


Abstract
By reducing their metabolism, dipeptidyl peptidase 4 inhibition (DPP4I) enhances the effects of numerous peptides including neuropeptide Y1–36 (NPY1–36), peptide YY1–36 (PYY1–36), and SDF-1α. Studies show that separately NPY1–36, PYY1–36 and SDF-1α stimulate proliferation of, and collagen production by, cardiac fibroblasts (CFs), preglomerular vascular smooth muscle cells (PGVSMCs), and glomerular mesangial cells (GMCs), particularly in cells isolated from genetically hypertensive rats. Whether certain combinations of these factors, in the absence or presence of DPP4I, are more profibrotic than others is unknown. Here we contrasted 24 different combinations of conditions (DPP4I, hypertensive genotype and physiologic levels [3 nM] of NPY1–36, PYY1–36, or SDF-1α) on proliferation of, and [3H]-proline incorporation by, CFs, PGVSMCs, and GMCs. In all three cell types, the various treatment conditions differentially increased proliferation and [3H]-proline incorporation, with a hypertensive genotype + DPP4I + NPY1–36 + SDF-1α being the most efficacious combination. Although the effects of this four-way combination were similar in male versus female CFs, physiologic (1 nM) concentrations of 2-methoxyestradiol (2ME; nonestrogenic metabolite of 17β-estradiol), abolished the effects of this combination in both male and female CFs. In conclusion, this study demonstrates that CFs, PGVSMCs, and GMCs are differentially activated by various combinations of NPY1–36, PYY1–36, SDF-1α, a hypertensive genetic background and DPP4I. We hypothesize that as these progrowth conditions accumulate, a tipping point would be reached that manifests in the long term as organ fibrosis and that 2ME would obviate any profibrotic effects of DPP4I, even under the most profibrotic conditions (i.e., hypertensive genotype with high NPY1–36 + SDF-1α levels and low 2ME levels).
SIGNIFICANCE STATEMENT This work elucidates combinations of factors that could contribute to long-term profibrotic effects of dipeptidyl peptidase 4 inhibitors and suggests a novel drug combination that could prevent any potential profibrotic effects of dipeptidyl peptidase 4 inhibitors while augmenting the protective effects of this class of antidiabetic agents.
Introduction
The pharmacology of dipeptidyl peptidase 4 (DPP4) inhibitors (DPP4Is), a class of antidiabetic drugs, is complex. Although inhibition of DPP4 increases incretin levels and thereby augments insulin release (McIntosh et al., 2005), DPP4Is also elevate concentrations of other biologically active DPP4 substrates (Mentlein, 1999; Gorrell, 2005; Mulvihill and Drucker, 2014); and this likely contributes to the net effects of DPP4Is. For example, neuropeptide Y1–36 (NPY1–36) and peptide YY1–36 (PYY1–36), which are potent endogenous agonists of Gi-coupled Y1 receptors (Y1Rs) (Michel et al., 1998; Berglund et al., 2003), are metabolized by DPP4 to neuropeptide Y3–36 (NPY3–36) and peptide YY3–36 (PYY3–36), respectively (Mentlein, 1999; McIntosh et al., 2005). Because PYY1–36 and NPY1–36 are potent Y1R agonists, whereas PYY3–36 and NPY3–36 are inactive at Y1Rs (Michel et al., 1998; Berglund et al., 2003), DPP4Is increase Y1R activation by impairing the metabolism of NPY1–36 and PYY1–36. In support of this concept, recent studies by Wilson and coworkers show that DPP4 inhibition increases plasma NPY1–36 yet decreases plasma NPY3–36 or PYY3–36 in hypertensive patients with type 2 diabetes (Wilson et al., 2019). Thus, DPP4Is may modify the biologic effects of endogenous NPY1–36 and PYY1–36. Indeed, our recent results show that via Y1Rs NPY1–36 and PYY1–36, but not NPY3–36 or PYY3–36, stimulate proliferation of, and collagen production by, cardiac fibroblasts (CFs), preglomerular vascular smooth muscle cells (PGVSMCs) and glomerular mesangial cells (GMCs) (Jackson et al., 2012; Zhu et al., 2015b); and these effects of NPY1–36 and PYY1–36 are enhanced by DPP4 inhibition (Jackson et al., 2012; Zhu et al., 2015b).
The chemokine SDF-1α is another important DPP4 substrate. SDF-1α, a 68 amino acid polypeptide, is a potent CXCR4-receptor agonist, which like Y1Rs are Gi-coupled (Busillo and Benovic, 2007). However, DPP4 cleaves two amino acids from the N terminus of SDF-1α, and this truncated form of SDF-1α is inactive at CXCR4 receptors (Wang et al., 2014). Our most recent results show that SDF-1α, via CXCR4 receptors, induces proliferation of, and collagen production by, CFs, PGVSMCs, and GMCs; and these effects are also enhanced by DPP4 inhibition (Jackson et al., 2017). Importantly, DPP4 inhibition increases SDF-1α levels in patients with type 2 diabetes (Fadini et al., 2010).
Progrowth and profibrotic effects of NPY1–36, PYY1–36 and SDF-1α on CFs, PGVSMCs, and GMCs depend in part on the blood pressure of the animals from which cells were obtained. For example, our previous results demonstrate that CFs, PGVSMCs, and GMCs harvested from spontaneously hypertensive rats (SHRs) are more responsive to the progrowth/profibrotic actions of NPY1–36, PYY1–36, and SDF-1α than are cells obtained from normotensive Wistar-Kyoto rats (WKYs) (Jackson et al., 2012, 2017; Cheng et al., 2013; Zhu et al., 2015b).
Because overactive CFs can contribute to cardiac fibrosis leading to heart failure (Brown et al., 2005) and proliferation of, and collagen production by, PGVSMCs and GMCs can lead to renovascular hypertrophy, glomerulosclerosis, renal fibrosis, and renal failure (Dubey et al., 1997), the effects of DPP4Is on growth responses to DPP4 peptide substrates must be thoroughly evaluated. An unanswered question is: How important are the combinatorial effects of NPY1–36, PYY1–36, SDF-1α, DPP4 inhibition, and genetic hypertension as stimulators of proliferation of, and collagen production by, CFs, PGVSMCs, and GMCs? This is a key question because in patients with cardiovascular diseases, some will have hypertension with coelevation of NPY1–36, PYY1–36, and SDF-1α. For example, in heart patients, SDF-1α levels are increased and are associated with heart failure, cardiac fibrosis, and all-cause mortality (Chu et al., 2010; Subramanian et al., 2014; Zuern et al., 2015). Simultaneously, the release of NPY1–36 from sympathetic nerves is augmented in heart failure (Hulting et al., 1990; Kaye et al., 1994). Also, PYY1–36 levels are elevated in advanced heart failure (Gouya et al., 2014). Indeed, Packer has proposed that DPP4Is cause heart failure by increasing levels of both NPY1–36 and SDF-1α, thus promoting central and peripheral sympathetic activation (Packer, 2018).
Together, the aforementioned findings suggest that combinatorial effects of small increases of NPY1–36, PYY1–36, and SDF-1α may have large effects on CFs, PGVSMCs, and GMCs that are further amplified by DPP4Is and hypertension. To test this concept, here we examined 24 different combinations of genotype, peptide treatments, and DPP4 inhibition on the activation of CFs, PGVSMCs, and GMCs. Our results confirm that the effects of NPY1–36, PYY1–36, SDF-1α, and DPP4 inhibition are context dependent due to combinatorial effects of these agents with genetic hypertension. Moreover, our studies show that a low, physiologic concentration of 2-methoxyestradiol (2ME), an antidiabetic and cardiorenal-protective metabolite of 17β-estradiol, abrogates the progrowth/profibrotic effects of the most efficacious combination of factors (i.e., DPP4 inhibition + genetic hypertension + NPY1–36 + SDF-1α). We hypothesize that these findings may help identify patients at greatest risk of adverse long-term consequences of DPP4Is and suggest that coadministration of 2ME might improve upon the beneficial effects of DPP4Is.
Materials and Methods
Materials.
Chemicals were from the following sources: SDF-1α (ProSpec, Rehovot, Israel); sitagliptin (Merck, Whitehouse Station, NJ); platelet-derived growth factor–BB (PDGF-BB), NPY1–36, and PYY1–36 (Sigma-Aldrich, St. Louis, MO); 2-methoxyestradiol (Steraloids, Newport, RI).
Animals.
Male and female SHRs and WKYs (approximately 12 weeks of age) were obtained from Charles River Laboratories (Wilmington, MA). The University of Pittsburgh Institutional Animal Care and Use Committee approved all procedures. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (eighth edition, 2011). Our rationale for employing SHR cells is threefold. First, our previous studies show that the progrowth and profibrotic effects of NPY1–36, PYY1–36, and SDF-1α on CFs, PGVSMCs, and GMCs are greater in cells harvested from SHRs (Jackson et al., 2012, 2017; Cheng et al., 2013; Zhu et al., 2015b). Second, we have previously published that both WKY and SHR CFs, PGVSMCs, and GMCs express both DPP4 protein and DPP4 activity and that there is no difference between WKY and SHR cells in this regard (Jackson et al., 2012; Zhu et al., 2015b). Third, SHRs express pathophysiological mechanisms and pharmacological responses similar to hypertensive patients with type 2 diabetes. For example, like patients with type 2 diabetes, SHRs have insulin resistance (Umeda et al., 2003) and increased oxidative stress (Chen et al., 2019); and like patients with diabetes, SHRs are highly responsive to angiotensin converting enzyme inhibitors (Kost et al., 1995).
Cell Cultures.
CFs, GMCs, and PGVSMCs were isolated from SHRs and WKYs and characterized as previously described in detail by us (Inoue et al., 1998; Zhu et al., 2015b; Zhu and Jackson, 2017).
Assessment of Cell Proliferation.
Each well of a 12-well plate was seeded with 5000 cells. Cells were maintained in phenol red-free DMEM/F12 containing 10% steroid-free FBS under standard tissue culture conditions. Three days later when cells were approximately 60%–70% confluent, cells were growth-arrested for 2 days in DMEM/F12 containing 0.4% bovine serum albumin. Next, cells were placed in DMEM/F12 containing a low concentration (25 ng/ml) of PDGF-BB and then treated every day for 4 days with various treatments. Cells were then harvested and cell number quantified, in a blinded fashion, using a Nexcelom Cellometer Auto T4 cell counter (Nexcelom Bioscience, Lawrence, MA).
Assessment of [3H]-Proline Incorporation.
Cells were prepared as described above with the exception that cells were allowed to achieve a confluent monolayer (usually 5 to 6 days after seeding). Then cells were made quiescent in DMEM supplemented with 0.4% bovine serum albumin. To initiate proline incorporation, growth-arrested cells were placed in DMEM with added PDGF-BB (25 ng/ml) and [3H]-L-proline (2 µCi/ml) and containing various treatments. After 36 hours, experiments were terminated by washing cells twice with phosphate-buffered saline (PBS) and twice with ice-cold trichloroacetic acid (10%). The precipitate was solubilized in 0.5 ml of 0.3 N NaOH and 0.1% SDS and radioactivity determined, in a blinded fashion, in the precipitate using a liquid scintillation counter.
Assessment of Release of Collagen I Into the Extracellular Compartment.
Each well of a six-well plate was seeded with 50,000 cells. Cells were maintained in phenol red-free DMEM/F12 containing 10% steroid-free FBS under standard tissue culture conditions. Three days later when cells were approximately 60% confluent, cells were growth-arrested for 2 days in DMEM/F12 containing 0.4% bovine serum albumin. Next, cells were placed in DMEM/F12 containing a low concentration (25 ng/ml) of PDGF-BB and then treated every day for 3 days with treatments. The conditioned medium was collected at 24, 48, and 72 hours, combined and assayed for collagen I. After 72 hours of treatment, cells were collected and pelleted by centrifugation and the supernatant was removed. Cells were then washed three times with PBS and then resuspended in PBS. Next, cells were lysed by ultrasonication four times, centrifuged at 1500g for 10 minutes at 2–8°C to remove cellular debris, and the supernatant was assayed for collagen I. Collagen I in both the conditioned medium and cell lysate was measured using the LSBio (Seattle, WA) Rat Collagen I ELISA Kit (catalog number, LS-F37378). Because membrane-delimited collagen I (i.e., collagen I within endoplasmic reticulum, Golgi apparatus, and secretory vesicles) would not be available for binding to capture and detection antibodies, the analysis of collagen I in cell lysate provides information related to predominantly cytosolic collagen I.
Protocol 1: Experimental Design and Statistics.
To clarify the “biological replicate,” our protocol design is illustrated in Fig. 1. The protocol entailed 12 batches of CFs, with each batch derived from a separate male rat (i.e., a total of 12 rats; six WKYs and six SHRs). In the case of GMCs or PGVSMCs, the protocol again entailed 12 distinct batches of cells; however, because sufficient numbers of GMCs and PGVSMCs cannot be obtained from one rat, each batch of cells was derived from a separate set of three male rats (i.e., a total of 12 sets of three rats; six sets of three WKYs and six sets of three SHRs). Thus, a single biologic replicate was a unique rat or a unique set of three rats. Cells from each WKY biologic replicate were seeded on a 12-well plate, and cells from each SHR biologic replicate were seeded on another identical 12-well plate. There were 12 different treatments for each plate (i.e., for each plate, each well of cells received 1 of 12 treatments). All plates received the same 12 treatments. Next, the plate of WKY cells and a plate of SHR cells were placed side-by-side in a cell incubator. After the indicated incubation time, either cell proliferation or [3H]-proline synthesis was measured (different set of cultures were required for the two outcome measures). We considered this one experiment. Next, the entire procedure was repeated five times on different occasions (i.e., sample size was n = 6 for WKY cells and n = 6 for SHR cells). Results were analyzed by a nested three-factor ANOVA in which one factor was genotype (fixed factor; levels were WKYs or SHRs), a second factor was plate number (nested under genotype), and the third factor was treatment (fixed factor; 12 treatment levels that were various peptide treatments [vehicle, NPY1–36, PYY1–36, SDF-1α, NPY1–36 + SDF-1α, PYY1–36 + SDF-1α] with or without the DPP4 inhibitor sitagliptin [Subbarayan and Kipnes, 2011]). Post hoc tests were conducted using the Bonferroni method to control for multiple comparisons. We used a sitagliptin concentration of 1 μM because we have previously shown that this concentration inhibits the metabolism of PYY1–36 to PYY3–36 in PGVSMCs and GMCs (Jackson et al., 2012). We used concentrations of NPY1–36, PYY1–36, and SDF-1α of 3 nM because with all three peptides, this concentration provides a low (just above threshold) stimulation of cell growth in WKY cells (Zhu et al., 2015b; Jackson et al., 2017). Also, the concentration of 3 nM approximates achievable in vivo tissue levels of NPY1–36, PYY1–36, and SDF-1α (Schmidt et al., 2005; Kuncová et al., 2011; Roux et al., 2012).

This figure illustrates the experimental design for Protocol 1. The protocol entailed 12 batches of CFs, with each batch derived from a separate male rat (i.e., a total of 12 rats; six WKYs and six SHRs). In the case of GMCs or PGVSMCs, the protocol again entailed 12 distinct batches of cells; however, because sufficient numbers of GMCs and PGVSMCs cannot be obtained from one rat, each batch of cells was derived from a separate set of three male rats (i.e., a total of 12 sets of three rats; six sets of three WKYs and six sets of three SHRs). Thus, a single biologic replicate is a unique rat or a unique set of three rats. Cells from each WKY biologic replicate were seeded on a 12-well plate, and cells from each SHR biologic replicate were seeded on another identical 12-well plate. There were 12 treatment groups for each plate (i.e., for each plate, each well of cells received 1 of 12 treatments). All plates received the same 12 treatments. Next, the plate of WKY cells and a plate of SHR cells were placed side-by-side in a cell incubator. After the indicated incubation time, either cell proliferation or collagen synthesis was measured. We considered this one experiment. Next, the entire procedure was repeated five times on different occasions (i.e., sample size was n = 6 for WKY cells and n = 6 for SHR cells). Pairs of plates performed at the same time are noted by vertical lines.
Protocol 2: Experimental Design and Statistics.
Figure 2 illustrates the design of Protocol 2. CFs from a specific male or female SHR (referred to as “male CFs” or “female CFs,” respectively) were seeded on a separate 12-well plate, and nine wells received one of nine treatments (vehicle, NPY1–36, SDF-1α, NPY1–36 + SDF-1α, sitagliptin, sitagliptin + NPY1–36, sitagliptin + SDF-1α, sitagliptin + NPY1–36 + SDF-1α, 2ME + sitagliptin + NPY1–36 + SDF-1α), and the two plates were placed together in a cell incubator. All plates received the same nine treatments. The concentrations of peptides and sitagliptin were the same as in Protocol 1. The concentration of 2ME (1 nM) was selected based on the physiologic levels of this 17β-estradiol metabolite (Zacharia et al., 2003; Barchiesi et al., 2006). After the indicated incubation time, cell proliferation was measured. We considered this one experiment. Next, the entire procedure was repeated four times on different occasions (i.e., sample size was n = 5 for male CFs and n = 5 for female CFs). The effects of treatments in male and female CFs were analyzed separately by three-factor ANOVA in which one factor was plate number (random factor), a second factor was peptide treatment (fixed factor; levels were vehicle, NPY1–36, SDF-1α, NPY1–36 + SDF-1α), and the third factor was sitagliptin treatment (fixed factor; without and with sitagliptin). The effects of 2ME on responses to sitagliptin + NPY1–36 + SDF-1α were analyzed by a two-factor ANOVA in which one factor was plate number (random factor) and the second factor was treatment group (vehicle, sitagliptin + NPY1–36 + SDF-1α, 2ME + sitagliptin + NPY1–36 + SDF-1α). Post hoc tests were conducted using the Bonferroni method to control for multiple comparisons.

This figure illustrates the design of Protocol 2. CFs from a specific male or female SHR were seeded on separate 12-well plates, and nine wells of each plate received one of nine treatments; and the two plates were placed together in a cell incubator. All plates received the same nine treatments. After the indicated incubation time, cell proliferation was measured. We considered this one experiment. Next, the entire procedure was repeated four times on different occasions (i.e., sample size was n = 5 for male CFs and n = 5 for female CFs). Pairs of plates performed at the same time are noted by vertical lines.
Protocol 3: Experimental Design and Statistics.
Protocol 3 was conducted in CFs obtained from five male SHRs. Two matching six-well plates of CFs were prepared from each SHR. One plate was treated with sitagliptin (1 μM) plus DPP4 substrates (NPY1–36 and SDF-1α; 3 nM each) and the other was treated with vehicle plus DPP4 substrates (control). For each group, the outcome assessment was intracellular (cytosolic) and extracellular (medium) collagen I levels, and the ratio of extracellular to intracellular collagen I. These values were obtained for each paired set of SHR CFs by averaging the results for the replicates on each plate. The results were statistically analyzed using a paired t-tailed Student’s t test (n = 5) because control and sitagliptin-treated cells were from the same biologic replicate (i.e., same SHR).
Results
Proliferation Studies in CFs.
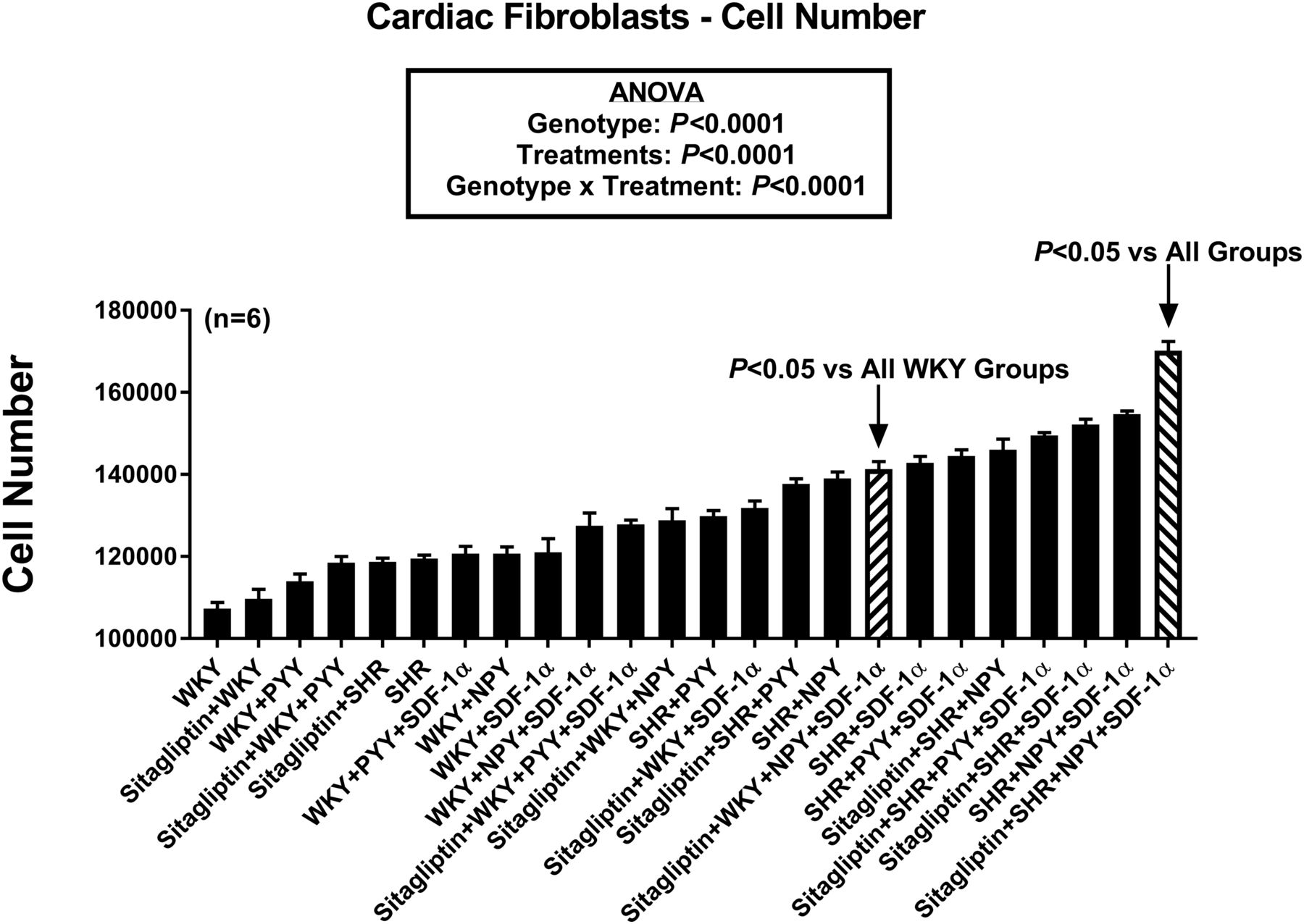
Figure 3 provides a summary of the effects of the 24 different treatments on proliferation of CFs. To facilitate visualization of the relative effects of the treatments on CF proliferation, the treatments are listed in order on the horizontal axis from least active (far left) to most active (far right). Statistical analysis by ANOVA revealed an overall effect of cell genotype (P < 0.0001) and treatments (P < 0.0001) on CF proliferation. Moreover, there was a significant genotype × treatment interaction (P < 0.0001). Bonferroni tests were conducted to compare all possible pairs and the results are shown in Supplemental Table 2 (which lists the results of all possible comparisons, whereas Supplemental Table 1 lists the group assignment numbers for Supplemental Table 2). Bonferroni tests showed that proliferation was significantly greater in WKY CFs treated with sitagliptin + NPY1–36 + SDF-1α compared with all other WKY CF groups and showed that compared with all of the other 23 groups, the most proliferative combination was SHR CFs treated with sitagliptin + NPY1–36 + SDF-1α. This analysis also demonstrated significantly increased proliferation in the order: WKY < SHR < SHR + NPY1–36 < SHR + NPY1–36 + SDF-1α < sitagliptin + SHR + NPY1–36 + SDF-1α; and WKY < SHR < SHR + PYY1–36 < SHR + PYY1–36 + SDF-1α < sitagliptin + SHR + PYY1–36 + SDF-1α.

Bar graph compares effects of 24 combinatorial treatments/conditions on the proliferation of cardiac fibroblasts. Values are means and S.E.M.s.
[3H]-Proline Incorporation Studies in CFs.
To assess the effects of the 24 different treatments on total collagen production, we measured the effects of treatments on [3H]-proline incorporation in CFs that were both confluent and quiescent as previously described (Barchiesi et al., 2002; Dubey et al., 2003a, 2010; Zhu et al., 2015b; Jackson et al., 2017). Because proline is abundant (enriched) in all isoforms of collagen, this approach permits an assessment of total collagen production. Also, by conducting these experiments in confluent/quiescent cells, incorporation of [3H]-proline in noncollagen proteins could be minimized and increases in [3H]-proline incorporation could be assessed at a constant cell number.
Figure 4 summarizes the relative effects of the 24 different treatment conditions on [3H]-proline incorporation by CFs. As with Fig. 3, the results are organized in Fig. 4 (as well as in Figs. 5–8) in order of treatment efficacy starting from the least active (left) to the most active (right) treatments on the horizontal axis. As with CF proliferation, statistical analysis by ANOVA showed that genotype (P < 0.0001) and treatments (P < 0.0001) significantly increased [3H]-proline incorporation by CFs and that there was a significant interaction between genotype and treatments (P = 0.0010). Bonferroni tests were conducted to compare all possible pairs and the results are shown in Supplemental Table 3. Bonferroni tests showed that [3H]-proline incorporation was significantly greater in WKY CFs treated with sitagliptin + NPY1–36 + SDF-1α compared with all other WKY CF groups and showed that compared with all of the other 23 groups, [3H]-proline incorporation was greatest in SHR CFs treated with sitagliptin + NPY1–36 + SDF-1α. This analysis also demonstrated significantly increased [3H]-proline incorporation in the order: WKY < SHR < SHR + NPY1–36 < SHR + NPY1–36 + SDF-1α < sitagliptin + SHR + NPY1–36 + SDF-1α; and WKY < SHR < SHR + PYY1–36 < SHR + PYY1–36 + SDF-1α = sitagliptin + SHR + PYY1–36 + SDF-1α.

Bar graph compares effects of 24 combinatorial treatments/conditions on proline incorporation, an index of collagen production, by cardiac fibroblasts. Values are means and S.E.M.s.
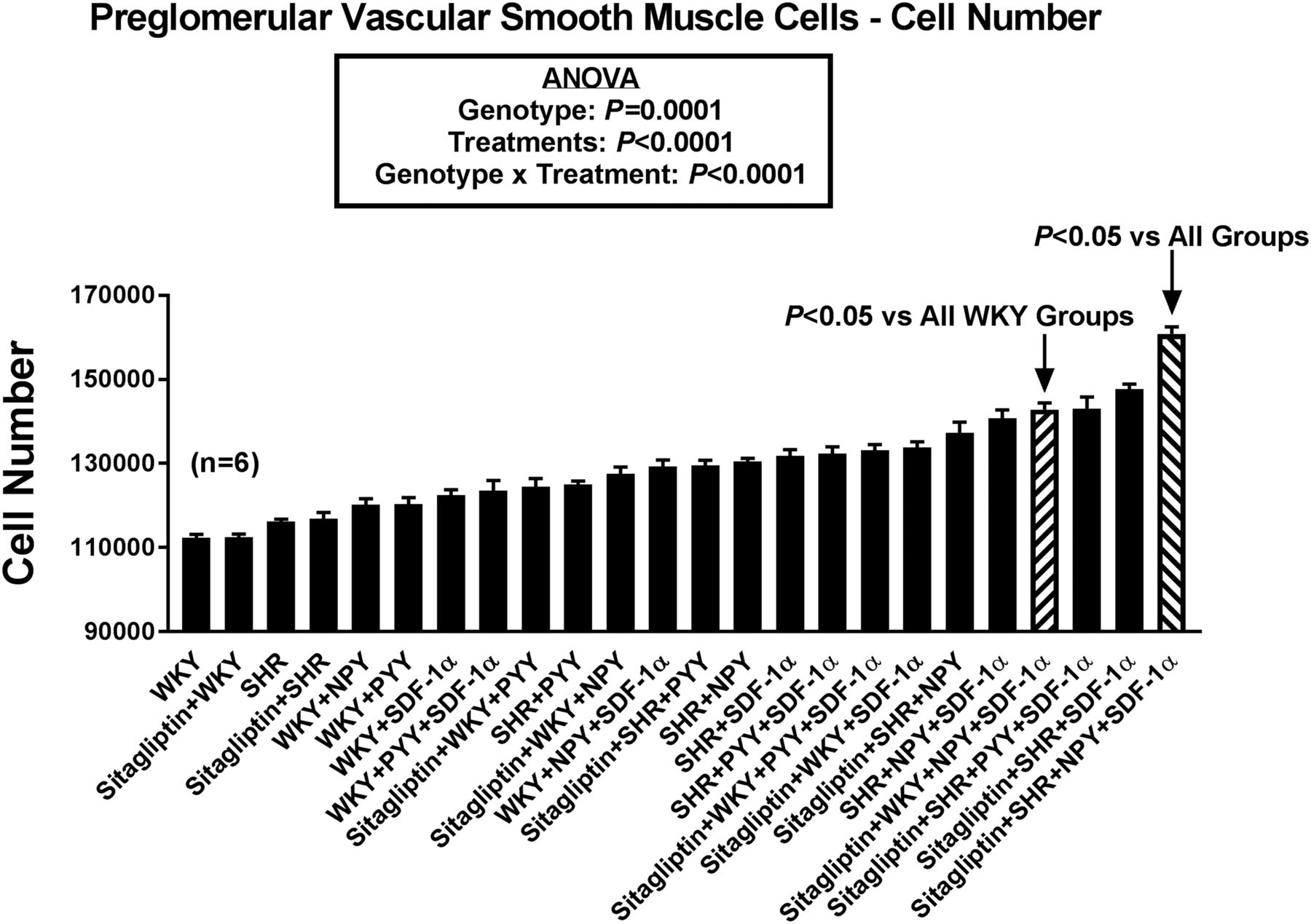
Bar graph compares effects of 24 combinatorial treatments/conditions on the proliferation of preglomerular vascular smooth muscle cells. Values are means and S.E.M.s.

Bar graph compares effects of 24 combinatorial treatments/conditions on proline incorporation, an index of collagen production, by preglomerular vascular smooth muscle cells. Values are means and S.E.M.s.

Bar graph compares effects of 24 combinatorial treatments/conditions on the proliferation of glomerular mesangial cells. Values are means and S.E.M.s.

Bar graph compares effects of 24 combinatorial treatments/conditions on proline incorporation, an index of collagen production, by glomerular mesangial cells. Values are means and S.E.M.s.
Proliferation Studies in PGVSMCs.
Figure 5 provides a summary of the effects of the 24 different treatments on proliferation of PGVSMCs. Statistical analysis by ANOVA revealed an overall effect of cell genotype (P = 0.0001) and treatments (P < 0.0001) on PGVSMC proliferation. Moreover, there was a significant genotype × treatment interaction (P < 0.0001). Bonferroni tests were conducted to compare all possible pairs and the results are shown in Supplemental Table 4. Bonferroni tests showed that proliferation was significantly greater in WKY PGVSMCs treated with sitagliptin + NPY1–36 + SDF-1α compared with all other WKY PGVSMC groups and showed that compared with all of the other 23 groups, the most proliferative combination was SHR PGVSMCs treated with sitagliptin + NPY1–36 + SDF-1α. This analysis also demonstrated significantly increased proliferation in the order: WKY = SHR < SHR + NPY1–36 < SHR + NPY1–36 + SDF-1α < sitagliptin + SHR + NPY1–36 + SDF-1α; and WKY = SHR < SHR + PYY1–36 < SHR + PYY1–36 + SDF-1α < sitagliptin + SHR + PYY1–36 + SDF-1α.
[3H]-Proline Incorporation Studies in PGVSMCs.
Figure 6 summarizes the relative effects of the 24 different treatment conditions on [3H]-proline incorporation by PGVSMCs. ANOVA showed that genotype (P < 0.0001) and treatments (P < 0.0001) significantly increased [3H]-proline incorporation by PGVSMCs and that there was a significant interaction between genotype and treatments (P = 0.0011). Bonferroni tests were conducted to compare all possible pairs and the results are shown in Supplemental Table 5. Bonferroni tests showed that [3H]-proline incorporation was significantly greater in WKY PGVSMCs treated with sitagliptin + NPY1–36 + SDF-1α compared with all other WKY PGVSMC groups and showed that compared with all of the other 23 groups, [3H]-proline incorporation was greatest in SHR PGVSMCs treated with sitagliptin + NPY1–36 + SDF-1α. This analysis also demonstrated significantly increased [3H]-proline incorporation in the order: WKY < SHR < SHR + NPY1–36 < SHR + NPY1–36 + SDF-1α < sitagliptin + SHR + NPY1–36 + SDF-1α; and WKY < SHR < SHR + PYY1–36 < SHR + PYY1–36 + SDF-1α = sitagliptin + SHR + PYY1–36 + SDF-1α.
Proliferation Studies in GMCs.
Statistical analysis by ANOVA (Fig. 7) showed that genotype (P < 0.0001) and treatments (P < 0.0001) significantly increased proliferation of GMCs and that there was a significant interaction between genotype and treatments (P < 0.0001). Bonferroni tests were conducted to compare all possible pairs and the results are shown in Supplemental Table 6. Bonferroni tests showed that proliferation was significantly greater in WKY GMCs treated with sitagliptin + NPY1–36 + SDF-1α compared with all other WKY GMC groups and showed that compared with all of the other 23 groups, proliferation was greatest in SHR GMCs treated with sitagliptin + NPY1–36 + SDF-1α. This analysis also demonstrated significantly increased proliferation in the order: WKY < SHR < SHR + NPY1–36 < SHR + NPY1–36 + SDF-1α < sitagliptin + SHR + NPY1–36 + SDF-1α; and WKY < SHR < SHR + PYY1–36 < SHR + PYY1–36 + SDF-1α < sitagliptin + SHR + PYY1–36 + SDF-1α.
[3H]-Proline Incorporation Studies in GMCs.
Figure 8 summarizes the relative effects of the 24 different treatment conditions on [3H]-proline incorporation by GMCs. ANOVA showed that treatments (P < 0.0001) significantly increased [3H]-proline incorporation by GMCs and that there was a significant interaction between genotype and treatments (P = 0.0006). Bonferroni tests were conducted to compare all possible pairs and the results are shown in Supplemental Table 7. Bonferroni tests showed that [3H]-proline incorporation was significantly greater in WKY GMCs and SHR GMCs treated with sitagliptin + NPY1–36 + SDF-1α compared with all other GMC groups except each other and the SHR + NPY1–36 + SDF-1α group.
Proliferation Studies in Female versus Male CFs.
In general, the results described above indicate that NPY1–36 and SDF-1α separately stimulate cell proliferation and [3H]-proline incorporation and that the two combined are more efficacious than each separately. Our results also indicate that inhibiting DPP4 with sitagliptin further augments the effects of NPY1–36 plus SDF-1α, as does a hypertensive genetic background.
Another important biologic variable is sex. To explore whether NPY1–36 and SDF-1α stimulate proliferation in female cells, we examined the effects of these two peptides alone and in combination in female CFs. In these studies, we used SHR CFs because CFs with a hypertensive genetic background respond more robustly than cells from normotensive animals. We conducted these experiments in both the absence and presence of sitagliptin to determine whether in female cells the effects of these peptide treatments are augmented by inhibition of DPP4. For comparison, we included a new group of male CFs in the same experimental series with the female CFs.
Figure 9 summarizes the effects of NPY1–36 alone, SDF-1α alone, and NPY1–36 + SDF-1α in female SHR CFs without (Fig. 9A) and with (Fig. 9B) sitagliptin. Figure 9, C and D show comparable data for male SHR CFs. Importantly, in female SHR CFs, analysis by ANOVA revealed a significant effect of the peptide treatments (P < 0.0001) and sitagliptin (P < 0.0001) on CF proliferation and demonstrated a significant interaction between peptide treatments and sitagliptin (P = 0.0026). Also, the combination of NPY1–36 + SDF-1α was more effective with regard to stimulating proliferation compared with each of the peptides per se. Overall, the effects of NPY1–36, SDF-1α and NPY1–36 + SDF-1α in the absence and presence of sitagliptin were similar in female and male CFs.

Bar graphs compares the pro-proliferative effects of neuropeptide Y1–36 alone, SDF-1α alone, and NPY + SDF-1α in female cardiac fibroblasts from SHRs without (A) and with (B) sitagliptin. (C and D) Comparable data for male SHR CFs are shown. Values are means and S.E.M.s. The letters a, b, c, and d indicate significantly different from control, NPY-treated, SDF-1α–treated, and NPY + SDF-1α-treated cells, respectively.
This experiment with female and male CFs confirms that the combination of DPP4 inhibition, NPY1–36 and SDF-1α is a powerful stimulus of SHR CF proliferation and that sex chromosomes per se do not affect the pro-proliferative effects of this combination. However, these experiments were performed in the absence of female hormones. Our previous studies show that 17β-estradiol is a potent inhibitor of cell proliferation (CFs, GMCs and vascular smooth muscle cells) induced by FBS and that this effect of 17β-estradiol is mediated entirely by the conversion of 17β-estradiol to 2-methoxyestradiol (2ME) (Dubey et al., 1998, 2000, 2001, 2003a,b, 2004, 2007; Xiao et al., 2001; Zacharia et al., 2001, 2002, 2003; Barchiesi et al., 2002, 2006, 2010; Dubey and Jackson, 2009; Rigassi et al., 2015), a major metabolite of 17β-estradiol. Whether physiologic concentrations of 2ME abrogate the combined effects of DPP4 inhibition, NPY1–36, and SDF-1α is an open question. To address this question, we compared the effects of vehicle-treated (control) SHR CFs versus SHR CFs treated with sitagliptin + NPY1–36 + SDF-1α versus SHR CFs treated with 2ME + sitagliptin + NPY1–36 + SDF-1α. As shown in Fig. 10, 1 nM of 2ME completely prevented the pro-proliferative effects of sitagliptin + NPY1–36 + SDF-1α in both male and female SHR CFs.

Bar graphs compares the pro-proliferative effects of neuropeptide Y1–36 (NPY) plus SDF-1α plus sitagliptin in male (A) and female (B) cardiac fibroblasts from SHRs in the absence and presence of 1 nM of 2-methoxyestradiol (2ME). Values are means and S.E.M.s. The letters a and b indicate significant difference from control and 2ME-treated cells, respectively.
Assessment of Release of Collagen I Into the Extracellular Compartment by Sitagliptin.
After synthesis at and simultaneous transport into the rough endoplasmic reticulum, collagens are processed in the Golgi apparatus and compartmentalized into secretory granules that under exocytosis, which results in the release of collagens into the extracellular compartment (Rodriguez-Feo et al., 2005). Because [3H]-proline is incorporated into collagens at the rough endoplasmic reticulum, [3H]-proline incorporation permits assessment of total collagen production, regardless of collagen isoform or location within the membrane-delimited secretory pathway. However, [3H]-proline can be incorporated into noncollagen proteins. Although removal of noncollagen proteins from collagen proteins can be accomplished using a multistep procedure (Peterkofsky and Diegelmann, 1971; Peterkofsky, 1972), this method relies upon pan-antibodies and requires a large number of sample manipulations. These drawbacks decrease sensitivity and increase variability. An alternative method, and the one employed here, is to measure [3H]-proline incorporation in confluent/quiescent cells to minimize [3H]-proline incorporation into noncollagen proteins. However, confluent/quiescent cells produce less collagen (Peterkofsky, 1972) and secrete only approximately 5% of synthesized collagen (Peterkofsky, 1972). Consequently, assessment of secreted collagen using [3H]-proline incorporation in confluent/quiescent cells is problematic. Here we applied an alternative method using a sandwich ELISA assay to measure specifically the effects of sitagliptin on secreted collagen I. Collagen I was specifically targeted because this collagen isoform is one of the most abundantly expressed in cardiac fibroblasts (Namba et al., 1997). In these experiments, collagen I was measured in both cell lysates (which measures free collagen within the cell that is not membrane delimited and therefore can bind to the capture and detection antibodies) and conditioned medium (which measures collagen secreted into the extracellular compartment). The ratio of extracellular to intracellular collagen I was calculated as an index of secretion efficiency. Shown in Fig. 11, in cells treated with DPP4 substrates, sitagliptin significantly decreased intracellular (cytosolic) collagen I (Fig. 11A; P = 0.0034), increased extracellular collagen I (Fig. 11B; P = 0.0130), and increased the ratio of extracellular to intracellular collagen (Fig. 11C; P = 0.0058).

Bar graphs show the effects of sitagliptin on collagen I secretion by cardiac fibroblasts from spontaneously hypertensive rats that were cotreated with DPP4 substrates (neuropeptide Y1–36 and stromal cell–derived factor-1α). Sitagliptin decreased intracellular (likely cytosolic) collagen I (A) while simultaneously increasing extracellular levels of collagen I (B). Thus, the ratio of extracellular to intracellular (cytosolic) collagen I was markedly increased (C), suggesting increased efficiency of collagen I packaging and secretion via exocytosis. Values are means and S.E.M.s.
Discussion
The pharmacological effects of DPP4Is are context dependent (Jackson, 2017). For example, whether DPP4Is increase, decrease, or have no effect on blood pressure in rats is strain dependent (Jackson et al., 2015). Similarly, in metabolic-syndrome patients, DPP4Is can induce antihypertensive or prohypertensive effects depending on the degree of ACE inhibition (Marney et al., 2010). Our working hypothesis is that the context-dependent effects of DPP4Is are due to the fact that DPP4 metabolizes many biologically active peptides (Mentlein, 1999; Gorrell, 2005; Klemann et al., 2016). Consequently, the overall effects of DPP4Is depends on the complex milieu of DPP4 substrates. Indeed, studies in humans indicate that DPP4Is can alter the physiologic responses to several DPP4 substrates including substance P (Devin et al., 2014), growth hormone-releasing hormone (Wilson et al., 2018) and NPY1–36 (Hubers et al., 2018).
Our laboratory has been investigating whether DPP4 substrates can influence the proliferation of, and collagen production by, CFs, PGVSMCs, and GMCs. This work revealed that DPP4 inhibition enhances the progrowth effects of NPY1–36 and PYY1–36 (mediated via Y1 receptors) (Jackson et al., 2012; Zhu et al., 2015b), as well as the progrowth effects of SDF-1α (mediated by CXCR4 receptors) (Jackson et al., 2017). In these studies, our focus was on the proliferation of, and collagen production by, CFs, PGVSMCs, and GMCs because such responses by these cell types can lead to cardiac and renal fibrosis and dysfunction. The clinical relevance of our findings is underscored by the facts that several randomized clinical trials, observational studies and US Food and Drug Administration evaluation support the conclusion that DPP4Is increase the risk of heart failure (Packer, 2018).
Our previous results with NPY1–36, PYY1–36, and SDF-1α suggest that the effects of DPP4 inhibitors on cellular activity of CFs, PGVSMCs, and GMCs may depend on the combination of effects of multiple stimuli including combinations of peptides and genetic background. Thus, the explicit goal of the present study was to evaluate this hypothesis by conducting a careful head-to-head comparison of cell proliferation and collagen production in CFs, PGVSMCs, and GMCs under highly controlled conditions in which cells were exposed to combinations of treatment conditions.
Important aspects of our experimental design were: 1) the selection of concentrations of NPY1–36, PYY1–36, and SDF-1α; 2) the utilization of cells from both genetically normotensive and hypertensive animals; 3) the rigorous adherence to appropriate biologic replicates; and 4) the assessment of both total cellular collagen in nonproliferating cells, as well as secreted collagen I in proliferating (active) cells. The concept we addressed was that even with very low concentrations of DPP4 substrates, under the appropriate combinatorial conditions (including genetic hypertension) cellular activity can be markedly influenced by DPP4Is. This, we felt, would model the in vivo condition in which several factors combined can have biologically, and possible clinically, meaningful effects that are much larger than the effects of any one given factor. Here we chose 3 nM because with all three peptides, this concentration provides a threshold stimulation of cell growth in WKY cells (Zhu et al., 2015b; Jackson et al., 2017) and approximates achievable in vivo tissue levels of NPY1–36, PYY1–36, and SDF-1α (Schmidt et al., 2005; Kuncová et al., 2011; Roux et al., 2012). The fact that we were able to detect significant effects at these low concentrations speaks to the potency and likely in vivo importance of these peptides in stimulating cellular activity, particularly when combined with additional treatment conditions.
A major finding of the current study is that in all three cell types there were accumulative effects of treatments such that the combinations of treatments increased cellular activity more than individual treatments. In general, in all three cell types, cellular activity was increased in a step-wise fashion by hypertensive genotype, the addition of NPY1–36 or PYY1–36 to the hypertensive genotype, the further addition of SDF-1α to the hypertensive genotype treated with NPY1–36 or PYY1–36 and finally by the addition of sitagliptin to the hypertensive genotype treated with NPY1–36 or PYY1–36 in the presence of SDF-1α.
In general, the most efficacious stimulus of proliferation and collagen production (assessed by [3H]-proline incorporation) was the combination of a hypertensive genotype cotreated with sitagliptin + NPY1–36 + SDF-1α. As noted, our method of [3H]-proline incorporation does not allow for an accurate assessment of secreted collagens. Importantly, however, we observed a statistically significant sitagliptin-induced increase in extracellular collagen I levels (measured by a sandwich ELISA) in SHR CFs conditioned with the DPP4 substrates NPY1–36 and SDF-1α. In these experiments, sitagliptin also markedly increased the ratio of extracellular to intracellular (cytosolic) collagen I. This suggests that activation of collagen I synthesis by DPP4Is plus DPP4 substrates improves the efficiency of collagen I packaging by the ER/Golgi system and secretion into the extracellular compartment, with reduced “spillage” of collagen I into the cytosol.
The relevance of our finding is highlighted by the facts that in heart patients SDF-1α levels are increased and are associated with heart failure, cardiac fibrosis, and all-cause mortality (Chu et al., 2010; Subramanian et al., 2014; Zuern et al., 2015). Studies in mice reveal that antagonism of the CXCR4 receptor (receptor for SDF-1α) promotes myocardial tissue repair and reduces myocardial scarring after myocardial infarction (Wang et al., 2019). The significance of our findings is also supported by the fact that the release of NPY1–36 from sympathetic nerves is augmented in heart failure (Hulting et al., 1990; Kaye et al., 1994) and in patients with chronic kidney disease, plasma NPY is an strong predictor of cardiovascular events (Zoccali et al., 2019). Moreover, often patients with heart disease have the comorbidity of hypertension. Consequently, the setting of a hypertensive genotype combined with elevated levels of NPY1–36 and SDF-1α is a realistic, and likely, scenario.
In addition to hypertension with elevated levels of NPY1–36 and SDF-1α, heart patients often have type 2 diabetes as an additional comorbidity. Thus, such patients would be candidates for treatment with DPP4Is. The current study suggests that it is patients with this combination of conditions (hypertension with elevated levels of NPY1–36 and SDF-1α) that may be at highest risk of adverse effects due to chronic administration of DPP4Is. With regard to the heart, the present data suggest that DPP4Is may have direct adverse effects on the myocardium by promoting proliferation of, and collagen production by, CFs. As reviewed by Packer (2018), the deleterious cardiac effects of DPP4Is may also be indirect. In this regard, DPP4Is augment the actions of SDF-1α, NPY1-36, and substance P in the central nervous system to increase sympathetic nerve traffic. This may, via β-adrenoceptor activation, elicit cardiomyocyte injury. Because sympathetic activation would also release NPY1–36 into the myocardium, this would reinforce the direct adverse actions of DPP4Is as described herein. Thus, these indirect (via the CNS) and direct (via stimulation of CFs) mechanisms may interact to increase the risk of DPP4I-induced adverse cardiac effects.
Sex is another important biologic variable that may influence proliferation of, and collagen production by, cardiac and renal cells. Thus, it is conceivable that the effects of DPP4Is in females with genetic hypertension and elevated levels of NPY1–36 and SDF-1α may be different than the effects in males. To test this, we compared the effects of NPY1–36 versus SDF-1α versus NPY1–36 + SDF-1α in female and male SHR CFs treated or not with sitagliptin. These experiments revealed that female CFs are responsive to these progrowth stimuli.
Although our in vitro findings indicate that sex chromosomes per se do not influence the responsiveness of CFs to DPP4Is, NPY1–36, SDF-1α, or their combination, these experiments were performed in cell culture medium devoid of sex hormones. This leaves open the question as to whether in vivo sex hormones protect premenopausal females from the growth-promoting effects of DPP4Is, NPY1–36, SDF-1α, or their combination. Our previous studies show that 17β-estradiol, the major ovarian estrogen, is a potent inhibitor of CF, GMC, and vascular smooth muscle cell proliferation induced by serum. Moreover, these effects of 17β-estradiol are mediated by the conversion of 17β-estradiol to 2ME (Dubey et al., 1998, 2000, 2001, 2003a,b, 2004, 2007; Xiao et al., 2001; Zacharia et al., 2001, 2002, 2003; Barchiesi et al., 2002, 2006, 2010; Dubey and Jackson, 2009; Rigassi et al., 2015), a major nonestrogenic metabolite of 17β-estradiol. Therefore, it is conceivable that because of endogenous 2ME, premenopausal women are resistant to the growth-promoting effects of DPP4 inhibition, NPY1–36, SDF-1α, or their combination. Consistent with the notion that 2ME protects against the progrowth effects of DPP4 inhibition, NPY1–36, SDF-1α, or their combination, we observed that a very low concentration (1 nM) of 2ME completely blocked the progrowth effects of the most efficacious growth-promoting combination (i.e., genetic hypertension + DPP4 inhibition + NPY1–36 + SDF-1α).
Previous studies by us and others show that 2ME and its metabolic precursor 2-hydroxyestradiol (2HE) are antidiabetic and exhibit antifibrotic and antihypertensive effects in various models of hypertension and related cardiovascular and renal injury and fibrosis. In this regard, 2HE reverses the metabolic syndrome (Tofovic et al., 2001); in mice on a high-fat diet, genetic deficiency, or inhibition of COMT (key enzyme for 2HE to 2ME conversion) leads to insulin resistance and 2ME treatment improves metabolic derangements (Kanasaki et al., 2017); and 2ME improves glucose tolerance in db/db mice (Yorifuji et al., 2011). Furthermore, in vivo, 2ME and/or 2HE attenuate cardiac, renal, lung, and systemic fibrosis induced by profibrotic toxins (puromycin, bleomycin), NO deficiency or by angiotensin II (Tofovic et al., 2009; Zhu et al., 2015a; Maayah et al., 2018; Salah et al., 2019). Finally, 2ME reduces blood pressure in rats with genetically, metabolic syndrome-, NO deficiency-, DOCA-, or Ang II-induced hypertension (Bonacasa et al., 2008; Yuan et al., 2013; Maayah et al., 2018; Salah et al., 2019). Therefore, the combination of 2ME and DPP4Is may be a highly effective strategy to manage type 2 diabetes and prevent its sequelae.
Although the present study explores conditions and aspects of DPP4 inhibition that may contribute to unwanted effects of this class of drugs, there is strong evidence that DPP4Is also promote beneficial effects, likely by augmenting cardioprotective hormones such as glucagon-like peptide-1 (GLP-1) (Esposito et al., 2017). As recently reviewed by Nistala and Savin (2017), there is abundant preclinical evidence indicating that DPP4Is protect the heart, vasculature, and kidneys, at least in rodent models of obesity, diabetes and hypertension. For example, DPP inhibition protects against renal injury (including fibrosis) in diabetic, eNOS knockout mice (Gangadharan Komala et al., 2016), mice with ureteral obstruction (Min et al., 2014), and streptozotocin-induced diabetes CD-1 mice (Shi et al., 2015); DPP inhibition also prevents cardiac fibrosis in Dahl salt sensitive rats (Esposito et al., 2017). Nonetheless, two recent, large, randomized, placebo-controlled multicenter studies with linagliptin show that in patients with type 2 diabetes, DPP4 inhibition does not provide cardiovascular or renal benefits (Rosenstock et al., 2019a,b). Avogaro and Fadini (2018) conclude that DPP4Is have pleiotropic effects mediated by diverse DPP4 substrates that are offsetting, thus accounting for the discrepancies between animal and human (involving >40,000 patients with type 2 diabetes) studies. This situation is described by Jackson as “context-dependent” effects of DPP4Is (Jackson, 2017). Indeed, DPP4Is may even exert antifibrotic indirect and direct effects that promote wound healing (Marfella et al., 2012; Long et al., 2018) and reduce fibrosis in systemic sclerosis (Soare et al., 2020).
There are several limitations of the present study. First, the experiments were carried out in vitro. Given the large number of groups under study and the variability associated with in vivo experiments, the questions addressed in the present study could not be practically approached with in vivo experiments. Nonetheless, extrapolation to patients must be taken with circumspection and more focused in vivo experiments, guided by the present findings, are required. Second, here we concentrated on what we judged as three of the most important “off-target” DPP4 substrates, namely NPY1–36, PYY1–36, and SDF-1α, that could be potential druggable targets for improving DPP4I pharmacotherapy. However, there are at least 50 DPP4 substrates, some of which are considered physiologic (e.g., substance P), others pharmacological (Mulvihill and Drucker, 2014). In addition, DPP4 physically interacts with a number of binding partners (e.g., adenosine deaminase, CXCR4, integrin β1, sodium-hydrogen exchanger-3, CD45, fibronectin, collagen, caveolin-1, mannose-6-phosphate/insulin-like growth factor II receptor) (Kanasaki, 2018). Thus, the number of possible permutations involving DPP4 substrates and binding partners is extremely large. Again, the goal of the present study was limited to permutations involving hypertension, NPY1–36, PYY1–36, SDF-1α, and DPP4. We acknowledge that many other permutations of DPP substrates (particularly substance P) and binding partners may influence the net effect of DPP4Is (and hence contribute to “context dependence”). Finally, although the SHR has been used in >17,000 published studies, the appropriate genetic control for this strain remains controversial, although most investigators use the corresponding WKY strain.
In summary, our study demonstrates that CFs, PGVSMCs, and GMCs are activated by combinations of conditions, the most powerful of which is the coexistence of NPY1–36, SDF-1α, DPP4 inhibition, and a hypertensive genotype. We hypothesize that as these progrowth conditions accumulate, a tipping point may be reached that manifests in the worst case as cardiac or renal dysfunction and in the best case as offsetting the beneficial effects of DPPIs. This could explain why some clinical studies confirm an increased risk of adverse cardiac effects of DPP4Is (Scirica et al., 2014; Zannad et al., 2015; McMurray et al., 2018), whereas others do not (Green et al., 2015; Gantz et al., 2017). Our experiments suggest that the full beneficial effects of DPP4Is could be maximized by combining DPP4Is with 2ME.
Authorship Contributions
Participated in research design: Jackson, Gillespie, Tofovic.
Conducted experiments: Gillespie.
Performed data analysis: Jackson.
Wrote or contributed to the writing of the manuscript: Jackson, Tofovic.
Footnotes
- Received October 25, 2019.
- Accepted January 30, 2020.
The work was supported by the National Institutes of Health [DK091190, HL069846, DK068575, HL109002, and DK079307].
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- CF
- cardiac fibroblast
- DPP4
- dipeptidyl peptidase 4
- DPP4Is
- dipeptidyl peptidase 4 inhibitors
- GMCs
- mesangial glomerular cells
- 2HE
- 2-hydroxyestradiol
- 2ME
- 2-methoxyestradiol
- NPY
- neuropeptide Y
- PDGF-BB
- platelet-derived growth factor–BB
- PGVSMC
- preglomerular vascular smooth muscle cell
- PYY
- peptide YY
- SDF-1α
- stromal cell–derived factor 1α
- SHR
- spontaneously hypertensive rat
- WKY
- Wistar-Kyoto rat
- Y1R
- Y1 receptor
- Copyright © 2020 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}