


Abstract
The enzyme geranylgeranyl diphosphate synthase (GGDPS) synthesizes the 20-carbon isoprenoid geranylgeranyl pyrophosphate, which is used in geranylgeranylation reactions. We have demonstrated that GGDPS inhibitors in multiple myeloma (MM) cells disrupt Rab geranylgeranylation, leading to inhibition of monoclonal protein trafficking, induction of the unfolded protein response pathway (UPR), and apoptosis. We have previously reported preclinical studies with the GGDPS inhibitor VSW1198, which is a mixture of homogeranyl/homoneryl triazole bisphosphonates. Additional structure-function efforts have led to development of the α-methylated derivatives RAM2093 (homogeranyl) and RAM2061 (homoneryl). As little is known regarding the impact of olefin stereochemistry on drug properties in vivo, we pursued additional preclinical evaluation of RAM2093 and RAM2061. In MM cell lines, both isomers induce activation of UPR/apoptotic markers in a concentration-dependent manner and with similar potency. Single-dose testing in CD-1 mice identified a maximum tolerated i.v. dose of 0.5 mg/kg for RAM2061 and 0.3 mg/kg for RAM2093. Liver toxicity was the primary barrier to dose escalation for both compounds. Disruption of geranylgeranylation in vivo was confirmed after multidose administration of either compound. Pharmacokinetic studies revealed plasma terminal half-lives of 29.2 ± 6 (RAM2061) and 22.1 ± 4 hours (RAM2093). Relative to RAM2061, RAM2093 levels were significantly higher in liver tissue but not in other tissues. Using MM.1S flank xenografts, we observed a significant reduction in tumor growth in mice treated with RAM2061 relative to controls. Collectively, these studies reveal olefin stereochemistry-dependent effects on GGDPS inhibitor biodistribution and confirm the in vivo efficacy of this novel therapeutic approach.
SIGNIFICANCE STATEMENT These studies reveal olefin stereochemistry-dependent effects on the in vivo properties of two novel triazole bisphosphonate inhibitors of geranylgeranyl diphosphate synthase and demonstrate the therapeutic potential of this class of inhibitors for the treatment of multiple myeloma.
Introduction
Multiple myeloma (MM) is characterized by the expansion of malignant plasma cells in the bone marrow. Despite advances in treatment, MM remains fatal. In 2019, an estimated 32,000 new cases of MM will be diagnosed, and nearly 13,000 individuals will die from the disease. Owning to their high secretory rate, MM cells express elevated levels of unfolded protein response (UPR) mediators to aid in the clearance of misfolded proteins and therefore are uniquely dependent on efficient protein handling for their survival (Obeng et al., 2006). Targeting this dependency could lead to the development of novel therapeutic strategies.
Rab proteins are a member of the Ras small GTPase superfamily and are responsible for mediating various aspects of intracellular trafficking processes. Regulation of Rab activity occurs through a post-translational modification termed geranylgeranylation. The enzyme geranylgeranyl diphosphate synthase (GGDPS) generates the 20-carbon isoprenoid donor termed geranylgeranyl pyrophosphate (GGPP). Geranylgeranyl transferase II (GGTase II) catalyzes the addition of the isoprenoid chain to the carboxy terminal of Rabs via interaction with a Rab escort protein. In the absence of prenylation, Rab proteins are mislocalized and nonfunctional (Gomes et al., 2003). Inhibition of Rab geranylgeranylation serves as a novel mechanism to disrupt protein trafficking in the cell. We have pursued the development of GGDPS inhibitors, which deplete cellular pools of GGPP, thus disrupting the activity of GGTase II and preventing prenylation of Rabs (Zhou et al., 2014; Wills et al., 2015, 2017; Matthiesen et al., 2016, 2018; Foust et al., 2016; Allen et al., 2017; Haney et al., 2017).
In MM, inhibition of Rab prenylation results in impaired monoclonal protein secretion, activation of the UPR, and apoptosis (Holstein and Hohl, 2011; Born et al., 2013). We previously characterized VSW1198, a novel and highly potent GGDPS inhibitor (Fig. 1A) (Wills et al., 2015; Haney et al., 2018). Preclinical evaluation of VSW1198 revealed a metabolically stable compound that exhibited broad tissue distribution and a favorable plasma terminal half-life of 47.7 hours (Haney et al., 2018). VSW1198 is a mixture of olefin isomers (3:1 homogeranyl/homoneryl) (Wills et al., 2015). Interestingly, whereas the homoneryl isomer is approximately three times more potent than the homogeranyl isomer, the two isomers interact in a synergistic manner to inhibit GGDPS (Allen et al., 2017). Computational modeling studies revealed that although both isomers can bind the enzyme simultaneously, the homoneryl is preferred at the farnesyl diphosphate binding site, whereas the homogeranyl is preferred at the GGPP site (Allen et al., 2017). The finding that the olefin isomers of VSW1198 synergistically interact to inhibit the target enzyme is unique and reveals insight into drug design, but the use of a mixture is not ideal from a drug-development perspective; it would be preferable to have a lead inhibitor that is a single isomer. We have therefore pursued additional structure-function work and recently reported a series of derivatives of VSW1198 that contain a methyl substitutent at the α-carbon position (Matthiesen et al., 2018) (Fig. 1A). The homoneryl isomer RAM2061 has an IC50 of 86 nM and lowest effective concentration in MM cells of 25 nM (Matthiesen et al., 2018). The homogeranyl isomer RAM2093 has similar activity with an IC50 of 125 nM and a cellular lowest effective concentration of 20 nM (Matthiesen et al., 2018).

GGDPS inhibitors induce the UPR and apoptosis in MM cells. (A) Chemical structures of GGDPS inhibitors (VSW1198, RAM2093, and RAM2061). (B) Immunoblot analysis showing protein levels of apoptotic markers (cleaved caspases 3 and 8) and UPR markers [ATF4, phosphorylated-eIF2α (p-eIF2α) and eIF2α] in human MM cell lines treated with or without RAM2061 or RAM2093 for 48 hours. β-tubulin is shown as a loading control. Immunoblots are representative of three independent experiments. (C) quantitative real-time-polymerase chain reaction analysis of mRNA expression levels of UPR markers (ATF4, CHOP, IRE1α, and PERK) in human MM cell lines treated with or without RAM2061 or RAM2093 for 48 hours. Data represent fold change normalized to control (n = 3 biologic replicates). Data are displayed as mean ± S.E.M., *P < 0.05; **P < 0.01; ****P < 0.001, Statistical calculations were performed using the Bio-Rad CFX manager 3.1 software).
Because of their potency and ability to induce the UPR and apoptosis of MM cells in vitro, we pursued in vivo preclinical evaluation of these novel GGDPS inhibitors, including dose-finding, pharmacokinetic (PK) analysis, and biodistribution studies. Our data demonstrate that although the two isomers have equivalent anti-MM activity in vitro, the homoneryl isomer RAM2061 has a higher maximum tolerated dose (MTD) and lower hepatic drug levels across multiple time points after injection compared with the same dose of the homogeranyl isomer RAM2093. In addition, treatment with RAM2061 slowed tumor growth in MM flank xenografts. Collectively, these studies help advance the development of clinically relevant GGDPS inhibitors for the treatment of MM and provide novel insight into the impact of olefin stereochemistry on biodistribution of these inhibitors.
Material and Methods
Chemicals.
RAM2061 and RAM2093 (Matthiesen et al., 2016) were kindly provided by Professor David Wiemer at the University of Iowa. Purity of the assayed compounds was determined as ≥95% by high-performance liquid chromatography and verified by nuclear magnetic resonance (Matthiesen et al., 2018).
Cell Culture.
MM.1s and RPMI-8226 cells were obtained from American Type Culture Culture (Manassas, VA). Cells were grown in media supplemented with 10% heat-ina/ctivated fetal bovine serum, glutamine, and penicillin-streptomycin at 37°C and 5% CO2. Mycoplasma testing was performed using MycoAlert mycoplasma detection kit (Lonza, Rockland, ME).
Quantitative Real-Time Polymerase Chain Reaction.
Cells were incubated for 48 hours in the presence or absence of drugs. The E.Z.N.A. HP total RNA kit from Omega was used for RNA isolation. RNA (1 μg) was reverse-transcribed to cDNA using the i-Script cDNA synthesis kit (Bio-Rad, Hercules, CA). cDNA, gene-specific primers (Supplemental Table 1), and i-Taq Sybr green super mix (Bio-Rad) were mixed according to manufacturer’s instruction. Quantitative real-time-polymerase chain reaction reactions were performed in duplicate in a CFX96 real-time machine (Bio-Rad). Data analysis, including statistical calculations, was accomplished using the Bio-Rad CFX manager 3.1 software. Gene expression was normalized to that of β-actin.
Immunoblot.
RPMI-8226 (0.5 × 106 cells/1 ml) and MM.1S (1 × 106 cells/1 ml) cells were incubated for 48 hours in the presence or absence of drugs. Cells were washed with phosphate-buffered saline (PBS) and lysed in RIPA buffer supplemented with protease and phosphatase inhibitors (1% sodium deoxycholate, 0.15 M NaCl, 0.1% SDS, 1% (v/v) Triton X-100, 0.05 M Tris HCl, pH 7.4). Mouse tissue samples were homogenized in T-PER (Invitrogen) supplemented with protease inhibitors using a handheld tissue homogenizer. The BCA method was used to quantify protein concentration. Protein (15 μg for cell lysate, 30 μg for tissue homogenate) was run on an SDS-PAGE gel and transferred to polyvinylidene difluoride membranes. Blots were incubated overnight in primary antibody and for 1 hour in secondary antibody. Protein was visualized using an electrochemiluminescence detection kit and a Bio-Rad Chemidoc MP imaging system. Antibodies used for immunoblot analysis can be found in Supplemental Table 2.
Animals.
Female CD-1 and NOD-SCID mice between the ages of 6 and 8 weeks were purchased from Charles River (Wilmington, MA). All animals were kept in the University of Nebraska Medical Center (UNMC) animal facility, which maintains a temperature of 23–25°C, relative humidity of 50%–70% and 12-hour light/dark cycles. The UNMC Institutional Animal Care and Use Committee approved all studies (protocol no. 16-132-11-FC).
Determination of Maximum Tolerated Dose.
CD-1 mice were injected by intravenous administration with RAM2061 or RAM2093 (dissolved in PBS and filter-sterilized, 100 μl of total volume) . We performed standard dose escalation procedures using the up-and-down method. To conserve mice, one mouse was injected at a time, and the dosage was increased 1.8 times until toxicity was detected. If the first mouse survived and showed no signs of toxicity after 7 days, then the second mouse would receive a higher dose. If the first mouse became moribund, the second mouse would receive a lower dose. The lowest dose that did not produce signs of toxicity was increased by 1.15 times until the upper limit was reached. To confirm MTD, five mice were injected and followed up over 14 days. Blood analysis was performed at days 7 and 14. Animal weight was recorded weekly, and histopathological analysis of organs was performed after euthanasia.
Blood Analysis.
Blood samples were obtained through cheek bleeds (approximately 150 μl) or postmortem from the heart. For dose-finding experiments, five mice were used per group. Blood was stored in lithium-heparin coated tubes. Samples were analyzed using an Abaxis Vetscan2 instrument (Allied Analytic, Tampa, FL) and the preventive profile plus rotor. Complete blood count measurements were obtained via whole blood using a HemaVet 950 (Drew Scientific, Miami Lakes, FL). These instruments provided the normal ranges of laboratory values (Tables 1–4). Creatine kinase and lactate dehydrogenase levels were measured using colorimetric kits obtained from BioAssay systems (catalog no. D2DH-100 and ECPK-100).
Blood chemistry panel for single dose testing of RAM2061 and RAM2093
Blood was collected at 7 and 14 days post-injection (mean ± S.D., n = 5 mice per group).
Complete blood cell count analysis for single-dose testing of RAM2061 and RAM2093
Blood was collected 14 days postinjection (mean ± S.D., n = 5 mice per group).
Blood Chemistry panel for RAM2061 and RAM2093 multidose testing
Blood was collected after 4 wk of drug administration (mean ± S.D., n = 5 mice per group, with exception of RAM2093 once-weekly group--n=3).
Complete blood cell count analysis for multidose testing of RAM2061 and RAM2093
Blood was collected after 4 wk of drug administration (mean + S.D., n = 5 mice per group, with exception of RAM2093 once-weekly group--n=3).
Histology.
Mouse organs were fixed in formalin for 24–96 hours, stored in 70% ethanol, embedded in paraffin, sectioned, and stained (H&E) using standard methods at the UNMC Tissue Science Facility.
PK Study and Liquid Chromatography-Tandem Mass Spectrometry Conditions.
In brief, mice were injected via tail vein with 0.3 mg/kg of RAM2061 or RAM2093 (dissolved in PBS and filter sterilized). Blood was collected at 11 time points (5 and 30 minutes; 1, 2, 4, 6, 8, 24, 48, 72, and 168 hours) either by cheek bleed or postmortem from the heart and plasma separated by centrifugation. Tissues were collected at six time points (2, 8, 24, 48, 72, and 168 hours). Plasma (25 µl) and tissue homogenate (100 µl) were used for quantitation of analytes. Calibration and quality control samples were separately prepared for RAM2061 and RAM2093 by spiking 10 µl of appropriate calibration stock of RAM2061 and RAM2093 in blank biomatrix and 10 µl of internal standard solution (RAM1147, 1.0 µg/ml) was added. Ammonium carbonate buffer (10 mM, 400 μl) with 0.1% NH4OH was added to dilute calibration, quality control, and study samples. Plasma and tissue samples were extracted using solid-phase extraction (SPE) and derivatized on cartridge using diazomethane (100 μl). The solid-phase extraction was carried out using Strata X-AW, 30 mg extraction cartridge (30 mg/1 ml; Phenomenex, Inc, Torrance, CA). Cartridges were preconditioned with 1 ml of methanol, followed by 1 liter of 10 mM ammonium carbonate buffer with 0.1% NH4OH. Samples were loaded to the cartridges and washed with 1 ml of 10 mM ammonium carbonate buffer with 0.1% NH4OH and then dried for 10 minutes. Derivatization was initiated by adding 100 μl of diazomethane solution to each cartridge and allowed to percolate through the sorbent by gravity. The cartridge was dried under a stream of nitrogen gas and analytes eluted with 2 ml of methanol. The eluents were collected in glass tubes and dried under nitrogen in a water bath at 50°C. The dry residues were reconstituted in 200 µl of 0.1% acetic acid-methanol-acetonitrile mixture (65:17.5:17.5) and centrifuged at 13,000g and 5 µl supernatant was injected into the liquid chromatography-tandem mass spectrometry (LC-MS/MS). The LC-MS/MS conditions were as previously described (Chhonker et al., 2018). The MS/MS system was operated at unit resolution in the multiple reaction monitoring mode using precursor ion → product ion combinations of 478.20 → 296.20 m/z for both RAM2061 and RAM2093.
The PK parameters of RAM2061 and RAM2093 in plasma and tissues were calculated using noncompartmental analysis with Phoenix WinNonlin 6.3 (Pharsight Corporation). The area under the concentration-time curve (AUC0-∞) was calculated using the linear trapezoidal method from 0 to tlast with extrapolation from tlast to infinity based on the observed concentration at the last time point divided by the terminal elimination rate constant (λ). The terminal half-life (t1/2) was calculated as 0.693/k. Clearance (Cl) and the volume of distribution of the elimination phase (Vd) were calculated as dose/AUC0-∞ and dose/k*AUC0-∞, respectively. Mean tissue concentrations were calculated and expressed as ng/g tissue.
In Vitro Metabolism Using Human Liver Microsomes.
The human liver microsomes (HLMs) metabolic stability experiments were performed as previously described (Haney et al., 2018). All samples were processed and derivatized according to our previously developed methods, and supernatant was injected for LC-MS/MS analysis (Chhonker et al., 2018). Testosterone served as the positive control; RAM2061/RAM2093 without NADPH was used as a negative control.
Plasma Protein Binding.
Plasma protein binding was assessed using the Rapid Equilibrium Dialysis Device system (Thermo Scientific, Rockford IL). Briefly, spiked samples were prepared by adding RAM2061 or RAM2093 at two concentrations (1 and 10 μg/ml) to mouse plasma (500 μl). In addition, mouse plasma samples from the PK studies (2 and 8 hours postinfusion samples) were assessed. The spiked plasma sample (300 μl) or the PK plasma sample (100 μl) was added to the donor chamber, and PBS (containing 100 mM sodium phosphate and 150 mM sodium chloride, pH 7.4) was added to the buffer chamber. The plate was sealed and incubated at 37°C on an orbital shaker at 100 rpm for 5 hours. Aliquots (50 μl) were removed from the sample and buffer chambers and mixed with an equal volume of buffer or blank plasma, respectively. Calibration and quality control samples were prepared using equal volumes of blank plasma and buffer. All matrix-matched samples were prepared as described and analyzed by LC-MS/MS. Plasma stability and nonspecific binding of RAM2061 and RAM2093 were assessed for 5 hours at 37°C to ensure that protein binding data were not affected by plasma stability or binding to the dialysis membrane. Propranolol and atenolol were used as high and low protein-bound positive controls. The stability, device recovery, and percent bound were calculated using eqs. 1–3: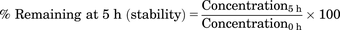 (1)
(1) (2)
(2) (3)
(3)
Myeloma Xenografts.
NOD-SCID mice were subcutaneously inoculated in the flank with MM1.S cells (10 million cells in 0.1 ml of sterile saline mixed 1:1 with Matrigel). When tumors became palpable (approximately 2 weeks), mice were randomly divided into treatment (0.08 mg/kg i.v. twice weekly of RAM2061, n = 10) and control groups (PBS i.v. twice weekly, n = 10). Tumor volume was recorded three times per week using a caliper. Mice were euthanized when tumors reached 2000 mm3. The following equation was used to calculate tumor volume: 4π/3 × (width/2)2 × (length/2). One mouse from the RAM2061-treatment group was excluded from the analysis because of intramuscular tumor growth.
Statistics.
Two-tailed t tests were used to calculate statistical significance between groups. The Bonferroni correction was used to adjust for multiple comparisons. The Kaplan-Meier method was used to estimate survival distributions, and differences in survival between groups were compared with the log-rank test. P < 0.05 was considered statistically significant. Power calculation for mouse studies of tumor growth gives 83% power with n = 10 mice per group to detect a 200 mm3 difference in tumor growth with group S.D. = 100 mm3. Linear mixed models were used to look at changes in tumor burden over time. Tumor burden was modeled on the log10 scale. The model included fixed effects for group, day, and the group × day interaction, and day is modeled as a continuous variable. A random slope and intercept were fit for each mouse. SAS software version 9.4 was used for data analysis (SAS Institute Inc., Cary, NC).
Results
RAM2061 and RAM2093 Induce the Unfolded Protein Response and Apoptosis in MM Cells.
We assessed the ability of the GGDPS inhibitors to induce the UPR and apoptosis in RPMI-8226 and MM.1S cells. Incubation with either RAM2061 or RAM2093 induces the cleavage of caspases 3 and 8 in a concentration-dependent manor (Fig. 1B). Furthermore, both inhibitors promote the upregulation of the UPR-associated proteins ATF4 and phosphorylated eIF2α (Fig. 1B). We also observed upregulation of the UPR mediators ATF4, CHOP, IRE1α, and PERK at the mRNA level in MM cell lines after 48 hours incubation with either RAM2061 or RAM2093 (Fig. 1C). Collectively, these data demonstrate that RAM2061 and RAM2093 induce the activation of the UPR and apoptosis in human MM cell lines with similar potency.
Determination of the Maximum Tolerated Dose of RAM2061 and RAM2093.
Dose-escalation studies were performed in CD-1 mice. The MTD for RAM2061 was determined to be 0.5 mg/kg intravenously. This is the same MTD as was observed with our previously characterized compound, VSW1198 (Haney et al., 2018). Interestingly, RAM2093 had a lower MTD of 0.3 mg/kg. Histopathological examination of the livers from animals treated with doses greater than the MTD revealed panlobular hepatocyte swelling, hepatocyte cell death, and invasion of inflammatory cells (Fig. 2). Examination of other tissues, including spleen, kidney, and brain, revealed no pathologic findings (Supplemental Fig. 1). Analysis of blood samples at 7 days postinjection demonstrated transient elevation in liver transaminases (ALT and AST) as the main pathologic finding (Table 1). At 14 days postinjection, the ALT and AST levels returned to near normal levels. Mice given 0.3 mg/kg of RAM2061 showed less elevation in liver transaminase levels than those given the equivalent dose of RAM2093 (Supplemental Table 3). Complete blood counts were largely normal at both 7 (data not shown) and 14 days postinjection (Table 2). Neither compound induced weight loss over 14 days (Supplemental Fig. 2). No effects on kidney function or on cardiac markers were observed (Table 1). These findings suggest liver toxicity as the primary barrier to dose escalation for both GGDSPS inhibitor compounds.

The effects of single doses of RAM2061 and RAM2093 on liver morphology. Liver H&E sections (original magnification, 200×) from mice after intravenous injection with 0.3 mg/kg RAM2093, 0.5 mg/kg RAM2093, 0.5 mg/kg RAM2061, and 1.3 mg/kg RAM2061. A normal liver is shown as a control. Tissues were collected 14 days postinjection.
Determination of the Maximum Tolerated Repeat Dosing.
We evaluated twice-weekly and once-weekly dosing schedules for both GGDPS inhibitors. For RAM2061, the maximum tolerated repeat dosing was determined to be either 0.1 mg/kg twice weekly (2 weeks on, 1 week off, 2 weeks on) or 0.2 mg/kg once weekly (continuous dosing for 4 weeks). For RAM2093, the maximum tolerated twice-weekly dose was 0.05 mg/kg twice weekly. At the dosing level of 0.15 mg/kg weekly of RAM2093, treatment was discontinued prematurely in two of five mice owing to liver toxicity. Examination of blood and tissues revealed liver toxicity as the only pathologic finding (Fig. 3; Table 3). No significant cytopenias were observed, and no loss of weight was observed for either the once-weekly or twice-weekly dosed animals (Supplemental Fig. 3; Table 4).

The effects of repeat dosing of RAM2061 and RAM2093 on liver morphology. Liver H&E sections (original magnification, 200×) from mice after multiple intravenous injections with RAM2061 (0.1 mg/kg 2×/wk or 0.2 mg/kg 1×/wk) or RAM2093 (0.05 mg/kg 2×/wk or 0.15 mg/kg 1×/wk). A normal liver is shown as a control. Tissues were collected 35 days postinitiation of treatment.
In Vivo Disruption of Geranylgeranylation.
To evaluate the in vivo on-target effects of the GGDPS inhibitors, immunoblot analysis of unmodified Rap1a was performed. Rap1a is a representative geranylgeranylated protein, and the use of a primary antibody that detects only the unmodified form of Rap1a allows for identification of disruption of protein geranylgeranylation. Liver, spleen, and kidney lysates were generated from mice injected with repeated doses (either once or twice weekly) of RAM2061 or RAM2093. Immunoblots demonstrate accumulation of unmodified Rap1a in all three tissues; the highest abundance was in the liver (Fig. 4). No accumulation of unmodified Rap1a was observed after a single dose of either RAM2061 or RAM2093 (Supplemental Fig. 4). This finding is consistent with the half-life of Ras-related small GTPases being approximately 24 hours (Holstein et al., 2002; Ramalho et al., 2002); thus, repeat dosing is required to achieve sustained depletion of GGPP, resulting in accumulation of newly synthesized unmodified protein.

RAM2061 and RAM2093 disrupt protein geranylgeranylation in vivo. Immunoblot analysis of unmodified Rap1a in liver, spleen, and kidney tissues isolated from mice injected either twice weekly (A) or once weekly (B) with RAM2061 or RAM2093. β-tubulin is shown as a loading control. Lysates generated from U266 human myeloma cells treated with lovastatin were used as a positive control for unmodified Rap1a protein.
Pharmacokinetic and Biodistribution Studies.
CD-1 mice were injected i.v. with 0.3 mg/kg RAM2061 or RAM2093. Blood samples were collected at 11 time points (5, 30 minutes, 1, 2, 4, 6, 8, 24, 48, 72, and 168 hours). RAM2061 or RAM2093 were isolated from plasma samples and detected using LC-MS/MS. The PK parameters are shown in Table 5. Both compounds were detected in the plasma up to 72 hours postinjection (Fig. 5), and the plasma elimination terminal half-lives (t1/2) were determined to be 29.2 ± 6 and 22.1 ± 4.2 hours for RAM2061 and RAM2093, respectively.
Pharmacokinetic parameters of RAM2061 and RAM2093

Measurement of RAM2061 and RAM2093 in mouse plasma samples. Plasma concentrations (nanograms per milliliter) of RAM2061 and RAM2093 after a single dose (0.3 mg/kg i.v.) (mean ± S.D., n = 5 mice).
Tissue samples (liver, lung, kidney, brain, spleen, and bone marrow) were collected at various time points (2, 8, 24, 48, 72, and 168 hours) postinjection, and drug levels were measured. Both GGDPS inhibitors showed the highest tissue accumulation in the liver and the lowest accumulation in the brain (Fig. 6, A and B). At 168 hours postinjection, RAM2093 was detected in all tested organs; RAM2061 was present in every organ except the kidney. Importantly, both compounds demonstrated stable bone marrow concentrations over 72 hours postinjection (Fig. 6C).

Tissue distribution of RAM2061 and RAM2093. Tissue concentrations (nanograms per gram) over time for RAM2061 (A) and RAM2093 (B) after a single dose (0.3 mg/kg IV). (C) Concentration of RAM2061 and RAM2093 in the bone marrow (nanograms per milliliter). (D) Liver levels of RAM2061 and RAM2093 (nanograms per gram). Data are shown as mean ± S.D. (n = 5 mice, *P < 0.05; **P < 0.01; ****P < 0.001, two-tailed student t test with Bonferroni correction).
Comparison of tissue distribution patterns between the olefin isomers revealed marked differences in hepatic drug levels. RAM2093 levels were approximately 2- to 3-fold greater than RAM2061 levels from a period of 2–72 hours postinjection (Fig. 6D). These results are consistent with the toxicity studies demonstrating increased hepatic toxicity with RAM2093 compared with RAM2061. RAM2093 levels were higher in the kidney at several time points; overall, no differences were observed between the two isomers with respect to lung or brain levels (Supplemental Fig. 5). There was a trend toward increased levels of RAM2093 in the spleen relative to RAM2061; however, no histopathologic findings were noted in the spleens of mice treated with RAM2093.
Metabolic Stability in HLMs.
The in vitro metabolic stability of RAM2061 and RAM2093 was determined using HLMs. RAM2061 and RAM2093 showed complete metabolic stability over a period of 60 minutes (Fig. 7). In comparison, the positive control testosterone was rapidly metabolized. These data, in conjunction with the tissue distribution studies, suggest both GGDPS inhibitor compounds are metabolically stable.

RAM2061 and RAM2093 display metabolic stability in HLMs. Metabolic elimination profiles (% turnover or amount remaining vs. incubation time) for reaction with RAM2061, reaction with RAM2093, negative control (reaction without NADPH), and in-house positive control (testosterone). Data are shown as mean ± S.D. (n = 3).
Plasma Protein Binding.
Plasma protein binding for RAM2061 and RAM2093 were assessed in vitro. Both compounds were stable for the duration of the study. Device recovery was complete with no nonspecific binding to the dialysis membrane. Controls drugs propranolol (93.5% ± 1.7%) and atenolol (5.2% ± 1.1%) were within the acceptable limits reported in literature. Both RAM2061 and RAM2093 were highly bound to mouse plasma proteins, with greater than 93% of both compounds bound at both concentrations evaluated (Table 6). Protein binding was concentration independent for the two concentrations evaluated and similar for both isomers. Similar results were obtained when plasma samples from the PK studies were analyzed, with greater than 93% of both compounds bound to plasma proteins at the two tested time points (2 and 8 hours postinfusion).
Plasma protein binding of RAM2061 and RAM2093 in mouse plasma (n = 3, mean ± S.D.)
RAM2061 Slows Tumor Growth In Vivo.
To determine whether RAM2061 could inhibit MM tumor growth in vivo, MM1.S cells were inoculated in the flanks of NOD/SCID mice. When tumors became palpable (approximately 2 weeks post injection), mice were divided into a treatment arm (0.08 mg/kg i.v. RAM2061 twice weekly) or a control group (PBS twice weekly). No loss of weight was observed during treatment (Supplemental Fig. 6). As shown in Fig. 8A, treatment with RAM2061 significantly slowed tumor cell growth relative to the controls. Furthermore, mice treated with GGDPS inhibitor had longer overall time until sacrifice compared with control animals (Fig. 8B). Immunoblot analysis confirmed disruption of protein geranylgeranylation in the tumors of mice treated with RAM2061 (Fig. 8C). These results confirm the in vivo efficacy of our novel GGDPS inhibitor.

RAM2061 slows MM tumor growth in vivo. (A) Tumor growth curves for MM.1S flank xenografts. Mice were treated twice weekly with either solvent only control (PBS, n = 10 mice) or RAM2061 (n = 9 mice). Data are displayed as mean ± S.E.M. A linear mixed model was used for statistical analysis. (B) Kaplan-Meier curve for MM1.S flank xenografts showing time to sacrifice. Mice were euthanized when tumors reached 2000 mm3. (C) Immunoblot analysis of unmodified Rap1a in tumors collected from the treated animals. β-tubulin is shown as a loading control. Densitometric analysis of unmodified Rap1a (normalized to β-tubulin) in tumors of the treated animals normalized to untreated (control) is shown as relative intensity (Rel Int).
Discussion
We have previously reported preclinical studies with a potent GGDPS inhibitor, VSW1198 (a mixture of homogeranyl/homoneryl triazole bisphosphonates), which demonstrated the agent’s metabolic stability, prolonged terminal half-life, systemic distribution, and confirmed in vivo disruption of geranylgeranylation (Haney et al., 2018). Generation of VSW1198 derivatives led to the development of RAM2061 and RAM2093, which have equivalent GGDPS inhibitory potency in vitro (Matthiesen et al., 2018). These compounds allowed us to determine the impact of olefin stereochemistry on the PK and pharmacodynamic properties of our triazole-based GGDPS inhibitors. Whereas several clinically used agents are chiral and there is precedence for enantiomers having different relative activities against the same target, as well as different PK profiles (Eriksson et al., 1995, 2001; Fischer et al., 2014), there is no literature precedence for whether olefin isomers of a drug would have distinct PK/biodistribution properties. Our studies reveal that although the two isomers have equivalent potency in vitro, the MTD of the homoneryl derivative RAM2061 is greater than the homogeranyl derivative RAM2093. Biodistribution studies that used the same dose of the two inhibitors revealed higher hepatic drug levels across multiple time points for RAM2093 relative to RAM2061, with otherwise similar levels observed in other tissues.
For all three of our lead triazole-based GGDPS inhibitors (VSW1198, RAM2061, RAM2093), the dose-limiting toxicity is hepatic in nature. The single MTD for RAM2061 was 0.5 mg/kg i.v., which is identical to that of previously characterized VSW1198 (Haney et al., 2018); however, the MTD of RAM2093 was lower at 0.3 mg/kg, and the multidose MTD of RAM2093 was also lower than that of RAM2061. Comparison of tissue distribution patterns found that, relative to RAM2061, RAM2093 was present at approximately 2.5-fold higher concentrations in the liver up to 72 hours postinjection. The in vitro HLM studies did not show evidence of hepatic metabolism of either isomer. Whereas HLM testing is a well established method for in vitro screening for phase 1 metabolism, it is possible that alternative metabolic pathways are differentially affecting the two RAM compounds. Of note, comparison of plasma protein binding did not detect any differences in the rate of binding between the two isomers. In addition, although it is possible that there could be nonspecific binding of the inhibitors to various tissues, the results of the biodistribution studies reveal very similar levels in the nonhepatic tissues. Collectively, these results suggest that the stereochemistry of the olefin isomers contributes to differences in hepatic uptake, potentially through differential affinity to hepatic transporters, or via hepatic clearance. The mechanism by which this occurs warrants further study.
Our previous studies suggest that GGDPS inhibitor-induced liver toxicity is not a consequence of the triazole moiety but is likely related to GGDPS inhibition itself (Haney et al., 2018). Whether the hepatic toxicity is a consequence of disruption of function of GGTase I substrates versus GGTase II substrates versus perturbation in isoprenoid levels remains to be determined. In a study in which SCID mice were treated with daily intraperitoneal dosing with a GGTase I inhibitor (P661A6), mild transaminase elevation at ≥2.32 mg/kg dosing without histopathological alteration was reported (Lu et al., 2009). A phase 1 study of the GGTase I inhibitor GGTI-2418 in patients with solid tumors was conducted (O’Dwyer et al., 2010). No dose-limiting toxicities were observed, and no grade 3/4 liver function test abnormalities were reported. The most common adverse events were fatigue, diarrhea, neutropenia, and pruritus. There are limited data available regarding the in vivo use of GGTase II inhibitors. 3-PEHPC is a carboxyphosphonate derivative of risedronate which inhibits GGTase II (Coxon et al., 2001, 2005). In one study, the 5T2MM syngeneic model of MM was used, and animals were treated with 3-PEHPC (0.125 mg/kg, SC, twice weekly) to assess effects on bone disease (Lawson et al., 2008). No toxicity data were included.
Previous reports on the in vivo efficacy of GGDPS inhibitors are limited. One study found that the GGDPS inhibitor, GGOHBP, was able to reduce adrenal tumor burden by approximately 50% in mice (Reilly et al., 2015). Another study using the same GGDPS inhibitor reported that this agent slowed the development of metastasis in a preventative mouse model of prostate cancer (Reilly et al., 2017). Neither study monitored for toxicity apart from recording animal weight. More recently, a series of thienopyrimidine-based GGDPS inhibitors was reported (Lacbay et al., 2018). Preclinical studies with one of these inhibitors demonstrated antiplasma cell activity in vivo as determined by decreased serum monoclonal protein levels in a Vk*MYC transgenic mouse model that slowly develops clonal plasma cells producing monoclonal protein (Lacbay et al., 2018). In contrast to the triazole-based inhibitors presented in the current report, the thienopyrimidine-based inhibitor was found to undergo metabolism in liver microsome studies (estimated terminal half-life clearance of ∼2 to 3 hours) to require daily administration at higher doses (3 mg/kg via intraperitoneal injection) and to have bone affinity equivalent to zoledronic acid, which would be expected to limit sustained systemic distribution. Whereas the bone affinity of the triazole-based GGDPS inhibitors has not yet been determined, the PK and biodistribution studies provide evidence of prolonged systemic exposure. Thus, in contrast to the clinically used nitrogenous bisphosphonates (e.g., zoledronic acid), which are quickly distributed to the bones and kidneys and therefore rapidly eliminated from the circulation (Lin, 1996; Cremers et al., 2005, 2019), the triazole-based GGDPS inhibitors such as RAM2061 and RAM2093 have the potential for providing therapeutically meaningful systemic anticancer activity. Despite the high level of plasma protein binding, leading to free drug concentrations in the plasma that would be expected to be subtherapeutic (e.g., ∼10 nM at 2 hours post-single dose infusion), tissue exposure after repeated doses is sufficient to not only disrupt protein geranylgeranylation (Figs. 4 and 8) but also slow tumor growth in vivo (Fig. 8). The MM xenograft studies reported here, along with our recent studies demonstrating the in vivo efficacy of VSW1198 in several mouse models of pancreatic adenocarcinoma (Haney et al., 2019), confirm the utility of these triazole-based GGDPS inhibitors.
In conclusion, our studies demonstrate that RAM2061 and RAM2093 have prolonged terminal half-lives, systemic distribution, and metabolic stability and can effectively disrupt protein geranylgeranylation in vivo. Furthermore, these inhibitors have favorable bone marrow exposure, which is important for MM therapy. Although in vitro studies showed equivalent activity of the two isomers, the in vivo studies have allowed selection of RAM2061 as the lead GGDPS inhibitor to be evaluated further in preclinical studies, and future studies will focus on the efficacy of RAM2061 in other models of MM, as well as other malignancies. It will be important to determine whether novel drug-delivery strategies could further diminish hepatic uptake of RAM2061, thereby allowing further dose escalation and increased bone marrow exposure. Collectively, these studies illustrate the therapeutic potential of RAM2061 as a first-in-class GGDPS inhibitor.
Authorship Contributions
Participated in research design: Haney, Murry, Holstein.
Conducted experiments: Haney, Varney, Chhonker, Holstein.
Contributed new reagents or analytic tools: Chhonker, Murry.
Performed data analysis: Haney, Chhonker, Talmon, Smith, Murry, Holstein.
Wrote or contributed to the writing of the manuscript: Haney, Smith, Murry, Holstein.
Note Added in Proof—Research supported by the Nebraska Department of Health & Human Services [LB-506] was accidentally not included in the Fast Forward version published August 16, 2019. The funding footnote has now been corrected.
Footnotes
- Received April 4, 2019.
- Accepted August 15, 2019.
This work was supported by the National Institutes of Health [Grants R01CA172070, P30 CA036727] and the Nebraska Department of Health & Human Services [LB-506].
Portions of these data have been presented as abstracts at the following meeting: American Society of Hematology Annual Meeting 2018.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- BCA
- bicinchoninic acid
- CK
- creatine kinase
- GGDPS
- geranylgeranyl diphosphate synthase
- GGPP
- geranylgeranyl pyrophosphate
- GGTase
- geranylgeranyl transferase
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MM
- multiple myeloma
- MTD
- maximum tolerated dose
- PBS
- phosphate-buffered saline
- PK
- pharmacokinetic
- UPR
- unfolded protein response
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}