


Visual Overview

Abstract
Leonurine (LEO) is a bioactive small molecular compound that has protective effects on the cardiovascular system and prevents the early progression of atherosclerosis; however, it is not clear whether LEO is effective for plaque stability. A novel mouse atherosclerosis model involving tandem stenosis (TS) of the right carotid artery combined with western diet (WD) feeding was used. Apolipoprotein E gene–deficient mice were fed with a WD and received LEO administration daily for 13 weeks. TS was introduced 6 weeks after the onset of experiments. We found that LEO enhanced plaque stability by increasing fibrous cap thickness and collagen content while decreasing the population of CD68-positive cells. Enhanced plaque stability by LEO was associated with the nitric oxide synthase (NOS)-nitric oxide (NO) system. LEO restored the balance between endothelial NOS(E)- and inducible NOS(iNOS)-derived NO production; suppressed the NF-κB signaling pathway; reduced the level of the inflammatory infiltration in plaque, including cytokine interleukin 6; and downregulated the expression of adhesion molecules. These findings support the distinct role of LEO in plaque stabilization. In vitro studies with oxidized low-density lipoprotein–challenged human umbilical vein endothelial cells revealed that LEO balanced NO production and inhibited NF-κB/P65 nuclear translocation, thus mitigating inflammation. In conclusion, the restored balance of the NOS-NO system and mitigated inflammation contribute to the plaque-stabilizing effect of LEO.
SIGNIFICANCE STATEMENT LEO restored the balance between endothelial NOS and inducible NOS in NO production and inhibited excessive inflammation in atherosclerotic “unstable” and rupture-prone plaques in apolipoprotein E gene–deficient mice. The protective effect of LEO for stabilizing atherosclerotic plaques was due to improved collagen content, increased fibrous cap thickness, and decreased accumulation of macrophages/foam cells. So far, LEO has passed the safety and feasibility test of phase I clinical trial.
Introduction
Atherosclerosis, a progressive, chronic inflammatory disease characterized by the accumulation of fatty plaques within the arterial wall (Libby et al., 2016), remains a major health burden and is predicted to become the leading cause of mortality and morbidity worldwide (Baldassarre et al., 2000; Baigent et al., 2005). The rupture of vulnerable atheromatous plaque is one major cause of stroke and myocardial infarctions. Culprit lesions are characterized by thin fibrous caps, lipid-rich necrotic cores, and high inflammatory cell counts (Anderson and Morrow, 2017). Therefore, enhancing the stability of vulnerable plaques to inhibit plaque rupture is a promising strategy to prevent stroke and myocardial infarction in patients with atherosclerosis.
Evidence is accumulating that inflammation plays an important role in the progression of atherosclerosis. Uncontrolled inflammation renders atherosclerotic plaques as “unstable,” such that these plaques are prone to rupture or erosion, which may lead to thrombosis and myocardial infarction. The current therapies for coronary artery disease, such as lipid-lowering statins, do not effectively control plaque inflammation. Therefore, new antiatherosclerotic drugs are needed, especially those targeting inflammation. Therefore, therapeutic drugs based on inhibition of excessive inflammation with low toxicity are promising in treating atherosclerosis.
Leonurine (LEO; 4-guanidino-n-butyl-syringate, also known as SCM-198) is the alkaloid component of Herba leonuri. Because of its low content in Herba leonuri and the poor purity in extract, our team has chemically synthesized leonurine from syringic acid, and the product, LEO, is of 99% purity (Liu et al., 2009). LEO is a competitive drug candidate for the treatment of myocardial ischemia and cardiac insufficiency in rats (Liu et al., 2009; Loh et al., 2010; Shi et al., 2011). Recent studies confirmed the antiapoptotic (Xin et al., 2009), antioxidant (Liu et al., 2010b), anti-inflammatory (Liu et al., 2012), and microcirculation-improving (Loh et al., 2010) effects of LEO under different experimental conditions. Several studies conducted in rat models have suggested that LEO has potent protective effects in both the cardiovascular and cerebrovascular system (Liu et al., 2010a, 2013). Recent studies have confirmed that long-term administration of LEO safely improves lipid profiles in mammals, which indicates its potential usage for the prevention and treatment of atherosclerosis (Zhang et al., 2012) and that it may be an alternative for patients who do not tolerate statins (Suguro et al., 2018). The peroxisome proliferator-activated receptor γ/liver X receptor -α pathway is thought to be involved in the protective role of LEO against atherosclerosis (Jiang et al., 2017). Encouraged by emerging evidence of its effectiveness and safety in preclinical studies reported by our team and other laboratories, we have initiated clinical transformation research of LEO since last year. At present, it has passed the safety test of phase I clinical experiment. On the other hand, it is important to address safety and potential pharmacodynamic interactions of LEO (Norata and Catapano, 2012). In view of this, toxicity and pharmacokinetic characteristics of LEO were tested, and the result was encouraging (Zhu et al., 2018).
However, whether LEO is protective against atherosclerosis in omnivores and whether it influences plaque stability is still unknown. Therefore, in this study, we examined the effects of LEO on vulnerable plaque stability in ApoE−/− mice and the potential underlying mechanisms.
A novel mouse model of unstable plaque induced by the combination of Western diets (WDs) and tandem stenosis (TS) was recently introduced (Chen et al., 2013), providing a powerful tool for assessing the therapeutic potential of candidate drugs as well as revealing the specific mechanisms that contribute to plaques progression. In this model, the right carotid artery of ApoE−/− mice is subjected to low shear stress and high tensile stress, which leads to the formation of plaques with unstable phenotype. This animal model has been widely used ever since because of its strength at mimicking pathologic changes in life-threatening clinical cardiovascular events (Rashid et al., 2018).
Metabolic disturbance not only induces endothelial cell dysfunction at the onset stage of atherosclerosis but also weakens endothelial barrier in advanced atherosclerotic plaques. Endothelial cells at the atherosclerotic lesion site display features of sustained inflammation, which is closely related to metabolic disturbance and immune insults. Previous studies have shown that a decrease in the bioavailability of NO from endothelial NO synthase (eNOS) is the main cause of endothelial fracture. Here, we evaluate the effect of LEO on eNOS expression and activity through in vitro and in vivo studies.
Materials and Methods
The authors declare that all supporting data are available within the article (Supplemental Data).
Mouse Model of Plaque Instability with Tandem Stenosis.
Six-week-old male ApoE−/− mice (C57BL/6J background) were purchased from the Animal Center of Nanjing University and were housed in Fudan University Experimental Animal Center with a 12-hour light/dark cycle and free access to water. Mice at 6 weeks old were divided into five groups (n = 8–20 per group): normal diet control group, TS + WD (product # D12079B; Research diets) model group, low-dose LEO treatment group (10 mg/kg per day), medium-dose LEO treatment group (20 mg/kg per day), and high-dose LEO treatment group (40 mg/kg per day). LEO or vehicle was administered intragastrically once per day for 13 weeks. At the age of 12 weeks, mice were anesthetized by intraperitoneal injection of 1% sodium pentobarbital, and TS surgery of the right common carotid artery was introduced. Mice in the normal diet control group received sham operations.
At the age of 19 weeks, blood samples were collected from each mouse by puncturing the retro-orbital plexus with a capillary tube. The samples were then transferred into EDTA anticoagulant tubes. Plasma was separated by centrifugation (3500 rpm, 15 minutes, 4°C), rapidly frozen in liquid nitrogen, and then stored at −80°C. Mice were then anesthetized by intraperitoneal injection of 1% sodium pentobarbital. The sternum was cut, and the heart was exposed. Under physiologic pressure, the catheter was placed into the left ventricle, and general perfusion was performed successively with PBS buffer (pH 7.4) to flush out red blood cells, followed by 4% formalin solution for fixation. The carotid artery and the aortic arch were then carefully separated, dissected out, and photographed en face using a Fuji XT 20 camera with a Fuji XF 80mm F2.8 R LM OIS WR macro lens.
Scheme 1 shows the schedule of the animal experiment and the details of the TS surgery. The Animal Ethics Committees of Fudan University approved all animal experiments.
Arterial Tissue Sample Preparation and Histologic Analysis.
Left and right common carotid arteries, aortic arches, and aortas were dissected and fixed in 4% formalin solution. Samples for histologic analysis were then embedded in paraffin, and 6-μm-thick continuous sections were prepared. Standard H&E staining was used to show plaque structure. Masson’s trichrome staining and picrosirius red staining were used for collagen content detection. The percentage of specific plaque component was quantified as the positive stained area divided by the total area of the plaque. The ratio of the area of the fibrous cap to the maximum height of the damaged area (cap-to-lesion height ratio), which H&E and picrosirius red staining of plaque with unstable phenotype in the right carotid artery. Cap thickness was determined as the area of picrosirius red–stained cap viewed under polarized light showing the fibrous cap (yellow line). Lesion height was determined from H&E-stained sections (arrow). The relative cap thickness was defined as the ratio of the shoulder thickness at the center of the plaque divided by the maximum intimal thickness at the center of the plaque (Cheng et al., 2011). Macrophage content was quantified by CD68 immunohistochemistry. After paraffin removal and antigen repair, CD68 (ba3638, 1:100; Boster, China), eNOS (CST5880, 1:100), and CD31 (ab182981, 1:1000; Abcam) were incubated overnight at 4°C. The diaminobenzidine substrate was used for detection. Mouse IgG2b control antibody was used for specificity verification. An optical microscope (Olympus bx50 optical microscope) was used to obtain H&E and Masson’s trichrome staining images. A polarized light microscope (dfc450c) was used to obtain picrosirius red staining images. ImageJ was used to quantify the positive area and signal intensity.

Animal model establishment. Six-week-old ApoE−/− mice were fed a WD containing 22% fat and 0.15% cholesterol and allowed to drink water freely. At 12 weeks of age, animals were anesthetized, and tandem stenosis surgery was performed. A 150-μm-diameter needle (Ethicon 8-0, silk blue, w1782) was placed alongside the right common carotid artery. Successive ligations with 6-0 polyester knitted fiber were made 1 mm from the distal point of the carotid bifurcation and 3 mm from the first one. Then, the needle was removed to form partial vascular stenosis. The outer diameter of the needle was measured with a spiral micrometer. The animals were killed 7 weeks after the surgery. Lt., left; Rt., right.
Magnetic Resonance Imaging.
In vivo MRI was performed with an 11.7 T horizontal-bore animal scanner (BioSpin; Bruker, Ettlingen, Germany) and Paravision 5 software. The mouse was initially anesthetized with 3% isoflurane and maintained with 1%–2% isoflurane during the MRI signal acquisition. The mouse was placed in a custom-made cradle, which contained a heating pad with a temperature of approximately 37°C to sustain body temperature. Then, the mouse-containing cradle was placed in a 2.5-cm-diameter quadrature birdcage radio frequency coil in the MRI scanner. Respiration and heart rate were monitored with a balloon sensor and ECG trigger leads connected to an ECG/respiratory unit (Rapid Biomedical, Rimpar, Germany). The MRI protocols consisted of time-of-flight, T1-, and T2-weighted imaging. The total examination time including preparation and planning was approximately 2 hours per mouse. Time-of-flight series were obtained by a two-dimensional gradient echo sequence. Sequence parameters were as follows: TR = 15 milliseconds, TE = 2.5 milliseconds, flip angle = 20°, FOV = 2.56 × 2.56 × 2.56 cm3, acquisition matrix = 256 × 256 × 256, NA = 2, and total scan time = 18 minutes. T1- and T2-weighted imaging in sagittal and transversal orientations were performed by using a black blood multislice spin echo sequence. Black blood imaging was achieved by placing two saturation slabs below and above the neck region, with the inflow blood set to saturation. Sequence parameters for T1-weighted imaging were as follows: TR = 800 milliseconds, TE = 7.5 milliseconds, FOV = 2.56 × 2.56 cm2, acquisition matrix = 256 × 256, reconstruction matrix = 512 × 512, slice thickness = 0.5 mm, NA = 2, and total scan time = 8 minutes. Sequence parameters for T2-weighted imaging were as follows: TR = 2000 milliseconds, TE = 20 milliseconds, FOV = 2.56 × 2.56 cm2, acquisition matrix = 256 × 256, reconstruction matrix = 512 × 512, slice thickness = 0.5 mm, NA = 2, and total scan time = 20 minutes. T1- and T2-weighted scanning was triggered by respiratory signals to suppress motion artifacts (van Bochove et al., 2010).
Blood Lipid Analysis.
Total plasma cholesterol, LDL and HDL cholesterol, and triglyceride concentrations were measured with the Cobas Integra 400 plus auto analyzer using the standard procedure of enzymatic assay kits from Nanjing Jiancheng (China). Colorimetric changes were measured at 512 nm for total cholesterol and triglycerides or at 583 nm for HDL and LDL cholesterol.
Measurement of IL-6, VCAM-1, ICAM-1, and NO.
The level of VCAM-1, ICAM-1, and IL-6 were detected by ELISA kits according to the manufacturers’ instructions (Nanjing Jiancheng). The NO levels in the plasma or cell culture medium were determined with the Griess reaction (Biyuntian, China). Intracellular NO was measured with an NO probe (Biyuntian), photographed with a fluorescent microscope, and quantified by ImageJ.
Cell Culture.
Primary human umbilical vein endothelial cells (HUVECs) were purchased from Allcells (H-001F-C; China) and cultured in HUVEC Growth Medium (H-004; Allcells), and 100 U/ml of penicillin and 100 μg/ml were added into the culture medium. The HUVECs were cultured in a 37°C incubator with 5% CO2.
Cell Viability.
Cell viability was determined by cell counting kit-8 assay according to manufacturer’s instructions (CK04; DOJINDO, Japan). HUVECs were cultured in a 96-well culture plate and treated with different concentrations of LEO (0, 0.625, 1.25, 2.5, 5, 10, 20, 50, 100, 250, 500, or 1000 μM) for 24 hours. Cells were subsequently incubated with 10 μl CCK-8 solution at 37°C for 4 hours. The absorbance at 450 nm was measured with a microplate reader.
Western Blot Analysis.
HUVECs were lysed in radio-immunoprecipitation assay lysis buffer, which consisted of RIPA solution, protease inhibitors, and phosSTOP phosphatase inhibitors (04906845001; Roche Life Sciences). Tissue samples were homogenized and lysed in RIPA lysis buffer. The proteins were separated by acrylamide gel electrophoresis before being transferred to polyvinylidene fluoride membrane (Millipore). The membranes were blocked with 5% bovine serum albumin dissolved in 1× TBST buffer and incubated with primary antibodies at 4°C overnight. The next day, the membranes were washed in TBST and subsequently incubated with corresponding secondary antibodies conjugated with horseradish peroxidase for 1 hour at room temperature. The membranes were then washed again in TBST before enhanced chemiluminescence detection on X-ray films. Information for all the antibodies used is in the main resource table in Supplemental Material.
Immunofluorescence Staining.
Immunofluorescence was used to detect the expression and distribution of P65 in the HUVECs and carotid arteries. The cells were pretreated with 50 μg/ml of ox-LDL (Guangzhou Yiyuan, China) for 24 hours and then treated with LEO. The cells were fixed with 4% paraformaldehyde fixation solution (P0099; Beyotime, China) plus 0.1% Triton X-100 at room temperature for 15 minutes and then washed three times with PBS and blocked in blocking solution (P0102; Beyotime) for 1 hour at room temperature. Then, the cell samples were incubated with p-P65 antibodies overnight at 4°C. The next day, the samples were then washed in PBS three times and incubated with Alexa Fluor 654–labeled secondary antibody. The cell nuclei were counterstained with 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) for 5 minutes. Immunofluorescence images were collected with a Leica dmi8 immunofluorescence microscope (Leica, Germany) and merged with Leica Las image analysis software x (Leica). Paraffin sections of the carotid arteries were dewaxed and stained with the same antibodies that we used in the cell samples after antigen repair. The fluorescence intensity of nuclear P65 in the nucleus was analyzed by ImageJ.
Statistical Analysis.
The results were expressed as means ± S.E.M. GraphPad Prism 6 was used to analyze the data. Before data analysis, all variables were tested for normality and equal variance. The differences among multiple groups were analyzed by one-way ANOVA. The data in Table 1 were analyzed with Fisher’s exact test. P < 0.05 was considered statistically significant.
Plaque characteristics in vessel segment I
Results
Western Diet plus Tandem Stenosis of Right Carotid Artery Successfully Induced Unstable Atherosclerotic Plaques in ApoE−/− Mice and Retarded Plaque Progression.
Histologic analysis and MRI were used to verify the successful establishment of an unstable atherosclerotic plaque mouse model. For histologic analysis, arteries in the vicinity of the TS were separated into five segments in accordance with the literature (Chen et al., 2013) (Fig. 1A). Vessel segments I, II, and III were located at the right common carotid artery. Vessel segment IV was located at the left common carotid artery. Vessel segment V composed the whole aortic arch, including the proximal areas of segment I. A representative image of the general view around the TS area indicated the atherosclerotic plaque hemorrhage in segment I (Fig. 1F). H&E staining confirmed that vessel segment I displayed the typical characteristics of unstable atherosclerotic plaque. Figure 1D showed an intraluminal thrombosis. Figure 1I demonstrated thin caps in the shoulder region with large necrotic core. Disruption of fibrous caps (DFC) (Fig. 1J), intraplaque hemorrhage (IH) (Fig. 1K), and carotid artery dissection (Fig. 1L) were also found in segment I. Vessel segment II exhibited a plaque phenotype that contained intact fibrous cap with abundant cellular components inside the plaque and extensive outward remodeling (Fig. 1C). Vessel segment III displayed a stable atherosclerotic plaque phenotype that contained intact and cellular rich fibrous caps (Fig. 1B). Vessel segment IV was plaque-free and represented a healthy artery (Fig. 1G). Vessel segment V showed a stable atherosclerotic plaque (Fig. 1H). Because IH and DFC are two characteristic features of unstable rupture-prone human plaques, we examined the incidence rate of IH and DFC at the endpoint of our in vivo experiment. The incidence rate of IH was 60%, and the incidence rate of DFC was 75% in model mice (Table 1). Administration of 20 and 40 mg/kg per day of LEO reduced the incidence of both indicators.

Pathologic studies of the mouse model and the effect of LEO. Cross section of different vessel segments represented healthy nonatherosclerotic areas and areas of stable and unstable plaques (A). Schematic drawing of the different vessel segments investigated in the mouse model (B). Vessel segment III exhibited a stable atherosclerotic plaque containing a thick fibrous cap and small necrotic core (C). Vessel segment II often contained an intact thin fibrous cap with highly cellular content (D). (I–L) Vessel segment I represented an unstable/rupture-prone atherosclerotic plaque characterized by a large necrotic core (NC), disruption of fibrous caps (DFC), intraplaque hemorrhage (IH), intraluminal thrombosis (IT), and carotid artery dissection (CAD). (E) Vessel segment I was obtained from mice with LEO administration. (F) Representative gross anatomy of the neck area at the endpoint of the experiment. The ligation suture was white. (G) Vessel segment IV represented plaque-free healthy vasculature. (H) Vessel segment V contained the aortic arch and adjacent vessel segments. The plaque in vessel segment V was a stable one characterized by thick caps. Scale bars, 100 μm in (A–G) and (I–L) and 1000 μm in (H). (M) Time-of-flight 2D MRI reconstruction measurement confirmed carotid artery stenosis in the mouse model mice. The yellow arrow indicated the position of the ligation, and the red arrow represented the compensatory blood flow of adjacent vessels. F, feet; H, head; L, left; R, right.
A time-of-flight 2D MRI reconstruction measurement was also conducted to evaluate carotid artery stenosis in a time series. Figure 1M shows the representative reconstructed images obtained from preoperative model mice and from model mice postoperative at 1, 4, and 7 weeks. In particular, we found that at 7 weeks after the TS, the blood flow in the carotid artery on the TS side was seriously blocked, which resulted in lost signal in reconstructed 2D MRI images. Meanwhile, the compensatory blood flow in adjacent vessels was obvious. These results indicated that the stenosis deteriorated over time. LEO (40 mg/kg per day) reduced the severity of vascular stenosis (Fig. 1M). Supplemental Figure 1 was captured from 3D MRI rotation around the H-F axis from model mice 1 week after the surgery. Tandem stenosis could be easily observed.
We also examined the sagittal T1- and T2-weighted MRI of the carotid arteries. The signal of inflow blood was magnetically saturated, and therefore, the lumen of the carotid artery appeared black. Transversal T1- and T2-weighted images were shown in slices placed directly downstream to the proximal ligation, as indicated by the dashed lines (Fig. 2A) The intensity of the right carotid artery wall was higher than that of surrounding muscles, particularly in the T1-weighted images, which could be attributed to the presence of the atherosclerotic lesions (Fig. 2, C and F). In contrast, the signal of the left carotid artery vessel wall and the signal of the right carotid artery wall in the sham group were essentially isointense with surrounding muscles (Fig. 2, B and E). Treatment with LEO (40 mg/kg per day) retained partial lumen volume (Fig. 2, D and G). The improvement of the retained lumen volume in the right carotid arteries was obvious in enlarged T1-weighted images. The ratio of remaining lumen area in the right common carotid artery in transverse T1- and T2-weighted images was significantly increased after LEO treatment compared with the model group (Fig. 2, H and I). En face photographs of common carotid arteries and aortic arches from different groups were collected, and the percentage of atherosclerotic lesion area was analyzed. LEO significantly reduced the plaque area in a dose-dependent manner (Fig. 2J). Treatment with 40 mg/kg per day of LEO reduced plaque area by 25% (Fig. 2K). These results proved the antiatherogenic properties of LEO.

LEO slowed the development of atherosclerotic plaques. T1- and T2-weighted MRI of the neck region of the mouse (A). T1-weighted image in sagittal orientation. The position of the proximal ligation is indicated by the dotted line. The dashed line indicated the scanning area. Image size was 12 × 15 mm2 (B–D). T1- and (E–G) T2-weighted transversal slice of sham, model, and LEO-treated groups, respectively. Image size of (B–G) was 10 × 10 mm2. (H and I) The quantitative analysis of retained vascular lumen in T1- and T2-weighted MRI. (J) En face photographs of common carotid arteries and aortic arches from sham, model, and LEO-treated groups. (K) The quantitative analysis of the atherosclerotic lesion areas (*P < 0.05). D, dorsal; (L), enlarged image; L, left; LCCA, left common carotid artery; LEO-H, high-dose LEO; LEO-L, low-dose LEO; LEO-M, medium-dose LEO; R, right; RCCA, right common carotid artery; V, ventral.
LEO Improved the Lipid Profile and Attenuated Inflammation in ApoE−/− Mice.
Hyperlipidemia is the main factor in the development of atherosclerosis. Plasma lipid profiles are altered in patients with atherosclerosis and are usually improved after antiatherosclerotic treatment. We tested total cholesterol, triglycerides, LDL-C, and HDL-C levels in mouse plasma. We found that treatment with 40 mg/kg per day of LEO reduced total cholesterol, triglyceride, and LDL-C but did not affect HDL-C levels (Fig. 3). Excessive inflammation contributes to “unstable” and rupture-prone plaque formation. Thus, we examined both plasma and tissue levels of proinflammatory cytokines and adhesion molecules in vivo. The plasma level of IL-6 increased 3-fold in the model group, whereas both 20 and 40 mg/kg per day of LEO attenuated IL-6 elevation (Fig. 4A). For potential leukocyte adhesion, 20 mg/kg per day of LEO reduced plasma VCAM-1 levels (Fig. 4C). However, none of the three doses of LEO tested altered plasma ICAM-1 levels (Fig. 4B). Then, we detected the expression of the adhesion molecules CD62P, CD62E, ICAM-1, and VCAM-1 (Fig. 4D) in the mouse carotid arteries. We found that both 10 and 40 mg/kg per day of LEO reduced arterial CD62P levels (Fig. 4E). All three doses of LEO tested reduced arterial CD62E and ICAM-1 (Fig. 4, F and G). Both 20 and 40 mg/kg per day of LEO reduced arterial VCAM-1 levels (Fig. 4H). These results indicated that LEO protected against atherosclerosis at least partially through its anti-inflammatory effect.

The effects of LEO on lipid profiles in atherosclerotic ApoE−/− mice. LEO-treated groups received daily administration of LEO (10, 20, and 40 mg/kg), and the control and model groups received double-distilled water. The levels of (A) total cholesterol, (B) total triglycerides, (C) low-density lipoprotein, and (D) high-density lipoprotein were measured, and statistical analysis was made. Values are means ± S.E.M. (n = 5–8 per group). *P < 0.05. LEO-H, high-dose LEO; LEO-L, low-dose LEO; LEO-M, medium-dose LEO.

LEO attenuated atherosclerosis and inhibited inflammation in the mouse model. Model mice were treated with LEO (10, 20, 40 mg/kg, daily, oral administration) or vehicle (n = 8–13 each). (A–C) The level of IL-6, ICAM-1, and VCAM-1 in plasma. (D)The expression of adhesion molecules in aortas without or with LEO treatment were measured by Western blotting. (E–H) The quantitative analysis of tissue CD62P, CD62E, ICAM-1, and VCAM-1 protein levels, GAPDH protein was served as the internal control. (*P < 0.05). LEO-H, high-dose LEO; LEO-L, low-dose LEO; LEO-M, medium-dose LEO; N.S., not significant.
Treatment with LEO Enhances Plaque Stability.
Thin-capped fibroatheromas account for the majority of plaques at risk for rupture (Virmani et al., 2000; Wu et al., 2013). Decreased fibrous cap thickness is mainly due to a decline in the number of vascular smooth muscle cells (VSMCs), as VSMCs are important for extracellular matrix (ECM)-secreting cells. One major component of the ECM inside plaque is collagen, and its degradation may cause fibrous cap weakening (Virmani et al., 2000). Hence, we evaluated the effect of LEO therapy on cap thickness as well as the collagen content of plaques located in segment I. Continuous paraffin sections were stained with H&E, picrosirius red, and Masson’s dye. H&E staining in Fig. 5A revealed healthy artery structure in control mice, whereas all of the other samples from the model and LEO groups had plaques developed under the endothelium. Interestingly, mice treated with middle- and high-dose LEO had larger residual lumen and smaller plaques.

LEO promoted plaque stability in model mice by increasing fibrous cap thickness and plaque collagen content and by reducing macrophage/foam cell accumulation. Serial sections of the vascular segment I in model mice with or without LEO treatment were used for different staining methods. (A) The structure of plaques was observed by H&E staining. Lesion height was marked and measured (black arrows). (B) Representative picrosirius red staining viewed under polarized light. (C) Representative Masson’s trichrome staining to show the collagen content. Collagen was in blue, and the smooth muscle fibers were in red. (D) Representative CD68 staining to show the macrophage/foam cell accumulation. Data were presented as the proportion (%) of the total plaque area. (E) The quantitative analysis of fibrous cap thickness measured with polarized light microscopy. (F) Ratio of fibrous cap thickness to lesion height. (G and H) The quantitative analysis of the percentages of collagen content and CD68-positive areas. Scale bar, 50 μm. *P < 0.05 LEO-H, high-dose LEO; LEO-L, low-dose LEO; LEO-M, medium-dose LEO.
The picrosirius red staining results in Fig. 5B showed strong yellow-orange birefringence of thick type I collagen fibers and other types of collagens with relatively weak birefringence signal under polarized light. The shoulder thickness of the fibrous cap at the center of the plaque was marked with arrows. LEO treatment of 20 and 40 mg/kg per day significantly increased the thickness of the fibrous caps (Fig. 5E). The ratio of the fibrous cap thickness to the lipid core height reflects the progression of unstable plaques. LEO treatment of 20 and 40 mg/kg per day increased the ratio, which meant slower progression of unstable plaques (Fig. 5F).
Masson’s trichrome staining in Fig. 5C showed collagen and smooth muscle cells. Collagen was shown in blue, and smooth muscle cells were shown in red. The ratio of blue-stained areas to plaque areas reflects the collagen content in plaque. Collagen content was significantly higher in all three doses of LEO tested compared with the model group (Fig. 5G).
Massive accumulation of macrophages/foam cells inside plaque was correlated with large lipid core and unstable plaques. Figure 5D shows CD68 staining of accumulated macrophages/foam cells in plaques. Samples from the model group showed large positive areas. All three doses of LEO tested significantly reduced CD68-positive areas (Fig. 5H). These results indicated that LEO increases atherosclerotic plaque stability by improving collagen content and decreasing macrophage/foam cell accumulation.
LEO Increased eNOS Expression, NO Production, but Inhibited iNOS Expression and NF-κB Nuclear Translocation in Primary Cultured Endothelial Cells.
Endothelial dysfunction–related decreased NO bioavailability from eNOS contributes to atherosclerosis (Sharma et al., 2015). Endothelial dysfunction also facilitates abnormal monocytes penetrating through the endothelium, which transform into macrophages and eventually become foam cells. This inflammation reaction further aggravates plaque instability (Hansson et al., 2015). Here, we induced endothelial cell inflammation by challenging HUVECs with ox-LDL, according to Sun et al. (2018), and then treated cells with LEO. LEO at a concentration of 50 μM or less did not influence cell viability and showed no toxicity. A dose of 50 μM LEO was thus chosen for in vitro experiments according to its effectiveness and safety (Supplemental Fig. 2). ox-LDL increased both intracellular and secreted NO levels in supernatant. LEO (50 μM) further elevated NO content (Fig. 6, A and B). Since NO can be derived from both eNOS and iNOS catalysis, we examined the expression of these two enzymes (Fig. 6C). We found that inflammatory iNOS expression was significantly induced in the model group, and LEO treatment inhibited this elevation. Meanwhile, p-eNOS/eNOS level remained the same in model or LEO-treated groups when compared with the control groups. However, decreased eNOS and p-eNOS after ox-LDL challenging were restored by LEO treatment (Fig. 6D), indicating that the signaling pathway of eNOS was upregulated by LEO. These data indicated that iNOS-derived NO production in ox-LDL–challenged endothelial cells was suppressed by LEO treatment, whereas deteriorated eNOS function was restored by LEO.

LEO increased NO production and inhibited NF-κB nuclear translocation in vitro. (A) The fluorescence signal of NO probe in HUVECs. (B) Statistical analysis of NO content in cells and supernatant. (C) Representative photographs of Western blot of NOS and NF-κB pathways. (D) The quantification of phosphorylated and total level of eNOS and iNOS. (E) Quantitative analysis of the ratios of p-I-κBα/I-κBα, p-P65/P65, and p-P65/LaminB. LaminB (or GAPDH)was used as a nuclear (or cytoplasma ) internal reference. (F) Representative cell immunofluorescence images of the expression and nuclear translocation of p-P65 in HUVECs under different treatments. (G) Quantitative analysis of the fluorescence intensity of p-P65 in each group. Scale bar, 50 μm. *P < 0.05 vs. ox-LDL group.
The NF-κB pathway plays a key role in the inflammatory response. We tested both the phosphorylation of key negative regulator I-κBα and the phosphorylation and nuclear localization of key component P65 (Oeckinghaus and Ghosh, 2009). ox-LDL challenging activated the NF-κB pathway, which was confirmed by elevated I-κBα and P65 phosphorylation, and nucleus p-P65 expression. LEO treatment considerably reduced the activation degree of NF-κB pathway in ox-LDL–injured HUVECs (Fig. 6E). Cell immunofluorescence was used to monitor the expression level and location of p-P65 in the cytoplasm and nucleus. The intranuclear fluorescence intensity of p-P65 in the ox-LDL–challenged cells was obviously enhanced. Pretreatment with LEO effectively reduced the fluorescence intensity of p-P65 in the nucleus (Fig. 6F), which was consistent with the results obtained from Western blotting (Fig. 6G). These results indicated that the protective effect of LEO observed in vivo may be related to enhancing eNOS-derived NO production and inhibiting cellular inflammation in the endothelium.
LEO Increased eNOS Expression, NO Production, and Inhibited NF-κB Pathway In Vivo.
Based on the findings from in vitro experiments, we detected mouse plasma NO levels. Similar with in vitro studies, plasma NO level increased significantly in the TS model group and further increased after LEO treatment (Fig. 7A). We also detected eNOS and iNOS protein levels in aortas. LEO increased both eNOS and p-eNOS expression, whereas it decreased iNOS expression (Fig. 7, B and C). We also measured the expression and phosphorylation of NF-κB subunit P65 and negative regulator I-κBα in the aortas. Compared with the control group, the phosphorylation of P65 and I-κBα in the model group was both significantly increased, which indicated the activation of NF-κB signal. All three doses of LEO tested effectively reduced this activation. Nucleus P65 phosphorylation was also decreased after LEO treatment (Fig. 7, D and E). Fluorescence microscopy was used to observe nuclear translocation of P65 in aortas.

LEO increased NO production and inhibited NF-κB nuclear translocation in vivo. (A) Plasma NO level in model mice. (B) Representative photographs of Western blot of NOS. (C) The quantitative analysis of p-eNOS, eNOS, and iNOS expression in aortas. (D) Representative photographs of Western blot of the NF-κB pathway. (E) Quantitative analysis of the ratios of p-IκBα/IκBα, p-P65/P65, and p-P65/LaminB. LaminB was used as a nuclear internal reference. (F) Representative immunofluorescence images of p-P65 in mouse aortas. (G) Representative immunohistochemical staining of eNOS and CD31 in vessel segment I with or without LEO administration. Nucleus were counterstained with DAPI (blue). Scale bar, 20 μm. *P < 0.05 vs. TS + WD group. LEO-H, high-dose LEO; LEO-L, low-dose LEO; LEO-M, medium-dose LEO.
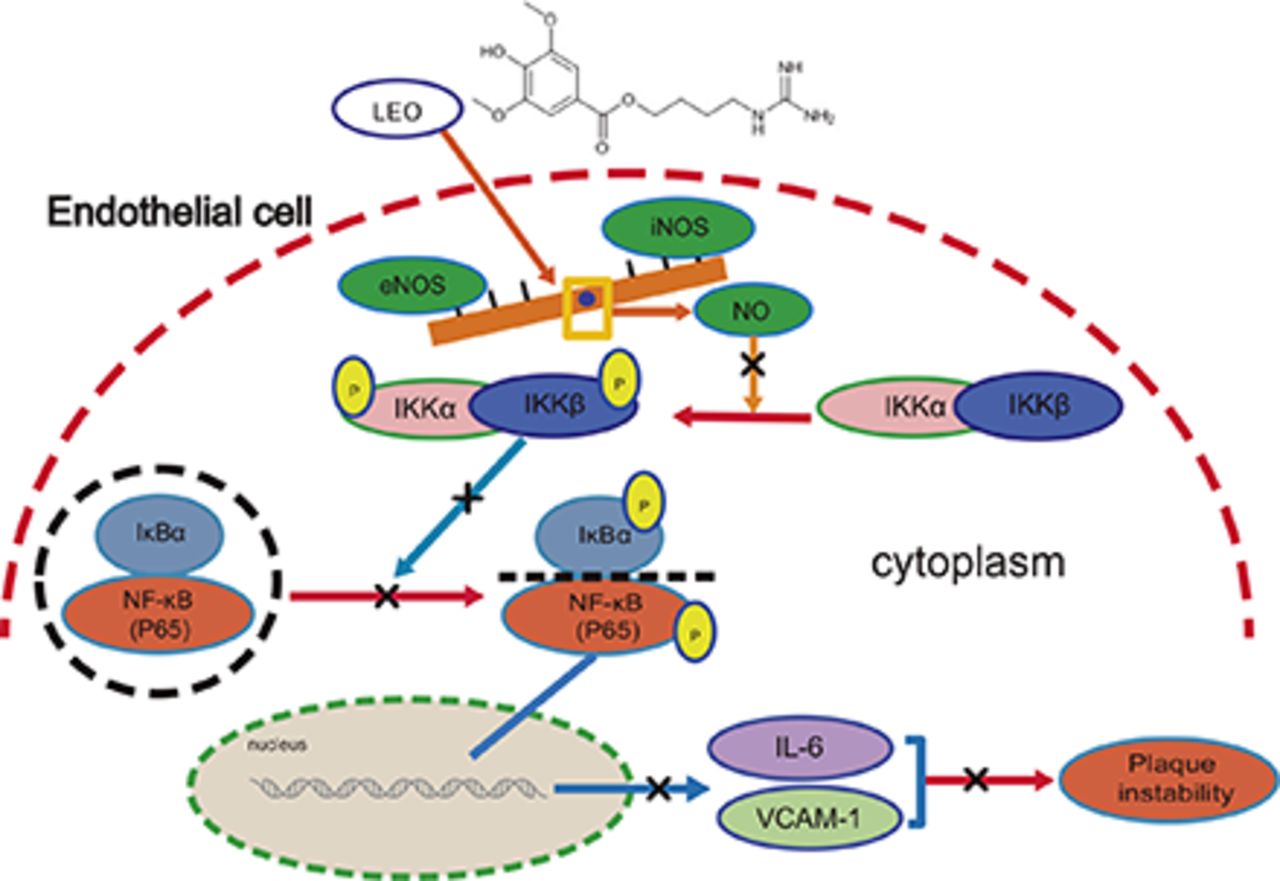
The intensity of phosphorylated P65 in aortas from model mice was obviously greater than that in the control group. LEO treatment of 50 mg/kg per day reduced nucleus p-P65 (Fig. 7F). Our results suggested that LEO could reduce the expression of P65 as well as block its translocation into nucleus, thus inhibiting the activation of the NF-κB pathway in atherosclerosis. Moreover, we observed immunohistochemistry of eNOS and CD31. Compared with the model group, LEO increased the expression of eNOS, which was consistent with the results of CD31, suggesting that eNOS expression in endothelial cells can be enhanced by LEO (Fig. 7G). Because of its anti-inflammation, eNOS/NO restoration, and plaque stabilization effects, LEO is expected to become a candidate drug for antiatherosclerosis therapy (Fig. 8).

Schematic diagram of the mechanism of the anti-inflammatory effect of LEO in endothelial cells. LEO inhibited the NF-κB signaling pathway by enhancing the production of NO from eNOS. LEO inhibited the production of NO from iNOS, thereby inhibiting the production of inflammatory transcription factors, such as IL-6 and VCAM-1. By these mechanisms, LEO increased plaque stability.
Discussion
Lipid metabolism disorder and inflammation are the main factors of atherosclerosis; moreover, increased vulnerability is prone to final rupture of atherosclerotic plaque, which can result in a severe cardiovascular event (Seneviratne et al., 2017). Given that LEO exhibits multiple inhibitory effects on proatherogenic factors, such as inflammation, hypercholesterolemia, and hyperlipidemia, in different animal models (Liu et al., 2012; Zhang et al., 2012), we postulate that LEO may exert antiatherogenic function and the composition of plaque, which determines the lesion stability. However, pharmacological effect and molecular function of LEO in atherosclerosis remains unknown. LEO is an alkaloid component from motherwort. We investigated the effect of LEO in the mouse model of atherosclerosis with plaque instability. Our data showed that the progression of atherosclerosis was significantly slower in mice treated with LEO. LEO treatment of 40 mg/kg per day increased the thickness of fibrous caps and the content of collagen in the plaques but decreased the population of CD68-positive cells. These novel observations indicated the plaque stabilization effect of LEO. Lipid regulation and inflammation inhibition also contributed to the protective role of LEO. The mechanism involved the regulation of eNOS activity and the shift of NO production from inflammatory iNOS to protective eNOS. These studies indicated that the eNOS-NO system contributed to an antiatherogenic effect of LEO via inflammation inhibition and plaque stabilization.
We tested three types of indicators in vivo to help determine the effect of LEO. Noninvasive MRI examination was performed to evaluate carotid artery stenosis in a time series. Plasma inflammatory biomarkers and lipid profiles were determined. Pathologic examinations were performed in both gross observation of en face plaques and histologic studies based on paraffin sections. Mice treated with 40 mg/kg per day of LEO retained more lumen volume in the right carotid arteries; subsided plasma IL-6 elevation; had reduced plasma total cholesterol, triglyceride, and LDL-C levels; and reduced plaque area in common carotid arteries and aortic arches. Taken together, these results provided solid evidence for LEO to be considered as a potential antiatherosclerosis drug.
Thin-capped fibroatheroma accounts for the majority of rupture-prone plaques. Decreased cap thickness is mainly due to a decline in the number of VSMCs resulting from apoptosis and the loss of ECM (van der Wal et al., 1994; Kolodgie et al., 2001; Sadeghi et al., 2010). Collagen is an important component of the ECM inside plaque; excessive degradation or insufficient production may cause lower collagen content and fibrous cap weakening (Schwartz et al., 2007). Thus, stable plaques usually have abundant collagen and high VSMC counts but low apoptosis levels and small lipid cores. In this study, we found that LEO increased cap thickness by enhancing collagen content and decreasing lipid core size.
Inflammation is a key driver of plaque rupture because it enhances collagen degradation and impairs fibrous cap formation (Schwartz et al., 2007; Hansson et al., 2015). Nonresolving inflammation drives the development of clinically dangerous atherosclerotic lesions by promoting sustained plaque inflammation, as well as the formation of large necrotic cores, thin fibrous caps, and thrombosis (Kasikara et al., 2018). In lieu of this evidence, therapeutic targeting of inflammation has been considered to be a complementary strategy to reduce LDL to lower the risk of atherosclerotic vascular disease. IL-1β has been proven to promote inflammation in atherosclerosis animal models by inducing the expression of leukocyte adhesion proteins, such as ICAM-1, E-selectin, and VCAM-1. The elevated expression of adhesion molecules contributes to the accumulation of leukocytes in the lesion area (Qamar and Rader, 2012; Welsh et al., 2017). In the present study, we observed attenuated production and expression of inflammatory factors, especially plasma IL-6 and VCAM-1. The tissue levels of ICAM-1 and VCAM-1 in the aortas were also significantly decreased after LEO treatment.
In vitro studies of ox-LDL–challenged HUVECs showed increased NO production and inflammation as evidenced by elevated expression of key proteins in the NF-κB signaling pathway. LEO further increased NO production but inhibited inflammation. ox-LDL is a widely used reagent that mimics the cell injury in atherosclerotic conditions in in vitro studies. Previous studies have reported that ox-LDL reduces NO concentration in endothelial cells (Lee et al., 2018). However, in our experimental settings, both intracellular NO content and NO concentration in the supernatant were increased after ox-LDL challenging. This discrepancy may come from the different concentrations of ox-LDL used in different studies. We used 50 μM ox-LDL, whereas Lee et al. used 100 μM ox-LDL. We observed that the exposure to ox-LDL led to the imbalanced activation of nitric oxide synthase (NOS) in HUVECs. ox-LDL promoted the activation of inducible isoforms of the enzyme (iNOS) and meanwhile inactivated the endothelial isoforms (eNOS). This kind of imbalanced activation accelerated the inflammatory process. Decreased eNOS and increased iNOS also existed in our mouse model. At the same time, we also found that the expression of eNOS was enhanced by LEO in immunohistochemistry. Interestingly, we observed the switch from iNOS activation to eNOS restoration after LEO treatment in both in vitro and in vivo studies. By inhibiting iNOS-mediated inflammation and restoring eNOS-mediated protection, LEO protected the endothelium from inflammatory damage and stabilized plaques.
It has been reported that NO inhibits the activity of NF-κB. We observed that exposure to ox-LDL activated the NF-κB signaling pathway. LEO treatment downregulated the phosphorylation level of two key proteins, P65 and I-κBα, in both in vivo and in vitro studies. In addition, LEO significantly decreased P65 expression level and its translocation into the nucleus. Therefore, the anti-inflammatory effect of LEO reported here may provide some evidence for its future usage in the clinic.
Collectively, the current study reveals the antiatherosclerosis property of LEO as a fine-tuning factor in the regulation of NO production and NF-κB activation. Administration of LEO induced NO production in vivo and in vitro. As an inhibitory factor, NO acts directly to inhibit the expression of P65, providing an additional layer to control the NF-κB signaling network. LEO upregulated eNOS-derived NO production and provided protection against atherosclerosis and inflammation by inhibiting NF-κB activity. Given the pivotal role of NF-κB in atherosclerosis and other inflammatory diseases, our study indicates the potential application value of LEO as a new therapeutic drug against atherosclerosis and inflammation.
Our study demonstrates that LEO enhances atherosclerotic plaque stability by increasing fibrous cap thickness and levels of collagen content and by decreasing macrophage/foam cell accumulation. However, there are several limitations. The intervening mechanisms of LEO to inhibit collagen degradation and macrophage/foam cell accumulation remain to be clarified. We only explored the phenotype and protective mechanism in the endothelium. The effect of LEO on other important cell types that are involved in plaque formation and stability, such as macrophages and smooth muscle cells, needs to be verified.
Acknowledgments
We would like to thank Zhao Zhonghua from Department of Pathology of Fudan University for his support on pathological experiments, Yang Baofeng from Institutes of Science and Technology for Brain-inspired intelligence of Fudan University for his technical support on MRI, and Zhang Yangwen from the School of Materials of Shanghai University for his technical support on polarized light microscopy.
Authorship Contributions
Participated in research design: Ning, Wang, Y. Huang, L. Zhu, Liang, Yu, Y.-Z. Zhu, Y.-C. Zhu.
Conducted experiments: Ning, Lin, Zhang, M.-Y. Li, Yang, Z.-M. Li, Y.-J. Huang.
Contributed new reagents or analytic tools: Y.-Z. Zhu, Y.-C. Zhu.
Performed data analysis: Ning, Chen.
Wrote or contributed to the writing of the manuscript: Ning, Wang, Zhang, Y.-C. Zhu.
Footnotes
- Received December 31, 2019.
- Accepted March 18, 2020.
↵3 K.N. and M.-J.W. contributed equally to this work.
This work was supported by grants from the Ministry of Science and Technology (2017YFE0120200), National Major Scientific and Technological Special Project (No. 2017ZX09301002), the National Natural Science Foundation of China (Grant 81670248, 81970361), funding of the Innovative Research Team of High-Level Local Universities in Shanghai, the Outstanding Clinical Discipline Project of Shanghai Pudong (PWYgy-2018-08) and the Program for Medical Key Department of Shanghai (ZK2019A10).
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ApoE−/−
- apolipoprotein E gene–deficient
- DFC
- disruption of fibrous cap
- ECM
- extracellular matrix
- eNOS
- endothelial NOS
- HDL
- high-density lipoprotein
- HUVEC
- human umbilical vein endothelial cell
- I-κB
- inhibitor of NF-κB
- ICAM-1
- intercellular cell adhesion molecule-1
- IH
- intraplaque hemorrhage
- IL-6
- interleukin 6
- LDL
- low-density lipoprotein
- LEO
- leonurine
- MRI
- magnetic resonance imaging
- NF-κB
- nuclear factor κB
- NOS
- nitric oxide synthase
- ox-LDL
- oxidized LDL
- p-
- phosphorylated
- TBST
- Tris-buffered saline/Tween 20
- TS
- tandem stenosis
- VCAM-1
- vascular cell adhesion molecule-1
- VSMC
- vascular smooth muscle cell
- WD
- Western diet
- TR
- Time of repetition
- TE
- Time of echo
- FOV
- Field of view
- NA
- Number of signal average
- iNOS
- inducible NOS
- RIPA
- radio-immunoprecipitation assay
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}