jusko, 2014; Bartelink et al., 2019).
One of the major challenges for antibody dose prediction is the complex target-mediated pharmacokinetics (PK), which can vary the receptor occupancy both spatially within tissues and over time. This is in contrast with small molecule pharmacokinetics that exhibit spatial gradients less frequently due to rapid diffusion. Small molecule organ uptake is primarily driven by drug lipophilicity (Poulin and Theil, 2002a,b), and tissue concentrations are more closely related to the plasma concentration, making predictions more robust. For antibodies targeted against solid tumors, receptor occupancy (RO) predictions are a cornerstone of assessing target engagement, and by extension pharmacodynamic response. Simplified receptor occupancy metrics that capture the predictions of complex mathematical models simulating in vivo antibody PK have been explored as a strategy to more accurately predict antibody-target engagement (Spilker et al., 2016; Glassman and Balthasar, 2017; Orcutt et al., 2017; Park et al., 2017; Stein and Ramakrishna, 2017; de Vries Schultink et al., 2018; Ahmed et al., 2019). Previously, Stein et al. have described a potency metric Average Free target concentration to Initial target concentration Ratio (AFIR), derived from a target-mediated drug-disposition compartmental model to predict antibody engagement with soluble/shed antigen in circulation (Stein and Ramakrishna, 2017). This metric was then extended using a physiologically based model that incorporates cell surface receptors in the tumor tissue in addition to the soluble/shed receptor species and renamed AFTIR (Average Free Tissue target to Initial target Ratio) (Ahmed et al., 2019). However, based on the model assumptions, AFTIR can only accurately capture receptor occupancy at near-saturating antibody doses and is less accurate at subsaturating dosing regimens, which are often clinically relevant, particularly for highly expressed targets.
Here we expand receptor occupancy calculations to be applicable at both saturating and subsaturating doses by deriving a mechanistic and analytical global AFTIR (gAFTIR) metric from the partial differential equations of a mechanistic Krogh cylinder model. (These approximate analytical solutions for different regimes are analogous to other engineering/mathematical simplifications, such as fluid flow approximations for turbulent versus laminar flow regimes.) All parameters employed in the AFTIR metric are readily measurable antibody kinetic parameters except for the free intratumoral antibody concentration, which is a function of the tumor microenvironment (e.g., tumor vascularity, vessel permeability) and drug parameters (e.g., dose, antibody affinity). The free antibody concentration is challenging both to measure experimentally and solve explicitly from analytical equations. Part of this challenge results from the nonlinearities present in antibody PK, primarily the local receptor binding equilibrium (high versus low affinity antibodies) and the tissue receptor saturation (high versus low antibody doses), which can result in drastically different quantitative and qualitative distribution of free antibody.
To overcome these limitations and achieve a robust prediction of RO while avoiding the need for more complex numerical simulations, we used dimensional analysis to define four qualitatively different “regimes” of antibody distribution, each with an accurate description of RO for those conditions. The original AFTIR metric describes RO for high doses approaching tissue saturation but does not accurately capture subsaturating doses. In contrast, a previous pseudo-steady state analysis of the partial differential equations of a Krogh cylinder model by Thurber and Wittrup (2012) provides a mechanistic estimate of free antibody concentration under subsaturating conditions but is not valid for saturating doses. In this work, we combine these approaches to develop a mechanistic framework for antibody pharmacokinetics that is valid under all dosing levels and antibody affinities. The framework is driven by two dimensionless numbers—a simplified version of the generalized Thiele modulus (Thurber et al., 2008a) (henceforth referenced here as just Thiele modulus) and a newly defined local saturation potential (Sp). These parameters determine the dosing regimen (saturating versus subsaturating and high affinity versus low affinity) to identify the valid mathematical expression for free antibody concentration, thereby providing a more universal receptor occupancy metric.
Materials and Methods
Computational Krogh Cylinder Model
The simulations for antibody uptake and distribution in vascularized tumors are based on a previously validated Krogh cylinder model of antibody distribution (Thurber and Wittrup, 2012). Briefly, a one-dimensional Krogh cylinder was used to capture radial concentration gradient given the permeability-limited uptake of antibodies i.e., no axial gradient along the length of the capillary (Fig. 1A). Plasma concentration of the antibody is captured as a biexponential equation. Antibody transport in the tumor interstitium is diffusion-driven given the elevated interstitial pressure limiting convective transport (Baxter and Jain, 1989; Baish et al., 2011). Bolus administration of the antibody results in extravasation of the antibody in the tumor, followed by intratumoral diffusion and binding to free target receptor. The antibody-target complex undergoes receptor-mediated endocytosis, as does the free target, which can also be recycled to the surface. The simulations track the concentration of free target as well as free, bound, and internalized antibody over both spatial location and time. Partial differential equations describe the extracellular diffusion of the constructs along the radius of the tumor, binding to target receptors, and receptor-mediated endocytosis (Supplemental Computational Model and Equations). Simulations were performed for three monoclonal antibodies used to treat solid tumors—ramucirumab (vascular endothelial growth factor receptor), cetuximab (endothelial growth factor receptor), and trastuzumab [human epithelial growth factor receptor 2 (HER2)], selected for the distinction in typical target expression. All parameters for base simulations used in the model (Table 1) are based on preclinical species (mice) and were gathered from the literature or measured independently (i.e., not fit to tissue distribution data). Simulations were performed using MATLAB (MathWorks, Natick, MA).

Pharmacokinetic models used to predict antibody-receptor occupancy. (A) Mechanistic Krogh cylinder model that captures systemic, intratumoral, and cellular pharmacokinetics to provide a realistic estimate of spatiotemporal average receptor occupancy regardless of antibody uptake and distribution. (B) Equilibrium compartment model that assumes plasma and tumor compartment are in equilibrium, which is valid at (super) saturating doses. (C) Mechanistic compartmental model that captures temporal antibody binding kinetics in the tumor compartment to estimate the antibody concentration in the tumor under subsaturating conditions.
Krogh cylinder model base parameters
Sensitivity Analysis
Parameter sensitivity analysis was performed to evaluate the accuracy and robustness of the gAFTIR metric described in this work. Global sensitivity analysis was performed by establishing a range of values for several model parameters (Table 2) and performing uniformly distributed random selection for each parameter for each run of the simulation, for a total of 2800+ Krogh cylinder simulations. The combination of parameters for each simulation was recorded and used to perform theoretical AFTIR calculations and quantitatively compare the theoretical calculations to the Krogh-simulated AFTIR. Local sensitivity analysis was performed by establishing base parameters for each of the three antibodies detailed in Table 1 and varying individual parameters from 0.001x to 1000x the base value, unless noted otherwise. Sensitivity analysis was performed using MATLAB (MathWorks) and visualized using Prism v9.4.1 (GraphPad Software Inc., San Diego, CA).
Global sensitivity analysis parameter
AFTIR Derivation
AFTIR is the ratio of free target available in the presence of drug to the initial free target available in the absence of drug (originally derived by Ahmed et al., 2019).
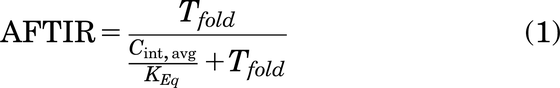 where Tfold is the fold change in target levels upon binding to the drug at saturating doses, due to target accumulation, Cint,avg is the average free interstitial antibody concentration in the tumor, and Keq is the equilibrium affinity simplified as
where Tfold is the fold change in target levels upon binding to the drug at saturating doses, due to target accumulation, Cint,avg is the average free interstitial antibody concentration in the tumor, and Keq is the equilibrium affinity simplified as
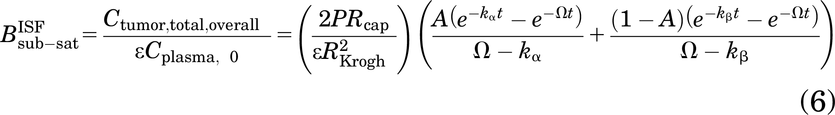
In many cases, target internalization is the same whether bound to antibody or not, leading to a Tfold = 1. However, in the event of up- or downregulation of the surface receptor in the presence of the antibody, these rates are likely different yielding Tfold ≠ 1 in the expression.
The AFTIR expression derived from the Krogh cylinder PDE model (Supplemental Computational Model and Equations) is nearly identical to that described by Ahmed et al. (2019), except here we expand the definition of Cint,avg, the interstitial antibody concentration at steady state. Cint,avg is dependent on many factors related to the tumor physiology, target properties, dose, and drug properties (Thurber et al., 2008a). While Cint,avg cannot be solved analytically or readily measured experimentally, approximate solutions can be obtained for specific conditions (i.e., certain “regimes” as discussed in detail in the Results). First, we show these approximate solutions, and then we describe the dimensionless numbers used to determine the relevant regimen based on the dose (saturating versus subsaturating) and affinity (high versus low relative affinity).
Approximate Cint,avg Solutions
Saturating Doses
Theoretical predictions for antibody receptor occupancy typically rely on the plasma antibody concentration. However, this approximation only holds true for (super) saturating doses, where the tumor uptake exceeds target binding and degradation of the drug (i.e., local tissue “clearance”), and the free interstitial concentration approaches that in the plasma (Fig. 1B). In such regimes, the linear plasma profile can be calculated using the biexponential equation
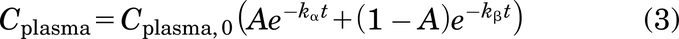 and the average plasma concentration over the first dosing window τ (Cplasma, average,first) can be calculated as
and the average plasma concentration over the first dosing window τ (Cplasma, average,first) can be calculated as
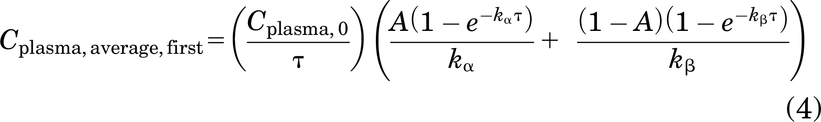 where Cplasma,0 is the plasma concentration after a bolus dose, τ is the dosing interval, A is the fraction of alpha phase redistribution, (1 – A) is the fraction of beta phase clearance, kα is the antibody redistribution rate constant, and β is the antibody clearance rate constant. These parameters can be estimated by fitting a biexponential curve to the mean/median antibody plasma PK data using commercially available software.
where Cplasma,0 is the plasma concentration after a bolus dose, τ is the dosing interval, A is the fraction of alpha phase redistribution, (1 – A) is the fraction of beta phase clearance, kα is the antibody redistribution rate constant, and β is the antibody clearance rate constant. These parameters can be estimated by fitting a biexponential curve to the mean/median antibody plasma PK data using commercially available software.
At steady state, the average plasma concentration (Cplasma, average,ss) can be calculated as (AUC0-∞)/τ (Bauer, 2008)
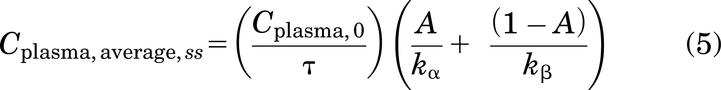
Note that the relevant Cplasma,ss values can also be estimated numerically from two-compartment model fits to antibody plasma PK data.
Subsaturating Doses
Under subsaturating antibody doses, which are often the case in clinical development of potent antibodies or highly expressed targets, assuming equilibrium between the tumor and plasma compartment concentrations is often not accurate. A previous pseudo-steady state analysis of the partial differential equations (PDE) of the Krogh cylinder model, approximated by a compartmental model in Thurber and Wittrup (2012), provides a mechanistic estimate of the total tumor antibody concentration (bound antibody plus free antibody) under subsaturating conditions. This estimate is not valid for saturating doses (simplified schematic shown in Fig. 1C), providing a complement to the assumption of saturation above. The underlying principle of the subsaturating dose analysis is that extravasation is the rate-limiting step in antibody uptake, which results in the total antibody uptake in the tumor being nearly independent of the distribution pattern. As such, it allows for the pseudo-approximation of a “well-mixed” tumor compartment for uptake, even though tumors are in fact not well-mixed compartments given the high interstitial pressure that prevents convective flow.
The approximation provides a mechanistic estimate of antibody concentration for subsaturating doses. In the context of AFTIR, this provides a mechanistic estimate of the “biodistribution coefficient” (i.e., fraction of drug from circulation found in the tumor interstitium at saturating doses) described by Ahmed et al. (2019), except at subsaturating doses here, and is calculated as
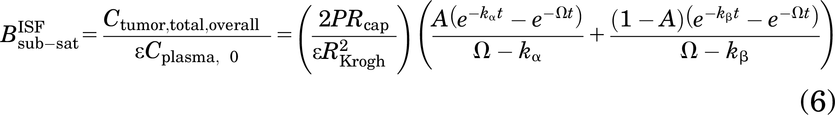 where Ctumor,total,overall is the overall total antibody concentration in the tumor (sum of free and bound antibody in the overall tumor volume), Cplasma,0 is the antibody plasma concentration after a bolus dose, ε is the void fraction in the tumor (accessible interstitial volume over total tumor volume), P is the vascular permeability, Rcap is the capillary radius, RKrogh is the Krogh cylinder radius (equivalent to half the intercapillary distance in the tumor), and 2Rcap/R2Krogh is the blood vessel surface area to volume ratio in the tumor. The cumulative local clearance rate, Ω, is a sum of two mechanisms of antibody loss from tumors—unbound antibody washing out of the tumor and bound antibody being internalized and degraded by cells
where Ctumor,total,overall is the overall total antibody concentration in the tumor (sum of free and bound antibody in the overall tumor volume), Cplasma,0 is the antibody plasma concentration after a bolus dose, ε is the void fraction in the tumor (accessible interstitial volume over total tumor volume), P is the vascular permeability, Rcap is the capillary radius, RKrogh is the Krogh cylinder radius (equivalent to half the intercapillary distance in the tumor), and 2Rcap/R2Krogh is the blood vessel surface area to volume ratio in the tumor. The cumulative local clearance rate, Ω, is a sum of two mechanisms of antibody loss from tumors—unbound antibody washing out of the tumor and bound antibody being internalized and degraded by cells
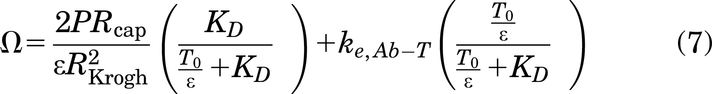 where T0 is the initial receptor concentration, ke,Ab-T is the antibody-receptor complex internalization rate, and KD is the binding affinity.
where T0 is the initial receptor concentration, ke,Ab-T is the antibody-receptor complex internalization rate, and KD is the binding affinity.

Note that progressively higher affinity (i.e., smaller KD) antibodies are often primarily eliminated by internalization (ke,Ab-T) and degradation (owing to tight binding and relatively slower dissociation rate, koff) while progressively lower affinity (i.e., larger KD) antibodies often wash out of the tumor intact (owing to slower binding/faster koff relative to ke,Ab-T) (Shih et al., 1994; Schmidt and Wittrup, 2009; Zhang et al., 2016).
To provide a single characteristic value for receptor occupancy, the average overall total antibody concentration in the tumor at steady state Ctumor total,average,overall,ss is calculated by taking the time-averaged total antibody (i.e., the integral of eq. 6 from 0 to τ divided by τ):

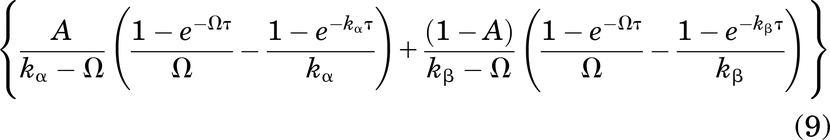 which yields an average interstitial total antibody concentration
which yields an average interstitial total antibody concentration
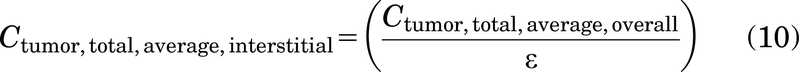
Note that eq. 9 uses Cplasma,max,ss instead of Cplasma,0 to approximate the total antibody concentration in the tumor at steady state rather than after the initial dose.
For subsaturating doses of a high affinity (i.e., small KD) antibody, the antibody distributes heterogeneously, exhibiting a “saturation front” moving through the tissue (i.e., binding site barrier; Saga et al., 1995). Outside of this saturation front, there is a negligible amount of antibody. Within this saturation front, though, the antibody is in excess of the target (hence saturation), so the free antibody tumor concentration term in the AFTIR metric can be approximated using the total antibody concentration in this region.
Conversely, for subsaturating doses of a low affinity (i.e., large KD) antibody, the antibody distributes evenly in the tissue with the antigen in excess. Therefore, the total tumor concentration of the antibody can be used to derive the average overall free antibody concentration in the tumor (Ctumor,free) using the KD, given by
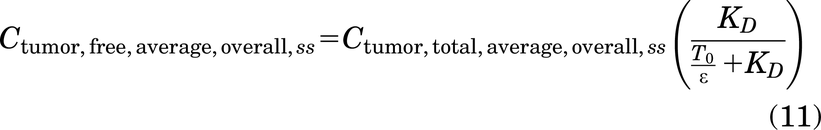 which yields an average interstitial free antibody concentration
which yields an average interstitial free antibody concentration

Eqs. (5), (10), and (12) mark the three main approximations for tumor interstitial antibody concentration that are used to define the gAFTIR. Note, the binding affinity used to calculate the tumor antibody concentration is based on in vitro measurements (antibody titration and bulk incubation with cells in suspension), and therefore gAFTIR is derived on a total antibody-antigen binding basis rather than individual binding arms/sites (i.e., two target sites per antibody).
Nondimensional Groups for Defining gAFTIR
The distinction between a saturating versus subsaturating dose is critical to determine the relevant antibody concentration to use in the AFTIR metric. Likewise, the determination of heterogeneous high affinity (i.e., small KD) antibody distribution versus homogeneous low affinity (i.e., large KD) antibody distribution impacts the qualitative and quantitative measure of RO. It is not the absolute affinity or dose that determines the behavior but rather the affinity and dose relative to other processes within the tissue. Therefore, it is the ratio of affinity and dose to these other rates that ultimately determines the behavior, resulting in two dimensionless numbers (i.e., two ratios)—the Thiele modulus (ϕ2) and the local saturation potential (SP).
Thiele Modulus
Tumor saturation is determined by a Thiele modulus, which described the fundamental ratio between local consumption of the antibody versus tumor uptake (Thurber et al., 2007). The formula for the Thiele modulus (Table 3) utilizes the steady state interstitial antibody concentration at the capillary wall, Ctumor,surf,free,int(Thurber et al., 2008b). Ctumor,surf,free,int is the tumor surface free antibody concentration determined by the incoming antibody flux across the capillary wall versus the antibody diffusion flux away from the capillary wall and can be calculated as
 where Bi is the mass transfer Biot number, a ratio of antibody extravasation across the capillary wall to the antibody diffusion rate, and is calculated as
where Bi is the mass transfer Biot number, a ratio of antibody extravasation across the capillary wall to the antibody diffusion rate, and is calculated as

Summary of nondimensional criteria
Large values for Bi result in concentration equilibration with the plasma, while small numbers (typically around 0.02 for antibodies) highlight a permeability-limited uptake.
Of note, the Thiele modulus is used in two distinct ways in this work. First, the calculation of the Thiele modulus for a given tumor radius RKrogh is used to determine if a saturating dose of antibody is administered, which determines the choice of Cint,SS for gAFTIR. Second, for a subsaturating dose of a high affinity (i.e., small KD) antibody, the Thiele modulus is used to determine the penetration distance of antibody within the tumor. Since ϕ2 = 1 corresponds to tumor saturation (i.e., the local antibody concentration is in excess over the target concentration) for high affinity antibodies, setting the Thiele modulus to a value of 1 allows for the calculation of the radius of saturation (Rsaturation) for a given antibody dose, target receptor concentration, binding affinity, and internalization rate (Supplemental Fig. 1).
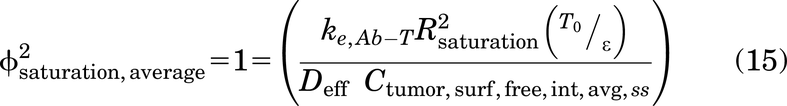 which yields
which yields
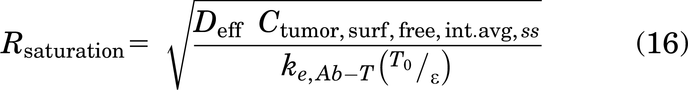
The radial saturation area correction term can be simplified to
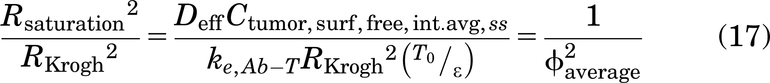
This is a necessary factor to account for penetration in the gAFTIR calculation for high affinity antibodies, as none of the Cint,SS approximations described earlier account for spatial variation in antibody concentration. For low affinity (i.e., large KD) antibodies, this is often not a concern as the affinity is weak enough to allow for homogeneous distribution without saturation.
Local Saturation Potential
Thurber et al. have previously described the effects of antibody affinity (KD) on tissue penetration (Thurber et al., 2008a), highlighting how high affinity (i.e., small KD) antibodies that exhibit the binding site barrier effect (Fujimori et al., 1989, 1990; Graff and Wittrup, 2003) typically exhibit KD ≪ Ctumor,surf,free,int, while low affinity (i.e., large KD) antibodies that can penetrate more homogeneously through the tumor exhibit KD ≫ Ctumor,surf,free,int. Ctumor,surf,free,int represents the free antibody concentration in the tumor at the capillary wall (driven by the vascular permeability, P, and intratumoral diffusion coefficient, Deff) and represents the free antibody concentration once binding equilibrium is achieved at the first cell layer. Therefore, the ratio of Ctumor,surf,free,int to the binding affinity KD, defined as local saturation potential (SP) here represents the potential for Ctumor,surf,free,int to saturate receptors on the first cellular layer next to the capillary (i.e., the source of antibody). This cellular saturation potential is distinct from the Thiele modulus, which captures the ability of the tumor antibody concentration to saturate receptors in the whole tumor. SP is reminiscent of the binding potential (Mintun et al., 1984), a combined measure of receptor density and ligand binding affinity used to quantify the binding kinetics for radioligands. However, the binding potential uses target concentration (due to target excess over trace amounts of radioligand) instead of the permeability-driven antibody concentration in the tumor interstitium, Ctumor,surf,free,int, in this work. Based on this analysis, it can be approximated that the ratio of Ctumor,surf,free,int to binding affinity (KD, which accounts for koff and kon) can aid in categorizing whether an antibody acts as a high affinity or low affinity antibody in the tumor and therefore determine the anticipated antibody accumulation and distribution pattern. If SP is greater than 1, the tumor surface free antibody concentration at the first cell layer (post-binding equilibrium) is larger than the KD. Thus, the antibody exhibits a binding site barrier effect; i.e., receptors on each cell layer will be saturated before the antibody diffuses to the next cell layer, resulting in heterogeneous/perivascular distribution. When this ratio is less than 1, the tumor surface free antibody concentration at the first cell layer (post-binding equilibrium) is smaller than the KD. The antibody acts as a low affinity (i.e., large KD) antibody and can diffuse through the tumor without needing to saturate all receptors at each cell layer. Together, the saturation potential and Thiele modulus determine the antibody binding regimen needed to estimate the free antibody concentration and receptor occupancy.
Results
Global Krogh Simulations Highlight Critical Parameters for RO Predictions
To aid in the development of a global AFTIR metric to predict receptor occupancy, we performed global sensitivity simulations using the Krogh cylinder model to highlight antibody and tissue parameters that are critical to RO predictions but may not be captured by the original AFTIR metric. We ran 2800+ simulations for randomly selected values within defined parameter sets across an eight-parameter space, namely antibody dose, dosing frequency, systemic beta phase clearance half-life, absolute receptor density, tumor vessel density, antibody association rate, antibody dissociation rate, and receptor internalization rate, all of which are known to influence antibody penetration (Thurber et al., 2008a). Global sensitivity analysis (Supplemental Fig. 2) shows that absolute receptor density, antibody dose, internalization rate, and systemic clearance half-life have a strong influence on receptor occupancy predictions regardless of combination of other parameters. Target receptor density (T0) is particularly noteworthy, as it is clearly a critical factor influencing receptor occupancy but is not present in the original AFTIR metric. However, the absolute receptor density influences the distribution pattern and steady state interstitial antibody concentration in the tumor, and accounting for it can greatly improve the accuracy of AFTIR.
Antibody Distribution Regimes Categorized by Nondimensional Groups
Antibody dose, plasma clearance half-life, absolute receptor density, and internalization rate, the four main parameters that were shown earlier to strongly impact receptor occupancy, collectively influence the accumulation and distribution of antibody in the tumor. Generally, antibody distribution patterns can be classified into regimes based on two factors: (1) binding affinity and (2) dose (Fig. 2).

Antibody distribution patterns in tumor tissue. Inset spheroid images reproduced from Thurber and Wittrup (2008). Antibody distribution can broadly be described by simplified regimes based on the affinity to the antibody and dose administered. Fluorescent images and spheroid diagrams show the inward tissue penetration seen with tumor spheroids (top of each panel), while the diagrams highlight the outward penetration from tumor blood vessels (bottom of each panel). Regimen 1 and 2: Under saturating doses, antibodies are in excess relative to receptor binding sites. Differences between high and low affinity become less relevant as the interstitial concentration of antibody far exceeds the molar concentration of receptors in the tissue, and all cells in the tissue are targeted regardless of the antibody distribution pattern (e.g., 7nM sm3E and 10nM shMFE). Regimen 3: With a subsaturating dose, high affinity antibodies (sm3E KD = 0.03 nM) typically exhibit the “binding site barrier” effect where receptors on each cell layer must be saturated before the antibody can diffuse to the next cell layer (e.g., 3 nM of sm3E). As the dose decreases below saturation (Regimen 1 → Regimen 3), the saturation front penetrates to a decreasing fraction of the tissue. Regimen 4: Lower affinity antibodies (shMFE KD = 8 nM) or rapidly diffusing fragments can penetrate more uniformly without needing to saturate any preceding cell layer, even under subsaturating doses (e.g., 3nM of shMFE). The saturating regimes (Panels 1 and 2) are accurately described by the original AFTIR metric. The current work extends this to describe RO on the regimes in Panels 3 and 4.
Typically, high affinity (i.e., small KD) and fast internalizing antibodies exhibit the “binding-site barrier” phenomenon, where the immobilization of the antibody on the receptor (and, subsequently, rapid internalization) is faster than diffusion, resulting in a perivascular penetration pattern where all receptors in each cell layer must be saturated before the antibody can diffuse to the next cell layer. This pattern of antibody distribution is exemplified by a subsaturating dose of sm3E, a high affinity (KD = 30pM) single chain variable fragment antibody against carcinoembryonic antigen (Graff et al., 2004; Thurber and Wittrup, 2008) (Fig. 2, Panel 3, 3nM sm3E). In contrast, antibodies with weaker binding affinity (i.e., large KD), faster diffusion, or slower cellular internalization rates are not immediately immobilized and can penetrate deeper in the tumor tissue without needing to saturate perivascular cell layers first. This antibody penetration pattern is exemplified by a subsaturating dose of shMFE, or an single chain variable fragment antibody against carcinoembryonic antigen (Begent et al., 1996; Thurber and Wittrup, 2008), but with lower binding affinity (KD = 8nM) (Fig. 2, Panel 4, 3nM shMFE). In vivo, both patterns of penetration can result in similar average antibody uptake [i.e., percent injected dose per gram (%ID/g)], because the total tumor uptake is limited by vascular permeability, not tissue distribution (Bhatnagar et al., 2014). However, a “well-mixed” compartmental PK model fails to capture spatial variations that can influence receptor occupancy predictions. For very large doses, the differences in distribution pattern become vanishingly small when the dose of antibody administered is saturating (i.e., the free antibody concentration is in excess over the target concentration), as seen with the distribution of higher doses of sm3E (7nM, Fig. 2, Panel 1) and shMFE (10nM, Fig. 2, Panel 2). These large doses have different intratumoral pharmacokinetics compared with subsaturating conditions. At saturating doses, antibody distribution in solid tumors can sufficiently be approximated by the original AFTIR metric, but the dose at which this inflection point of “saturation” occurs can vary (Supplemental Fig. 3), depending on both the antibody and the tumor tissue of interest.
Though qualitatively descriptive, this categorization alone does not provide quantitative benchmarks for distinguishing high versus low doses and affinities. In fact, there are no absolute doses or affinities that distinguish these different regimes, but it is the relative doses and affinities compared with other kinetic processes that determine the behavior of the biologic. Dimensional analysis has been used to relate the various parameters that determine the antibody pharmacokinetics described earlier (Thurber and Weissleder, 2011; Ferl et al., 2016; Evans and Thurber, 2022). In this work, we employ two nondimensional groups to select the relevant Cint,avg based on the antibody distribution regimen. As the dose increases, more receptors become occupied as uptake into the tumor increases relative to local “consumption” (internalization and degradation), and this ratio is described by the Thiele modulus. As the affinity increases (i.e., KD becomes smaller), antibodies bind at greater levels to the first cell layer that they contact, resulting in greater saturation of cells near blood vessels (or at the periphery of spheroids) relative to more distant cells. This ratio is described by the local saturation potential. The Thiele modulus and local saturation potential categorize different regimes of antibody distribution using well-validated computational models backed by experimental measurements (Schmidt and Wittrup, 2009). This serves as the basis for defining the relevant intratumoral antibody concentration to use in the AFTIR metric to robustly predict receptor occupancy for both low and high affinity antibodies at both saturating and subsaturating doses.
Mechanistic Derivation of Global AFTIR
A conceptual summary of gAFTIR in the different regimes is shown in Fig. 3. As the affinity and dose change, the distribution and saturation of receptors changes as depicted in the spheroid diagrams. Higher affinity (lower Kd) results in greater heterogeneity (e.g., Fig. 2, Panel 3), while higher doses increase the saturation level (e.g., Fig. 2, Panels 1 and 2). The base formula for the mechanistic gAFTIR derived from the Krogh cylinder model PDEs (eq. 1) is similar to Ahmed et al. (2019). However, the choice of intratumoral antibody concentration is determined based on mechanistic interdependence of dose, equilibrium affinity, and absolute receptor density, as reflected by the different regimes in Fig. 3.

Simplified scheme of mechanistically weighted gAFTIR. Tumor receptor occupancy is a function of binding at the relevant interstitial antibody concentration. This concentration varies depending on the affinity and dose of the antibody, specified by the regimen as determined by the relative binding affinity (SP) and the tumor saturation (Thiele modulus). These parameters can be plotted to describe different regimes (four quadrants on the right illustrated with spheroids). For high affinity antibodies, increasing doses result in a saturation front penetrating deeper into the tissue (Regimen 3) until it reaches all tissue at a saturating dose (Regimen 1). At subsaturating doses, the saturation front becomes more diffuse as the affinity is reduced until it distributes evenly in the tissue at a subsaturating level (moving from Regimen 3 to 4). If these lower affinity antibodies are dosed at higher levels, the antibodies occupy more receptors evenly throughout the tissue until saturating all cells (moving from Regimen 4 to 2). In Regimen 3, since the radius of the saturation front (Rsaturation) is less than the radius of the tumor (RKrogh), the calculated AFTIR must be normalized to the radial average.
Regimes 1 and 2: Saturating Dose
When the dose is saturating, the form of the receptor occupancy calculations is less influenced by differences in antibody binding affinity. In Fig. 3, Regimes 1 and 2 represent saturating antibody doses relative to target receptor density, which accounts for receptor occupancy using a well-mixed equilibrium compartment model regardless of antibody equilibrium affinity. These regimes can be identified mechanistically by a Thiele modulus cut-off of 1 (Thurber et al., 2007) (i.e., ϕ2average < 1; Supplemental Fig. 10), regardless of the value of SP. Here saturation refers to a stoichiometric excess of total antibody concentration relative to target concentration (rather than 100% receptor occupancy). Therefore, the tumor interstitium can equilibrate with the plasma concentration, and the interstitial free antibody tumor concentration (Cint,avg) can be approximated by the average plasma concentration (Cplasma,average) described in eq. 5.
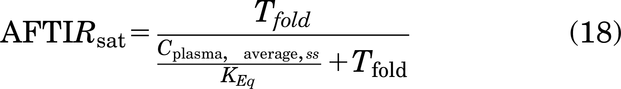
However, if the dose is reduced to the point that the total antibody concentration is no longer in excess, the pattern of distribution becomes dependent on the binding affinity. Likewise, local (tumor cell) internalization and degradation of the antibody result in free antibody concentrations below the plasma concentration, necessitating a more detailed compartmental model to describe the free antibody concentration.
Zone 3 and 4: Subsaturating Dose
Zones 3 and 4 represent subsaturating dose regimes where the assumption of equilibrium between tumor free interstitial antibody concentration and plasma concentration becomes invalid. Instead, tumor interstitial antibody concentration is estimated based on a mechanistic uptake model (Thurber and Wittrup, 2012). Subsaturating regimes exhibit ϕ2average > 1 (Supplemental Fig. 9). However, it is important to note the difference in patterns of subsaturation between high affinity (steep concentration gradient in Zone 3) and low affinity (relatively uniform concentration in Zone 4) antibodies.
For high affinity antibodies that exhibit the “binding site barrier” phenomenon (Zone 3), each cell layer must have all its receptors saturated before the antibody can penetrate to additional cell layers, i.e., SP > 1. Note that when SP > 1, a subsaturating dose does not imply that none of the cells in the tumor are saturated—rather it represents compartmentalized saturation where only a fraction of the tumor radius has cells saturated with the antibody, while the rest of the tumor has no receptors occupied (Supplemental Fig. 1). Consequently, the full tumor averaged AFTIR emerges when the local AFTIR calculation within the saturation front is scaled by the fractional area of the tumor that is saturated with the antibody. For this regimen (ϕ2average > 1, SP > 1) the available interstitial free antibody concentration can be approximated by the total interstitial antibody concentration (Ctumor,total,avg,int, eq. 10) because the antibody is assumed to be in excess relative to antigen within the saturation front. The whole tumor RO is then calculated by combining the free antigen within the saturation front and outside the saturation front.

Reducing the binding affinity manifests as an increasingly diffuse gradient. Eventually, the affinity becomes low enough that a “binding site barrier” no longer exists (i.e., Rsaturation ≥ RKrogh), and the local AFTIR does not require scaling to the saturation radius (Rsaturation). This is because as the affinity decreases, the distribution of the antibody becomes more homogeneous, effectively approaching a “well-mixed” compartment model approximation (i.e., SP < 1) where most of the cell layers in the tumor are partially targeted but not all receptors are saturated. Given the lack of a binding site barrier effect for low affinity antibodies as the dose is further decreased below Regimen 2, very little antibody is bound to the target receptor. In this subsaturating dose regimen (ϕ2average > 1, SP < 1), the free interstitial antibody concentration in the tumor (Ctumor,free,avg,int, eq. 12) best approximates the antibody concentration for calculating AFTIR.

Overall, the regimes highlight the mechanistic nuances in calculating receptor occupancy and can be summarized into one gAFTIR expression as follows, with the values for R and Cint,avg specified for each regimen, which in turn are identified by the quantitative combination of ϕ2average and SP (Fig. 3).
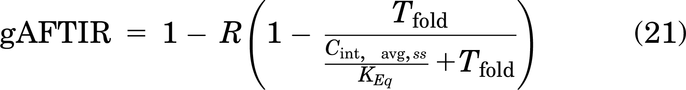
With the relevant antibody concentrations and quantitative criteria for the nondimensional groups defined for different regimes, we next focused on global and local sensitivity analysis for validation of the gAFTIR metric.
Validation of Mechanistic gAFTIR
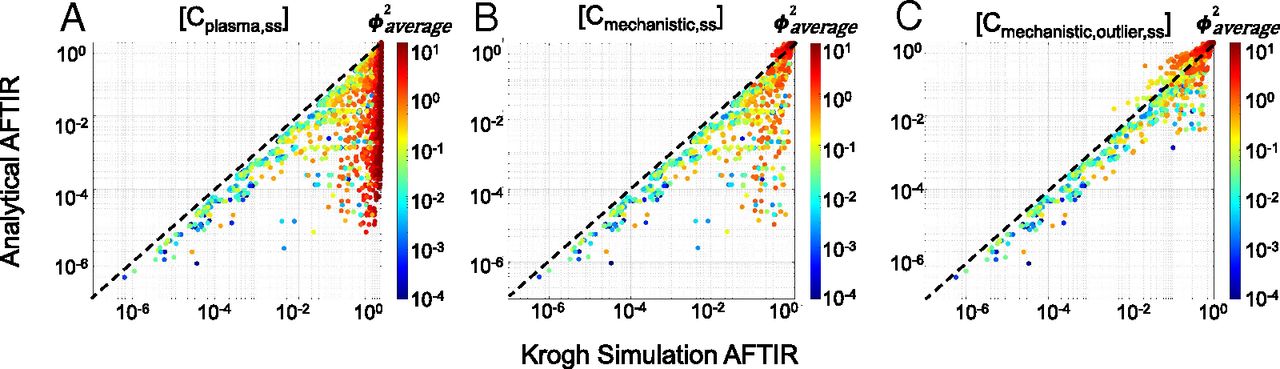
Having established the regimes, formulae, and quantitative criteria for the gAFTIR, we sought to validate the applicability of the algorithm. We mechanistically predicted the RO for the 2800+ global sensitivity simulations using the full partial differential equation simulations of antibody distribution and compared the results to the simplified (analytical) RO using gAFTIR (Cmechanistic) to quantify the improvement in accuracy. The receptor occupancy was averaged over both time and radius (distance from blood vessels) in the Krogh cylinder simulations for the simulation average free fraction of receptor. Qualitative comparison of the simulations shows a vast improvement in correlation between calculated (analytical approximation) and simulated RO (Fig. 4, Supplemental Fig. 4), particularly in the subsaturating cases (identified by ϕ2average > 1). However, a small cluster of outliers was observed in the lower right corner (Fig. 4B), indicating cases where gAFTIR is overpredicting receptor occupancy compared with the mechanistic Krogh cylinder simulations. Most scenarios in this outlier cluster exhibited SP > 1 and ϕ2average < 1, indicating a high affinity antibody in a saturating regimen. Further exploration highlighted these simulations to indeed be saturating for most of the dosing period. However, as the plasma concentration drops over time, the system switches for a saturating regimen to a subsaturating regimen toward the end of the dosing period (Supplemental Fig. 8), resulting in a mismatch between gAFTIR and simulated RO (Supplemental Fig. 5). To identify and correct for these outliers, any scenarios that presented ϕ2average < 1 but ϕ2trough > 1 (i.e., saturating at the average concentration but subsaturating at trough concentration, Supplemental Fig. 5) were flagged. The gAFTIR was averaged with the trough free fraction tissue target to initial target ratio (TFTIR) calculated using the tumor surface free antibody interstitial concentration (Ctumor,surf,free,int,trough,ss) and corrected to the trough saturation radius (Rsaturation,trough, which is calculated the same as eq. 17 but using Ctumor,surf,free,int,trough,ss instead).
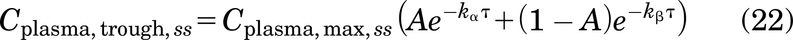
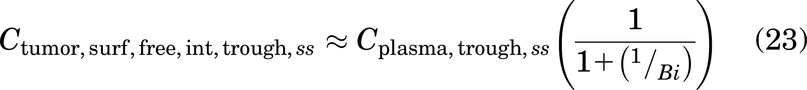
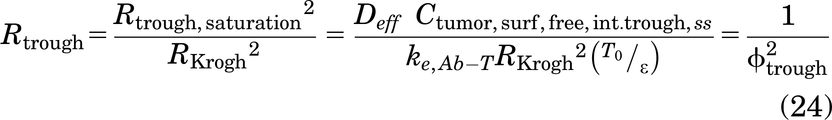


Global sensitivity analysis for validation of mechanistic gAFTIR. Comparison of spatiotemporal Krogh cylinder simulation AFTIR to (A) calculations using average plasma concentration, equivalent to AFTIR derived from a simple equilibrium compartment model, (B) calculations using mechanistic gAFTIR, and (C) calculations using gAFTIR including outlier corrections that correspond to a zone transition at the trough concentration, as identified using ϕ2trough.
Empirically, we observed that when ϕ2average> 0.5, a linear average of gAFTIR and TFTIRsurf was more applicable, while a logarithmic average was usually more appropriate when ϕ2average < 0.5. This correction resulted in further improvement in the accuracy of gAFTIR (Cmechanistic,outlier) (Fig. 4C). To provide a more quantitative metric of improvement, we calculated the residual error between simulated RO and calculated RO for all [Ctumor] calculation methods and found that using the gAFTIR with outlier corrections, approximately 70% and 83% of the calculated ROs were within ±0.05 and ±0.1 residual error margin respectively, compared with about 33% to 54% (±0.05) and about 41% to 66% (±0.10) for alternative approaches (Supplemental Fig. 4F), supporting that the mechanistically weighted gAFTIR has significantly improved RO prediction accuracy.
To further demonstrate improved matching between simulated RO and the mechanistic AFTIR, we performed a local sensitivity analysis for three antibody-receptor systems (Fig. 5) across each of the individual eight parameters varied in the global sensitivity analysis. Each column of Fig. 5 and Supplemental Fig. 7 represents the result of changing one parameter while holding the other parameters fixed, and the change in the parameter is listed on the x-axis of each plot. For example, fold-change of 1 in the “Dose” column indicates the original dose (1 mg/kg, except for τ and RKrogh which was 10 mg/kg) used in all simulations; fold-change of 10 indicates 10x original dose (i.e., 10 mg/kg), fold-change of 0.1 indicates 1/10th the original dose (i.e., 0.1 mg/kg), and so on while all other parameters are held constant. Only the τ and RKrogh columns represent absolute values for these parameters instead of a fold change. The error bars on the simulations represent the standard deviation in simulated RO per dosing period and tumor radius. Overall, a marked improvement in accuracy was observed with the mechanistic AFTIR (Fig. 5 for log scale, Supplemental Fig. 7 for linear scale). The calculations/simulations for kβ showed discrepancies when t1/2,β > τ (which indicates unrealistically frequent dosing on the order of days to weeks) for ultra-slow clearance rates on the order of months to years [rarely possible (Robbie et al., 2013)] due to the Krogh cylinder model not achieving steady state by 6 weeks. In these scenarios, we show that gAFTIR at the first dose closely matches the Krogh simulations after 6 weeks (Supplemental Fig. 6), indicating agreement when the Krogh simulations align with the time frame of gAFTIR.

Local sensitivity analysis with individual parameters (log scale). The global AFTIR calculations with outlier corrections (dark pink) is overall more accurate at reproducing the results from simulations (black) than either the uncorrected gAFTIR (purple) or using an interstitial antibody concentration only from specific regimes. When beta phase clearance half-life exceeds dosing frequency (marked by *), the comparison was excluded since the Krogh simulation run time was not long enough to achieve steady state (Supplemental Fig. 6).
It is important to note that the analytical (algebraic) calculations may not always perfectly match the simulated results—this would only be achievable if the partial differential equations for the tumor antibody concentrations were analytically solvable. However, this mechanistically weighted AFTIR metric provides a closer and more realistic estimate of in vivo receptor occupancy than calculations with any standalone steady state antibody concentration estimate (Ctumor,surf,free, Cplasma, Ctumor,total, or Ctumor,free).
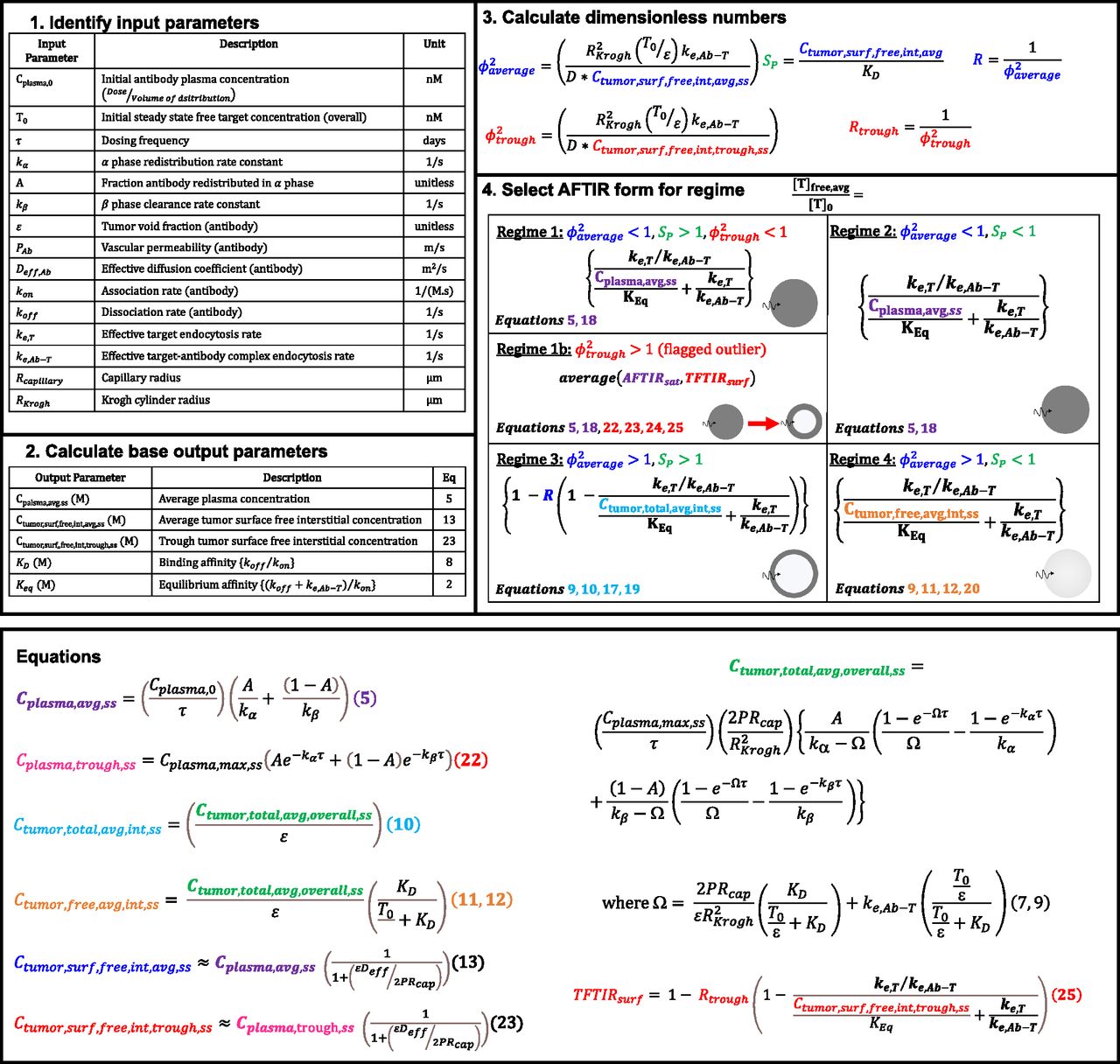
Practical Guide for Implementing gAFTIR
The gAFTIR metric provides more accurate calculations of receptor occupancy primarily by improving the estimation of the average (over space and time) interstitial antibody concentration in the tumor. However, it is tedious given the multiple regimes needed to describe the approximate solutions to these coupled, nonlinear, partial differential equations with time-dependent mixed boundary conditions in a cylindrical annulus. To aid the practical implementation of this regimen-based RO prediction, a step-by-step guide describing the calculations for RO is shown in Fig. 6, with detailed formulae for various calculations in the expanded AFTIR listed in the Methodssection. (Detailed derivations can be found in previous works by Thurber et al., 2007; Thurber and Wittrup, 2012). Additionally, an Excel spreadsheet interface is made available to automatically calculate the RO based on input parameters (Supplemental Materials: “Global_AFTIR_calculation_sheet.xlsx”).

Flowchart for calculating RO using global AFTIR, the average Thiele modulus (ϕ2average), local saturation potential (SP), and trough Thiele modulus (ϕ2trough), calculated from the input parameters and base output parameters, determine the antibody targeting regimen. Once the regimen is determined, the appropriate AFTIR metric is selected that incorporates the relevant free antibody concentration. Specific formulas for the antibody concentrations are found in the Methods section, and an Excel sheet is provided to automatically calculate these values (Supplemental Materials: “Global_AFTIR_calculation_sheet.xlsx”).
The first step is identifying relevant input parameters (both intrinsic and extrinsic). During development, easily modifiable parameters Cplasma,0 and τ are dependent on the dose (mg/kg) and dosing frequency, while antibody-intrinsic parameters A, kα, kβ, kon, koff, ke,Ab-T, PAb, Deff,Ab, and ε are predetermined by physicochemical and molecular properties of the antibody. A, kα, kβ can be estimated by fitting a biexponential function or a two-compartment model to plasma antibody PK data (see example calculations for Supplemental Fig. 11 in Supplemental Materials: “Global_AFTIR_calculation_sheet.xlsx”). Note that the biexponential function is only applicable for antibodies that exhibit linear pharmacokinetics. In cases where the antibody exhibits nonlinear pharmacokinetics (e.g., target-mediated drug-disposition; Mager and Jusko, 2001), or when estimating biexponential parameters is otherwise inconvenient, the steady state average plasma concentration can be estimated by other methods (e.g., population PK fitting/simulations) and directly input into subsequent formulae. Molecular parameters kon, koff, and ke,Ab-T can be experimentally measured in vitro and ex vivo using quantitative assays such as flow cytometry (Nessler et al., 2020), surface plasmon resonance (Hearty et al., 2012), and so on. PAb, Deff,Ab, and ε can be estimated based on the molecular weight/hydrodynamic radius of the antibody (Schmidt and Wittrup, 2009). Note that PAb, Deff,Ab, and ε are also influenced by the tissue properties (e.g., leaky vasculature, tumor extracellular matrix, cellular packing density, etc.) Tissue-intrinsic parameters like T0 and ke,T can also be measured in vitro and ex vivo using quantitative assays such as flow cytometry (Vasilyev et al., 2013; Khera et al., 2021). Note ke,T and ke,Ab-T are typically similar; however, in some cases, binding of the antibody to the target receptor can result in different effective internalization rates (e.g., decreased target recycling) and trigger up- or down-regulation of the receptor. Rcapillary (typically 8 µm) may vary by tumor type and from effects of vasodilation, while RKrogh can often be estimated by stereology of tumor histology images marking functional blood vessels, which provides the average intercapillary distance (i.e., 2 x RKrogh) (Yoshii and Sugiyama, 1988; Boyce et al., 2010).
The second step is calculating the base output parameters required for determining the antibody regimen (formulae for each can be found in the Methods section; auto calculated in the Excel sheet interface). The third step is calculating the dimensionless numbers ϕ2average, SP, and ϕ2trough using the base outputs and input parameters. The final step is identifying the regimen based on the combination of dimensionless numbers and calculating RO.
Discussion
Monoclonal antibodies represent one of the most rapidly growing class of drugs and have in recent decades emerged as a clinically and commercially successful approach to targeted therapy of cancer. Antibodies can exert their antitumor effects via several mechanisms, including inhibiting tumor growth signaling (Tan et al., 2006) and coengagement of tumor-infiltrating immune cells to activate antitumor immunity. The latter mechanism includes activating immune mechanisms such as natural killer cell mediated antibody-dependent cellular cytotoxicity, tumor-associated macrophage mediated phagocytosis, and dendritic cell response against shed antigen (Weiner et al., 2012). Implicit in the success of monoclonal antibody efficacy, particular for receptor antagonists, is successful and sustained target receptor engagement on the tumor cell surface. RO can be measured experimentally in discovery and preclinical settings via quantitative flow cytometry (Stewart et al., 2016), imaging (Zhang and Fox, 2012; Tang et al., 2019), and other sophisticated methods such as microfluidics (Chou et al., 2020).
Though efforts have been made to extrapolate receptor occupancy estimates from proxy sources such as circulating cells (Spilker et al., 2016), direct estimation of target engagement in situ in the clinic is extremely challenging and has prompted the use of increasingly sophisticated and mechanistic models to predict receptor occupancy via simulations. While these complex models, including the Krogh cylinder model used here, provide valuable insights on antibody penetration, they are not conducive for use by clinicians and nonmodelers who may not have the time or extensive experience to perform complex simulations. The AFTIR potency metric developed by Ahmed et al. (2019) presents a simple but powerful quantitative tool that can be used to guide recommended phase 2 dose predictions and rapidly predict target engagement for a range of molecular properties without the need for tedious simulations to expedite the drug design and development process. However, the AFTIR metric was developed with the assumption of “saturating” doses—though a reasonable assumption for some dosing regimens, it is difficult to determine when this assumption is valid. Furthermore, this assumption makes AFTIR predictions less useful at subsaturating doses, which are relevant for many clinical antibody dose decisions (Supplemental Fig. 11). Here we expand the existing AFTIR metric and present a mechanistically driven global AFTIR guided by the Thiele modulus and local saturation potential to predict receptor occupancy more reliably regardless of antibody dose, antibody pharmacokinetics, and tissue properties, providing a simple and universal guide to estimating target engagement for antibodies.
Antibody distribution is a complex phenomenon that varies both spatially and over time within a tumor. Any model is necessarily an approximation of the underlying processes governing drug distribution. However, a single value approximating the average receptor occupancy spatially and over time within the tumor can be a useful metric to guide dosing decisions. While the use of multiple regimes adds some additional complexity to the calculations, these results are nevertheless analytical expressions that can be easily calculated by hand or in a spreadsheet rather than numerically solving multiple partial differential equations. This general gAFTIR is more broadly applicable to estimates of receptor occupancy by not requiring an assumption of tumor saturation, which is often an unknown by itself.
We demonstrate through both global and local sensitivity analyses that absolute receptor density is a critical parameter that influences target engagement, and it is necessary to accurately predict receptor occupancy using the mechanistic gAFTIR. However, immunohistochemistry remains the gold standard for estimating clinical receptor density quantification despite being a semi-quantitative method due to the use of variable labeling protocols that can only provide relative expression levels and not quantify absolute receptor density on tumors. Thus, for the successful practical application of the mechanistic AFTIR metric, the utilization of quantitative tools like flow cytometry and mass cytometry that can quantify absolute receptor density (receptors/cell) with single-cell resolution is critical.
Though more accurate and universal than existing RO calculation metrics, the described mechanistic AFTIR does have limitations including a few outlier scenarios (Fig. 6) that are simulated to show significant unoccupied receptor (AFTIR > 0.1) but are predicted to have high receptor occupancy (AFTIR approximately 0.01–0.0001). Many of these discrepancies resulted from a transition from one regimen to another during the course of dosing (e.g., a transition from a saturating (ϕ2average < 1) to nonsaturating regimen at trough concentration (ϕ2trough > 1). However, the discrepancy is not delineated in all the outlier cases. By using simple cut-off values rather than smoother approaches (e.g., asymptotic matching), small changes in parameters for scenarios close to the regimen transitions can result in step-changes in the model assumptions and predictions. Likewise, the model is not valid for small molecule therapeutics that may be blood-flow limited or diffusion-limited in the tissue (Thurber and Weissleder, 2011; Bhatnagar et al., 2014).
For subsaturating doses, the pattern of antibody penetration and receptor occupancy becomes an important consideration for efficacy depending on the dominant mechanism of action (i.e., the pharmacodynamics). Low affinity antibodies can better penetrate the tumor, but better penetration comes at the cost of losing complete saturation of receptors on individual cell layers. High affinity antibodies are excellent at completely blocking receptors on targeted cell layers, but they may not be capable of targeting cells deep in the tumor at low doses. When saturating doses are not feasible, targeting more cell layers with partial receptor occupancy per cell layer might be beneficial for an antibody that functions primarily by activation of the immune system (i.e., agonists; Jung et al., 2022). Contrarily, complete saturation of at least a fraction of cells might be better for an antibody that mediates efficacy by blocking ligand-receptor signaling (antagonists), as even 1% of receptors occupied by the ligand can be sufficient for near complete activation of downstream growth signaling pathways (Wiley, 2018). These strategies can be independently pursued beyond just dosing and affinity considerations. For example, Bordeau et al., (2021) and Chen et al. (2022) have recently described a unique strategy to improve antibody distribution at subsaturating doses via transient competitive inhibition of antibody-antigen binding using an anti-idiotypic distribution enhancer. By coadministering an anti-trastuzumab single domain antibody that competitively binds to trastuzumab, the high affinity binding between trastuzumab-based therapeutics and HER2 is transiently disrupted, akin to epitope masking but independent of the tumor microenvironment, enabling more uniform distribution before dissociating and binding to HER2.
Accounting for distribution effects becomes even more important (and complex) when designing advanced therapeutics such as antibody-drug conjugates (ADC), which are designed to deliver cytotoxic payloads to cells in addition to antibody mechanisms of therapeutic action and are often limited by a relatively low maximum tolerated dose. High affinity ADCs administered at subsaturating doses are likely to exhibit increased efficacy when carrying a bystander payload that can efficiently compensate for perivascular antibody distribution of the ADC (Burton et al., 2019). Alternatively, a carrier dosing strategy, where the high affinity ADC is coadministered with the unconjugated antibody (Cilliers et al., 2018), may be another efficient method to exploit both growth signaling inhibition and cytotoxic payload delivery. However, in such a case it is important to ensure that the payload used is highly potent, so that dilution of the payload concentration per cell does not significantly impact efficacy (Ponte et al., 2021). If immune cell engagement is a primary mechanism of antibody-mediated efficacy, where even partial surface receptor saturation is sufficient to engage tumor immune cells, a low affinity antibody conjugated to an ultrapotent payload that uniformly targets all cells (but not all receptors on each cell) might ensure sufficient activation of immune cells throughout the tumor while also enabling efficient cytotoxic cell death from the ultrapotent payload like pyrrolobenzodiazepine or indolinobenzodiazepine that can mediate irreversible cell death at even extremely low intracellular concentrations (pM – nM) (Nessler et al., 2020; Ponte et al., 2021).
The analysis presented here specifically validates gAFTIR for intravenously dosed antibodies, though there is increasing clinical interest in subcutaneous antibody dosing. While we have not evaluated the translatability of the analytical approximations of complex PDE simulations that include the subcutaneous absorption and lymphatic distribution on the plasma compartment, this approach could readily be extended to other dosing routes of interest for monoclonal antibodies.
This mechanistic approach to refining receptor occupancy metrics opens avenues to employing a similar methodology in more sophisticated target engagement scenarios. For example, the mechanistic model can be expanded to include additional species such as endogenous ligand and shed antigen to mechanistically calculate the degree of ligand-blocking mediated by the antibody, both in the presence and absence of decoy shed antigen (Schmidt et al., 2019; Alaybeyoglu et al., 2021). This work can also be expanded to calculate the receptor occupancy of bispecific antibodies to rapidly estimate the degree of engagement between tumor cells and T-cells.
Conclusion
In conclusion, we have developed an improved and mechanistically validated metric to calculate the receptor occupancy of antibodies more rapidly and reliably regardless of distribution PK, dosing regimen, and tumor tissue properties. This universal guide can be used by quantitative scientists and clinical pharmacologists alike to gain valuable insights on target engagement by antibody-based drugs and aid in advancing the field toward mechanistically driven drug design and clinical dosing decisions.
Authorship Contributions
Participated in research design: Khera, Kim, Stein, Thurber.
Conducted experiments: Khera.
Contributed new reagents and analytical tools: Khera, Ratanapanichkich, Thurber.
Performed data analysis: Khera, Thurber.
Wrote or contributed to the writing of the manuscript: Khera, Kim, Stein, Thurber.
Footnotes
- Received December 7, 2022.
- Accepted March 30, 2023.
This work was supported in part by National Institutes of Health National Institute of General Medical Sciences [Grant R35-GM128819].
J.K. and A.S. were employed by Novartis at the time of this work. E.K., M.R., and G.M.T. have no actual or perceived conflict of interest with the contents of this article.
An earlier draft of this work is included in the thesis titled “Engineering Tumor Distribution of Antibody-Drug Conjugates” authored by E.K. in partial fulfillment of a doctoral degree from the University of Michigan (embargoed through April 2023).
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ADC
- antibody drug conjugate
- AFTIR
- average free tissue target to initial target ratio
- gAFTIR
- global average free tissue target to initial target ratio
- HER2
- human epithelial growth factor receptor 2
- PDE
- partial differential equation
- PK
- pharmacokinetics
- RO
- receptor occupancy
- TFTIR
- trough free fraction tissue target to initial target ratio
- Copyright © 2023 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}