


Abstract
Aucubin (AUB), which is extracted from Eucommia ulmoides Oliver seeds, has been found to possess anti-inflammatory and antiapoptotic properties. Recent studies have indicated that inflammation, oxidative stress, and apoptosis are involved in the pathophysiology of lipopolysaccharide (LPS)-induced cardiac dysfunction. Our study aimed to investigate the effect of AUB on LPS-induced acute cardiac injury. Male C57BL/6 mice were injected with LPS (one 6 mg/kg injection) to induce cardiac dysfunction without or with AUB pretreatment (20 or 80 mg/kg per day) for 1 week. We found that AUB ameliorated cardiac dysfunction, inflammation, oxidative stress, and apoptosis induced by LPS stimulation. Mechanistically, AUB inhibited LPS-induced oxidative stress by decreasing reactive oxygen species and thioredoxin interaction protein (TXNIP) levels. Moreover, AUB suppressed LPS-induced inflammation and apoptosis by reducing nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3)/apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC)/caspase-1 inflammasome formation. Overexpression of NLRP3 in cardiomyocytes attenuated the protective effects of AUB. Interestingly, NLRP3 deficiency ameliorated cardiac function and reduced the inflammatory response and oxidative stress after LPS insult in mice, whereas AUB could not further prevent LPS-induced cardiac dysfunction in NLRP3-deficient mice. In summary, AUB exerts a protective effect against LPS-induced inflammation, oxidative stress, and apoptosis in vivo and in vitro by regulating the TXNIP pathway and inactivating the NLRP3/ASC/caspase-1 inflammasome. Hence, AUB may be a promising agent against LPS-induced cardiac dysfunction.
SIGNIFICANCE STATEMENT Aucubin exerts a protective effect against lipopolysaccharide-induced cardiac dysfunction by regulating nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 inflammasome.
Introduction
Sepsis is a fatal clinical disease caused by infection that can lead to severe multiple organ dysfunction syndrome (Mayr et al., 2014). Sepsis-induced cardiomyopathy (SIC) is a global but reversible dysfunction that features left ventricular (LV) dilatation and LV ejection fraction (LVEF) reduction, and patients recover in 1 to 2 weeks (Sato and Nasu, 2015). Pathogen-associated molecular patterns released from infecting organisms and damage-associated molecular patterns released from dead and damaged cells bind immune receptors on inflammatory cells and heart cells, which causes excessive inflammation by activating nuclear factor κ-B (NF-κB) (Durand et al., 2017). Increased levels of proinflammatory cytokines and oxidative stress can induce mitochondrial damage, calcium regulation disorders, and endothelial dysfunction (Liu et al., 2017). Thus, suppression of inflammation and oxidative stress is considered a potential therapy for SIC.
Reactive oxygen species (ROS) activate the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome; activated NLRP3 plays a vital role in the pathophysiology of diabetic cardiomyopathy (Luo et al., 2014). Thioredoxin (TRX) interaction protein (TXNIP) is an endogenous inhibitor of the ROS-scavenging protein TRX that is released from the TXNIP–TRX complex and specifically binds to the NLRP3 inflammasome to activate and release caspase-1 and interleukin (IL)-1β (Koenen et al., 2011). The NLRP3 inflammasome is the best-characterized inflammasome that responds to pathogen-associated molecular patterns, damage-associated molecular patterns, and ROS. The inflammasome forms primarily through interactions between NLRP3 and apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and then recruits and activates procaspase-1 to produce activated caspase-1. Activated caspase-1 cleaves pro–IL-1β into active IL-1β to exert the effect of IL-1β–converting enzyme (Franchi and Núñez, 2012). Mature IL-1β is an important mediator of the recruitment of innate immune cells to sites of infection and the induction of additional proinflammatory mediators, and it contributes to the amplification of inflammation (Chen and Nuñez, 2010). Activation of NLRP3 inflammasome pathways in cardiovascular diseases, including hypertension, atherosclerosis, ischemic injury, SIC, and myocardial infarction, has been well demonstrated (Toldo et al., 2015; Kalbitz et al., 2016; Bullón et al., 2017; Hoseini et al., 2018). Previous studies suggested that NLRP3 inhibition attenuates cardiac dysfunction in sepsis (Zhang et al., 2014b; Kalbitz et al., 2016). Therefore, inhibition of NLRP3 signaling may be a therapeutic target for SIC.
Aucubin (AUB) can be isolated from Eucommia ulmoides Oliver seeds, and it exerts multiple pharmacological effects (Yang et al., 2017). There is reliable evidence that AUB lowers blood pressure (Zhang et al., 2014a), protects against liver injury (Chang, 1998), and exhibits anti-inflammatory (Jeong et al., 2002) and analgesic activity (Kim et al., 2014). Initially, AUB was reported to exert anti-inflammatory effects via inhibition of NF-κB signaling (Jeong et al., 2002; Park and Chang., 2004; Wang et al., 2015) and to prevent IL-1β – and tumor necrosis factor (TNF)-α–induced inflammation in many cell types, such as rat articular chondrocytes (Wang et al., 2015) and RAW 264.7 cells (Park and Chang., 2004). In addition, AUB treatment markedly decreases oxidative stress, which may be attributable to its functions in inhibiting ROS formation and reducing malondialdehyde (MDA) levels (Ho et al., 2005). AUB has been indicated to reduce apoptosis via activation of the estrogen receptor β pathway (Li et al., 2016). Our previous research demonstrated that AUB protects against pressure overload-induced cardiac hypertrophy, which supports the cardiac protective effects of AUB (Wu et al., 2018). However, the role of AUB in lipopolysaccharide (LPS)-induced cardiac dysfunction and the precise mechanisms remain unclear. In this study, we aimed to clarify the possible effects of AUB on LPS-induced cardiac dysfunction.
Materials and Methods
Animals and Animal Models.
All animal experimental protocols were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University (approval number: WHRM-2016-1012-012) and were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health. Male C57/B6 mice aged 8–10 weeks (23.5–27.5 g) were purchased from the Chinese Academy of Medical Sciences (Beijing, China). NLRP3-knockout (KO) mice were purchased from Jackson Laboratory of Nanjing University. The mice were randomly grouped into either normal saline injection groups or LPS-treated groups (receiving one 6 mg/kg injection of LPS) without or with AUB pretreatment [dissolved in sterile saline and administered by gavage; low dose (LD): 20 mg/kg body weight per day; high dose (HD): 80 mg/kg body weight per day] for 7 days. The control group mice were given equal volumes of sterile saline. After 6 hours, the mice were killed to obtain the hearts.
Echocardiography.
After the mice were anesthetized with 1.5%–2% isoflurane and placed on a 37°C temperature-controlled warming pad, cardiac function was detected with a MyLab 30CV ultrasound machine (Esaote SpA) and a 10-MHz linear array ultrasound transducer, as reported previously (Wu et al., 2016, 2018). The parameters measured included LV end-systolic dimension (LVESd), LV end-diastolic dimension (LVEDd), LV end-systolic posterior wall thickness, LV end-diastolic posterior wall thickness, LVEF, and LV fractional shortening (LVFS).
Histologic Analysis.
As reported previously (Wu et al., 2016; Xiao et al., 2017), the hearts were immediately placed in 10% potassium chloride solution to maintain the diastolic phase, fixed in formalin, and then embedded in paraffin. Each heart was cut into slices with thicknesses of 4 to 5 μm. To assess the extent of collagen deposition, picrosirius red (PSR) staining was performed. The percentage of collagen content was calculated by dividing the PSR-stained area by the total area. For immunohistochemistry, after xylene immobilization and alcohol concentration gradient dehydration, the citric acid method was used to repair the antigens of the heart sections. Then the slides were incubated with anti-CD68 antibody (ab125212; Abcam) at 1:100 dilution, anti-CD45 antibody (ab10558; Abcam) at 1:100 dilution, and anti–4-hydroxynonenal antibody (ab46545; Abcam) at 1:100 dilution overnight. Diaminobenzidine kit was used to develop the cardiac tissue. A light microscope (Nikon, Japan) was used to photograph the heart sections.
Real-Time Polymerase Chain Reaction and Western Blot.
TRIzol reagent (Invitrogen) was used to collect total RNA from LV tissues or cardiomyocytes. Oligo(dT) primers, a cDNA synthesis kit (Roche, Germany), and 2 μg RNA were used to obtain cDNA. A LightCycler 480 system and SYBR Green 1 Master Mix (Roche) were used to conduct real-time polymerase chain reaction. Target gene expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression.
Radioimmunoprecipitation assay buffer was used to extract proteins from cardiac tissues and cardiomyocytes, and a bicinchoninic acid protein assay kit was used to detect the protein concentration. The separated proteins were transferred to polyvinylidene fluoride membranes (Millipore), which were incubated with the following primary antibodies: Bax (2722; Cell Signaling Technology), Bcl-2 (2870; Cell Signaling Technology), cytochrome c (4272; Cell Signaling Technology), superoxide dismutase (SOD)2 (ab13533; Abcam), P67 (ab109366; Abcam), gp91 (ab129068; Abcam), TRX-2 (14907; Cell Signaling Technology), TXNIP (ab188865; Abcam), NLRP3 (ab214185; Abcam), ASC (ab47092; Abcam), c-caspase-1 (ab1872; Abcam), IL-1β (sc-52012; Santa Cruz), and GAPDH (2118; Cell Signaling Technology). On the second day, the membranes were washed three times with PBS and incubated with secondary antibodies for 1 hour. The membranes were then scanned and analyzed with a two-color infrared imaging system (Odyssey). The results were normalized to the GAPDH levels.
Neonatal Rat Cardiomyocyte Culture.
Neonatal rat cardiomyocytes (NRCMs) were obtained based on the methods in a previous study (Jiang et al., 2014) and incubated with DMEM/F12 (C11330; Gibco) supplemented with 10% FBS. To evaluate the effect of AUB on LPS-induced injury, NRCMs were seeded in six-well, 24-well, and 96-well plates for 48 hours. The cells were treated with LPS (10 μg/ml) with or without AUB (5, 15, or 45 μM) for 24 hours. To assess the role of NLRP3 in the effect of AUB, the cells were also infected with an adenovirus (Ad) engineered to overexpress NLRP3 (Ad-NLRP3; multiplicity of infection = 100; obtained from Vigene).
Cell-Counting Kit-8 Assay.
NRCMs cultured in a 96-well plate at 37°C and NRCM suspensions of 100 ml/well were prepared. Then the NRCMs were starved in serum-free medium. A volume of 10 μl cell-counting kit-8 solution was added to each well, and the plate was incubated in an incubator for 2 to 3 hours. Then the absorption was detected at 450 nm.
Detection of Oxidative Stress in Cardiomyocytes.
The 2,7-dichlorodi-hydrofluorescein diacetate was used to assess the expression of ROS based on the manufacturer’s instructions. NRCMs were seeded in 24-well plates for 48 hours. After discarding the medium and washing the cells three times with PBS, the medium was replaced and the cells were incubated with 2,7-dichlorodi-hydrofluorescein diacetate (10 μM) for 30 minutes at 37°C, while protected from light. A fluorescence microplate reader was used to detect immunofluorescence.
After discarding the medium and washing the cells three times with PBS, the NRCM suspensions were centrifuged, and the supernatant was discarded. Then the cells were collected. Total SOD, NADPH oxidase, and MDA assay kits were used to detect the activity of SOD and NADPH oxidase and the levels of MDA, respectively, according to the manufacturer’s instructions.
Terminal Deoxynucleotidyl Transferase–Mediated Deoxyuridine Triphosphate–Biotin Nick End–Labeling Staining.
Cardiac tissue slices and cultured cells were prepared. An apoptosis Fluorescein Detection Kit (Millipore) was used to detect apoptotic cells. Briefly, the slides and cells were treated with proteinase K (20 μg/ml) for 20 minutes and incubated with deoxyuridine triphosphate for 60 minutes. The cardiomyocyte nuclei were stained with 4′,6′-diamidino-2-phenylindole. Image-Pro Plus 6.0 was used to calculate the results.
Statistical Analysis.
All data are presented as the mean ± S.D. SPSS 13.0 software was used to analysis the results. An unpaired Student’s t test was used for comparisons between two groups. Comparisons among three or more groups were conducted using one-way ANOVA. A value of P < 0.05 was considered to have statistical significance.
Results
AUB Attenuates Inflammation in NRCMs in Response to LPS.
Cardiomyocytes were stimulated with LPS and treated with different concentrations of AUB (0, 5, 15, and 45 μM). Cell viability was not affected by AUB at any concentration (Fig. 1A). The LPS-induced decrease in cell viability was attenuated by AUB pretreatment in a concentration-dependent manner (Fig. 1B). AUB also inhibited LPS-induced inflammatory cytokine (IL-1β, IL-18, IL-6, TNF-α) transcription in a dose-dependent manner (Fig. 1, C–F). These data indicated that AUB suppressed the LPS-induced inflammatory response. We selected 45 μM AUB for use in the next experiments.

AUB attenuates LPS-induced cardiomyocyte inflammation. Neonatal rat cardiomyocytes were stimulated with LPS (10 μg/ml) and treated with AUB (0, 5, 15, or 45 μM). (A and B) Cell viability (n = 6). (C-F) mRNA levels of TNF-α, IL-1β, IL-18, and IL-6 (n = 6).*P < 0.05 vs. the PBS group. #P < 0.05 vs. the LPS group.
AUB Inhibits LPS-Induced Oxidative Stress and Apoptosis in NRCMs.
Excessive oxidative stress, such as that caused by ROS and TXNIP, can induce cell injury (Wang et al., 2017b; Hou et al., 2018). Therefore, ROS levels and oxidation/redox system (SOD, NADPH, and MDA) were evaluated. LPS increased the levels of ROS and production of the lipid peroxidation intermediate MDA. Compared with the control group, NADPH oxidase activity was increased in the LPS group, whereas SOD activity was reduced in the LPS group (Fig. 2, A–C). AUB ameliorated oxidative stress, as indicated by reduced ROS levels and MDA production, reduced NADPH activity, and increased SOD activity in the AUB–LPS group compared with LPS group (Fig. 2, A–C). Excessive oxidative stress induces cell injury, which ultimately causes cell death. Thus, we detected apoptosis by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate–biotin nick end–labeling staining. As shown in Fig. 2D, the LPS-induced increase in the cardiomyocyte apoptotic rate was inhibited by 45 μM AUB treatment (Fig. 2D). After LPS stimulation, the expression of the proapoptotic proteins Bax and cytochrome c was increased, whereas the expression of the antiapoptotic protein Bcl-2 was decreased. AUB decreased Bax expression levels but increased Bcl-2 expression levels, which subsequently reduced cytochrome c release (Fig. 2E).

AUB inhibits LPS-induced cardiomyocyte oxidative stress and apoptosis. (A and B) ROS levels and quantitative results (n = 6). (C) SOD and NADPH oxidase activity and MDA production (n = 6). (D) Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate–biotin nick end–labeling (TUNEL) staining and analysis of apoptosis in cardiomyocytes (n = 6). (E) Bax, Bcl-2, and cytochrome c expression and quantitative results (n = 6). *P < 0.05 vs. the PBS group. #P < 0.05 vs. the LPS group.
AUB Attenuates LPS-Induced Cardiac Dysfunction.
Mice subjected to LPS showed impaired cardiac function, as evidenced by increased LVEDd, LVEF, and LVFS. Both 20 and 80 mg/kg per day AUB improved cardiac function, as indicated by decreased LVEDd and increased LVEF and LVFS (Table 1).
Echocardiographic parameters in mice after LPS injection Values are means ± S.E.
AUB Inhibits the LPS-Induced Inflammatory Response In Vivo.
After LPS injection, mice exhibited a severe inflammatory response in the heart with increased CD45-labeled leukocyte and CD68-labeled macrophage infiltration (Fig. 3, A and B) and increased IL-1β, IL-6, and TNF-α transcription (Fig. 3C). Both LD and HD AUB pretreatment attenuated these inflammatory responses in a dose-dependent manner (Fig. 3, A–C). Fibrosis was detected by PSR staining, and there were no significant differences in collagen volume between SIC hearts and normal hearts (Fig. 3D).

AUB inhibits the LPS-induced inflammatory response in vivo. (A and B) Immunohistochemical staining and analysis of CD45 and CD68 in the heart (n = 10). (C) mRNA levels of TNFα, IL-1β, and IL-6 (n = 10). (D) PSR staining of LV collagen volume in heart tissues (n = 10). *P < 0.05 vs. the vehicle–normal saline (NS) group. #P < 0.05 vs. the vehicle–LPS group.
AUB Ameliorates LPS-Induced Oxidative Stress and Apoptosis In Vivo.
After LPS injection, severe oxidative stress, as evidenced by accumulation of the lipid peroxidation intermediate 4-hydroxynonenal (Fig. 4A), increased NADPH oxidase activity and MDA production and decreased SOD activity (Fig. 4B). Both LD and HD AUB reduced the excessive oxidative stress (Fig. 4, A and B). We also detected the expression levels of oxidants and antioxidant enzymes. As shown in Fig. 4C, the levels of antioxidant SOD2 and TRX were reduced in mouse hearts after LPS injection, whereas the levels of the oxidant enzymes NADPH oxidase, p67 phox, and gp91 phox were increased. HD AUB sharply increased the expression of the antioxidant enzymes and reduced the expression of the oxidant enzymes (Fig. 4C). Increased cardiomyocyte apoptosis was observed in mouse hearts in the LPS group compared with the control group (Fig. 4D). Both LD and HD AUB treatment reduced the apoptosis in mouse hearts (Fig. 4D). The expression of Bax and cytochrome c was increased in the LPS group, whereas that of Bcl-2 was decreased. AUB decreased Bax expression levels and increased Bcl-2 levels, which subsequently reduced cytochrome c release in mouse hearts following LPS injection (Fig. 4E). We also detected the effects of AUB treatment of different durations (1, 3, and 5 days) on LPS-induced cardiomyocyte apoptosis in mice and found that LPS-induced cardiac apoptosis decreased with increasing AUB treatment time in the LPS/AUB-treated group compared with the control group (Fig. 4F).

AUB ameliorates LPS-induced oxidative stress and apoptosis in vivo. (A) Immunohistochemical staining of 4-hydroxynonenal (4-HNE) (n = 10). (B) SOD and NADPH oxidase activity and MDA levels in the heart (n = 10). (C) SOD, TRX2, P67, and gp91 protein expression (n = 10). (D) Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate–biotin nick end–labeling staining and analysis (n = 10). (E) Bax, Bcl-2, and cytochrome c levels and quantitative results. *P < 0.05 vs. the vehicle–normal saline group. #P < 0.05 vs. the vehicle–LPS group. (F) Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate–biotin nick end–labeling staining and analysis (n = 6). *P < 0.05 vs. the vehicle–NS group.
AUB Regulates TXNIP/P65/NLRP3 Signaling.
To evaluate the underlying mechanism of the effect of AUB, oxidative stress and inflammation-related signaling were examined. We found that LPS increased inflammasome formation, as indicated by elevated TXNIP and NLRP3 expression and downstream ASC, c-caspase-1, and IL-1β expression in cardiomyocytes stimulated with LPS (Fig. 5, A and B). AUB (45 μM) decreased TXNIP expression and inhibited NLRP3 inflammasome formation in LPS-stimulated cardiomyocytes (Fig. 5, A and B). These data indicate that AUB exerts its protective effect against LPS insult in the heart by directly inhibiting the NLRP3 inflammasome in cardiomyocytes.

AUB regulates TXNIP/P65/NLRP3 signaling. (A and B) TXNIP, NLRP3, ASC, c- caspase-1, and IL-1 protein levels in cardiomyocytes stimulated with LPS (10 μg/ml) and AUB (45 μM) (n = 6). (C-F) TXNIP, NLRP3, ASC, c-caspase 1 and IL-1 quantitive results. *P < 0.05 vs. the PBS group. #P < 0.05 vs. the LPS group.
TXNIP/P65/NLRP3 Is the Target of AUB in Cardiomyocytes.
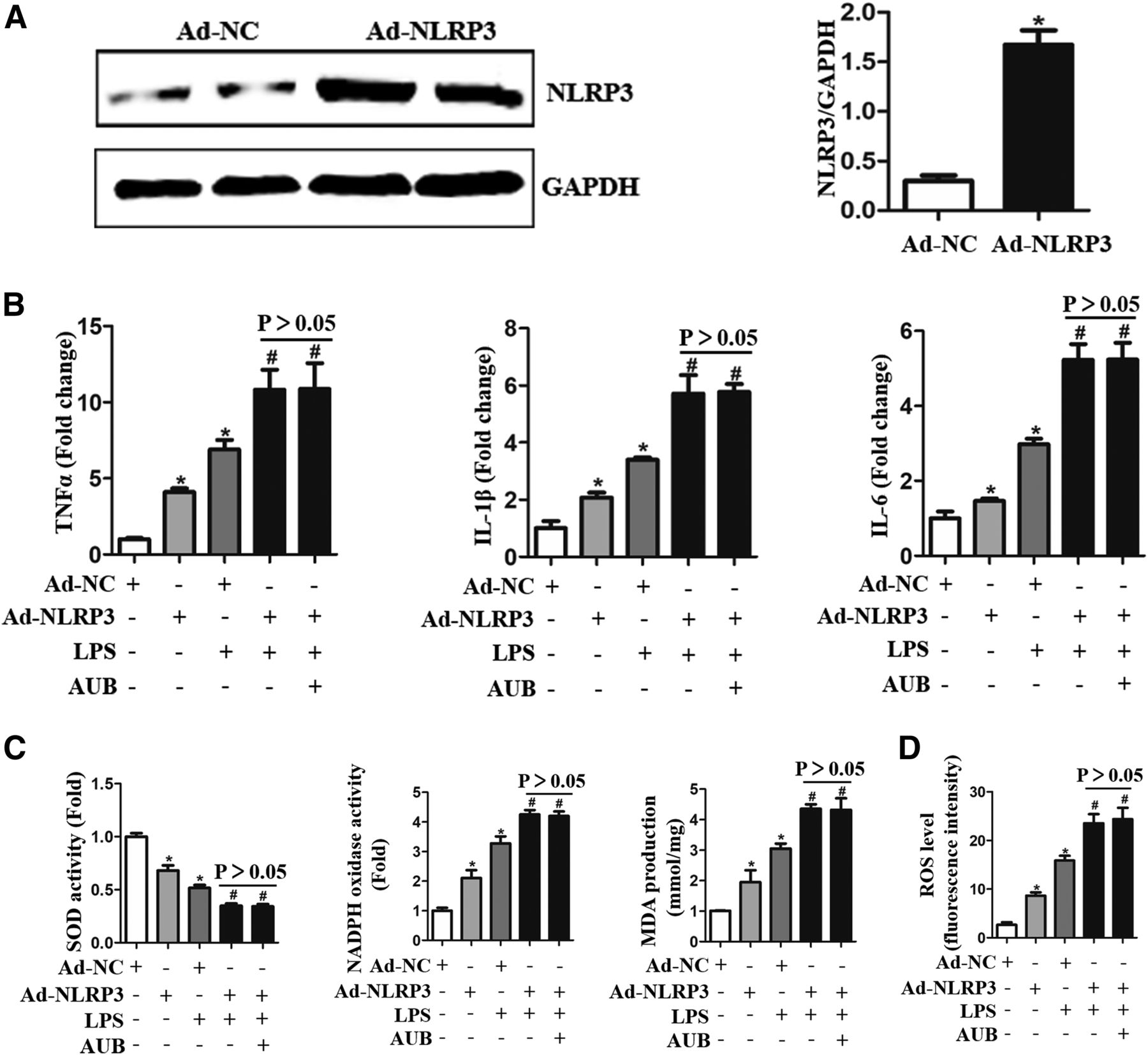
To further confirm the effect of AUB on NLRP3 signaling, cardiomyocytes were infected with Ad-NLRP3 to overexpress NLRP3 (Fig. 6A). NLRP3 overexpression deteriorated the LPS-induced inflammatory response and oxidative stress (Fig. 6, B–D). Interestingly, AUB could not exert its anti-inflammatory response or antioxidative stress effects against LPS insult in NLRP3-overexpressing cells. These results indicated that NLRP3 is the target of AUB.

NLRP3 is the target of AUB in cardiomyocytes. Cardiomyocytes were infected with NLRP3-expressing adenovirus (Ad-NLRP3), stimulated with LPS (10 μg/ml), and cultured with AUB (45 μM). (A) NLRP3 expression and quantitative results (n = 6). (B) mRNA levels of TNF-α, IL-1β, and IL-6 (n = 6). (C) SOD and NADPH oxidase activity and MDA production (n = 6). (D) ROS levels and quantitative results. *P < 0.05 vs. the Ad-negative control (NC)/PBS group. #P < 0.05 vs. the Ad-NC/LPS group.
The Effect of AUB on LPS-Insulted Heart in NLRP3 Deficiency Mice.
To obtain further evidence, NLRP3-KO mice (Fig. 7A) were pretreated with 80 mg/kg per day AUB for 7 days. Then the mice were injected with LPS. After 6 hours, improved cardiac function was observed in NLRP3-deficient mice compared with wild-type mice injected with LPS (Table 2). NLRP3 deficiency mimicked AUB pretreatment, as shown by decreased inflammation (Fig. 7, B–D), oxidative stress (Fig. 7E), and apoptotic rates in the NLRP3 mice compared with the wild-type mice (Fig. 7F). However, AUB pretreatment could not further amplify the protective effects of NLRP3 deficiency (Fig. 7, B–F).

NLRP3 deficiency mimics the effects of AUB in vivo. (A) Protein levels of NLRP3 in NLRP3-deficient mouse hearts (n = 4). (B and C) Immunohistochemical staining of CD45 and CD68 (n = 10). (D) mRNA levels of TNFα, IL-1β, and IL-6 (n = 10). (E) SOD and NADPH oxidase activity and MDA levels (n = 10). (F) Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate–biotin nick end–labeling staining and analysis of apoptosis (n = 10). *P < 0.05 vs. the NLRP3-KO/vehicle–normal saline group. #P < 0.05 vs. the wild type (WT)/vehicle–LPS group.
Echocardiographic parameters in NLRP3-KO mice after LPS injection Values are means ± S.E.
Discussion
LPS is a primary component of the bacterial cell wall and is a main cause of septic cardiac dysfunction that contributes to SIC (Hotchkiss and Karl, 2003). Recent studies have demonstrated that septic patients with cardiac dysfunction have higher mortality rates than those without cardiac involvement (Zaky et al., 2014). Considerable evidence indicates that inflammation, oxidative stress, and apoptosis are involved in the pathophysiological process of SIC (Flynn et al., 2010). Our previous study revealed that iridoid glycoside AUB inhibited ROS generation via regulation ofneuronal nitric oxide synthase/nitric oxide signaling (Wu et al., 2018). Numerous studies have confirmed that AUB exerts anti-inflammatory effects in many cell types (Park and Chang, 2004; Wang et al., 2015). Moreover, AUB treatment has been proven to attenuate oxidative stress and apoptosis (Ho et al., 2005; Wu et al., 2018). A previous study indicated that AUB inhibits oxidative stress by decreasing MDA levels and ROS generation and suppresses inflammation by reducing proinflammatory cytokine levels and p-NF-κB expression in the context of LPS-induced acute pulmonary injury (Qiu et al., 2018). Nevertheless, the effect of AUB on LPS-induced cardiac dysfunction has remained unclear. In the present study, we found that 1) AUB treatment attenuated LPS-induced inflammation, oxidative stress, and apoptosis in vitro and in vivo; 2) AUB improved LPS-induced cardiac dysfunction by inhibiting the of NLRP3 inflammasome; 3) NLRP3 overexpression abolished the protective effects of AUB in vitro; and 4) NLRP3-KO in mice in vivo mimicked AUB treatment, but AUB pretreatment could not produce further effects. Therefore, our findings identify AUB as a novel therapeutic agent for the treatment of LPS-induced cardiac dysfunction that exerts its effects by suppressing the NLRP3 inflammasome (Fig. 8.

Graphical abstract. Proposed mechanisms for the protective effect of AUB against LPS-induced cardiac dysfunction. AUB inhibits LPS-induced oxidative stress and apoptosis by reducing ROS production and TXNIP expression. Moreover, AUB suppresses LPS-induced inflammation and apoptosis by inactivating the NLRP3 inflammasome, thus improving cardiac dysfunction induced by LPS.
Cardiac dysfunction is the primary feature of SIC. Clinical studies incorporating echocardiographic detection of cardiac function in septic patients have shown that these patients exhibit ejection fraction decreases and diastolic and systolic dysfunction (Kimchi et al., 1984; Parker et al., 1990). One study used Doppler tissue echocardiography and found an increased E/e′ ratio (an indicator of diastolic function) in SIC patients (Brown et al., 2012). However, several studies have failed to observe increases in LVED volume index (Vieillard Baron et al., 2001; Zaky et al., 2014). Our study revealed decreased LVEF and LVFS and increased LVESd in SIC mouse hearts compared with normal hearts. However, LVEDd was not significantly different between SIC mice and normal mice. AUB increased LVEF and LVFS and decreased LVESd in SIC mice. These changes indicated that the preservation of cardiac function by AUB may have been attributable to the reduction in apoptosis in SIC mouse hearts. However, the cause of the diastolic dysfunction and increased volume index is not clear. Further studies are needed to confirm the mechanism by which diastolic function is altered during SIC.
Studies have indicated that mitochondrial damage during SIC development stimulates excessive ROS production; in turn, ROS production causes irreversible mitochondrial failure, which leads to multiple organ dysfunction and further initiates inflammatory responses (Cimolai et al., 2015). TNF-α, IL-1β, and IL-6 are also produced in excess and serve as the primary inflammatory mediators in the early stages of sepsis; these mediators depress cardiac function synergistically (Lv and Wang, 2016). Additionally, ROS overproduction induces lipid oxidation and protein oxidation, which further compromise the cell membranes integrity and cell function and lead to apoptosis (Celes et al., 2010; Cimolai et al., 2015). Many studies have suggested that AUB can protect against inflammation in mice (Wu et al., 2018), suppress ROS generation and downregulate NADPH expression in hepatic stellate cells (Lv et al., 2017), and inhibit apoptosis by enhancing estrogen receptor β signaling (Li et al., 2016). Consistent with these findings, we found that AUB decreased the transcription of inflammatory cytokines (TNF, IL-1β, IL-8, and IL-6) in LPS-stimulated mouse hearts and cardiomyocytes in our present study. Furthermore, AUB inhibited the expression of NADPH oxidase, increased the expression and activity of the antioxidant SOD, and reduced the levels of ROS and lipid peroxidation. In addition, we discovered that AUB reduced the apoptotic rate in vitro and in vivo. Hence, these anti-inflammatory, antioxidative, and antiapoptotic effects in the heart attenuated LPS-induced cardiac dysfunction.
The NLRP3 inflammasome includes NLRP3, ASC, and caspase-1. Once activated, the inflammasome promotes the maturation of IL-18 and IL-1β to cause inflammation and induces apoptosis by cleaving procaspase-1 to form caspase-1 (Hoseini et al., 2018). A previous study indicated that TXNIP binds NLRP3 directly and modulates its oligomerization to produce a signal (Luo et al., 2017). Additional research has revealed that activation of the NLRP3 inflammasome is induced by the triggering of ROS-dependent lipid peroxidation and endoplasmic reticulum stress (Wang et al., 2017a). ROS upregulates TXNIP expression, which successively triggers the primary and secondary steps of NLRP3 activation (Luo et al., 2017). Therefore, inhibition of TXNIP expression can attenuate cardiac dysfunction (Liu et al., 2014). Wang et al. (2014) demonstrated that NLRP3 inflammasome activation plays a vital role in coxsackievirus B3–induced myocarditis and that suppressing the NLRP3 inflammasome can ameliorate severe cardiac injury. Moreover, the NLRP3 inflammasome can be activated in LV cardiomyocytes during sepsis, and the absence of NLRP3 decreases both IL-1β and IL-6 (Kalbitz et al., 2016). These results are consistent with our findings that activation of the NLRP3 inflammasome in cardiomyocyte plays a key role in LPS-induced cardiac dysfunction. In this study, we found that AUB inhibited ROS production and TXNIP expression, consistent with the results of previous studies indicating that inhibition of NLRP3 attenuates cardiac dysfunction associated with sepsis (Luo et al., 2014; Kalbitz et al., 2016). Moreover, ROS increased TXNIP protein expression, to achieve the first steps of NLRP3 activation. The three subtypes of the TRX family are cytosolic (TRX1), mitochondrial trx (TRX2), and trx sperm-specific subtype (sp-TRX3). As antioxidants, TRX maintains the balance of sulfur oxygen–related redox state and plays a key role in the regulation of redox signals (Matsuzawa, 2017). Previous studies indicated that Trx2 is a major controller of mitochondrial ROS emission, particularly in the heart (Stanley et al., 2011; Aon et al., 2012). In this study, we found that AUB increased the expression of TRX2 in response to LPS. However, the redox status of TRX2 is still unknown. Due to the limitations of the study, further studies are needed. Interestingly, we discovered that NLRP3 overexpression totally abolished the protective effects of AUB in our in vitro study. In the same way, AUB pretreatment did not provide further protective effects in NLRP3-KO mice. These findings confirm that the protective effects of AUB against SIC rely on NLRP3 signaling. It has been reported that AUB inhibits antigen-induced TNF-α, IL-6, and NF-κB expression (Jeong et al., 2002). Furthermore, AUB suppresses oxidative stress by reducing MDA levels and ROS generation in the context of LPS-induced acute pulmonary injury (Qiu et al., 2018). Our previous study demonstrated that AUB protects against myocardial infarction–induced apoptosis by regulating the neuronal nitric oxide synthase/nitric oxide pathway (Yang et al., 2018). In the present study, AUB was found to alleviate LPS-induced inflammation, oxidative stress, and apoptosis via inhibition of TXNIP and NLRP3 inflammasome. Inflammation, oxidative stress, and apoptosis play vital roles in sepsis-induced cardiac injury (Flynn et al., 2010). The protective effect of AUB may improve sepsis-induced cardiac dysfunction. Thus, AUB may be a potential therapeutic agent for SIC. Mechanistically, AUB increases the expression of TRX and decreases the expression of TXNIP. Moreover, AUB may promote the combination of TXNIP and TRX, thereby reducing the release of TXNIP and inhibiting the activation of NLRP3. However, this hypothesis still needs to be confirmed by subsequent experiments.
Conclusion
The NLRP3 inflammasome is involved in LPS-induced inflammation, oxidative stress, and apoptosis. AUB protects against LPS-induced sepsis by preserving cardiac function and reducing ROS levels, and the expression of the TXNIP protein, thus inactivating NLRP3 inflammasomes (Fig. 8).
Authorship Contributions
Participated in research design: Duan, Tang, Wu.
Conducted experiments: Duan, Wu, Liu, Cai.
Performed data analysis: Xie, Hu.
Wrote or contributed to the writing of the manuscript: Duan, Yuan.
Footnotes
- Received May 15, 2019.
- Accepted August 12, 2019.
↵1 M.-X.D. and Y.Y. contributed equally to this work.
This work was supported by grants from the National Natural Science Foundation of China (81530012, 81700353, and 81700218), Hubei Province’s Outstanding Medical Academic Leader Program, National Natural Science Foundation of Hubei Province (2017CFB320), and the Fundamental Research Funds of the Central Universities (2042017kf0060).
Abbreviations
- Ad
- adenovirus
- ASC
- apoptosis-associated speck-like protein containing a caspase recruitment domain
- AUB
- aucubin
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HD
- high dose
- IL
- interleukin
- KO
- knockout
- LD
- low dose
- LPS
- lipopolysaccharide
- LV
- left ventricular
- LVEDd
- LV end-diastolic dimension
- LVEF
- LV ejection fraction
- LVESd
- LV end-systolic dimension
- LVFS
- LV fractional shortening
- MDA
- malondialdehyde
- NF-κB
- nuclear factor κ-B
- NLRP3
- nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3
- NRCM
- neonatal rat cardiomyocyte
- PSR
- picrosirius red
- ROS
- reactive oxygen species
- SIC
- sepsis-induced cardiomyopathy
- SOD
- superoxide dismutase
- TNF
- tumor necrosis factor
- TRX
- thioredoxin
- TXNIP
- TRX interaction protein
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}