


Abstract
Tamoxifen, raloxifene, and nafoxidine are selective estrogen receptor modulators (SERMs) reported to inhibit the catalytic activity of human aldehyde oxidase 1 (AOX1). How these drugs interact with AOX1 and whether other SERMs inhibit this drug-metabolizing enzyme are not known. Therefore, a detailed in vitro and in silico study involving parent drugs and their analogs was conducted to investigate the effect of specific SERMs, particularly acolbifene, bazedoxifene, and lasofoxifene on AOX1 catalytic activity, as assessed by carbazeran 4-oxidation, an AOX1-selective catalytic marker. The rank order in the potency (based on IC50 values) of AOX1 inhibition by SERMs was raloxifene > bazedoxifene ∼ lasofoxifene > tamoxifen > acolbifene. Inhibition of liver cytosolic AOX1 by bazedoxifene, lasofoxifene, and tamoxifen was competitive, whereas that by raloxifene was noncompetitive. Loss of 1-azepanylethyl group increased the inhibitory potency of bazedoxifene, whereas the N-oxide group decreased it. The 7-hydroxy group and the substituted pyrrolidine ring attached to the tetrahydronaphthalene structure contributed to AOX1 inhibition by lasofoxifene. These results are supported by molecular-docking simulations in terms of predicted binding modes, encompassing binding orientation and efficiency, and analysis of key interactions, particularly hydrogen bonds. The extent of AOX1 inhibition by bazedoxifene was increased by estrone sulfate and estrone. In summary, SERMs differentially inhibited human AOX1 catalytic activity. Structural features of bazedoxifene and lasofoxifene contributed to AOX1 inhibition, whereas those of acolbifene rendered it considerably less susceptible to AOX1 inhibition. Overall, our novel biochemical findings and molecular-docking analyses provide new insights into the interaction between SERMs and AOX1.
SIGNIFICANCE STATEMENT Aldehyde oxidase (AOX1) is a molybdo-flavoprotein and has emerged as a drug-metabolizing enzyme of potential therapeutic importance because drugs have been identified as AOX1 substrates. Selective estrogen receptor modulators (SERM), which are drugs used to treat and prevent various conditions, differentially inhibit AOX1 catalytic activity. Structural features of bazedoxifene and lasofoxifene contribute to AOX1 inhibition, whereas those of acolbifene render it considerably less susceptible to AOX1 inhibition. Our novel biochemical findings, together with molecular- docking analyses, provide new insights into the differential inhibitory effect of SERMs on the catalytic activity of human AOX1, how SERMs bind to AOX1, and increase our understanding of the AOX1 pharmacophore in the inhibition of AOX1 by drugs and other chemicals.
Introduction
Aldehyde oxidase is a member of the family of molybdo-flavoprotein, which requires molybdopterin and flavin adenine dinucleotide cofactors for its catalytic activity (Garattini and Terao, 2013). In humans, there is only one isoform of aldehyde oxidase (AOX1), and the transcript and protein are expressed primarily in the liver and adrenal gland, and a lesser extent in other tissues such as the kidney and lung (Moriwaki et al., 2001; Terao et al., 2016b). It is a phase I drug-metabolizing enzyme that catalyzes the oxidation of a broad variety of chemical scaffolds, such as aldehydes, azaheterocycles, and iminium ions (Pryde et al., 2010), and mediates the reduction of sulfoxides, N-oxides, and nitro compounds with the presence of an electron donor (Konishi et al., 2017; Paragas et al., 2017). Drugs such as methotrexate (Chladek et al., 1997; Kitamura et al., 1999), famciclovir (Rashidi et al., 1997), and idelalisib (Ramanathan et al., 2016) are substrates for human AOX1. The importance of AOX1 in drug metabolism is mainly due to its ability to metabolize azaheterocycles, which represent a common scaffold in drugs (Pryde et al., 2010). AOX1-catalyzed drug metabolism has led to failures in clinical trials because of its impact on drug clearance, resulting in unacceptable pharmacokinetic properties (Akabane et al., 2011) and renal toxicity (Diamond et al., 2010; Lolkema et al., 2015). Although knockout of Aox4 in mice is not lethal (Terao et al., 2009, 2016a), the consequence of knocking out other mouse Aox genes is not known. In the human, genetic polymorphisms in AOX1 affect its activity (Hartmann et al., 2012; Foti et al., 2017), and an association between AOX1 polymorphism and azathioprine dosing and response has been reported (Smith et al., 2009; Kurzawski et al., 2012).
Selective estrogen receptor modulators (SERMs) are drugs used to treat and prevent various conditions, including breast cancer and postmenopausal osteoporosis (Pickar et al., 2010). Chemical classes of SERMs, based on their backbone structures, include triphenylethylenes (e.g., tamoxifen, toremifene, ospemifene, droloxifene, clomifene), benzothiophenes (e.g., raloxifene, arzoxifene), tetrahydronaphthalenes (e.g., nafoxidine, lasofoxifene), indoles (e.g., bazedoxifene), and benzopyrans (e.g., acolbifene) (Dowers et al., 2006; DeGregorio et al., 2014; Patel and Bihani, 2018). Raloxifene, nafoxidine, and tamoxifen have been identified as inhibitors of human liver cytosolic phthalazine oxidation (Obach, 2004; Obach et al., 2004), which is catalyzed by AOX1 (Beedham et al., 1987). Bazedoxifene was approved by the European Medicines Agency as a single agent in 2009 (Conbrizar product information) and by the U.S. Food and Drug Administration in 2013 as a combination product with conjugated estrogens (Genazzani et al., 2015). Lasofoxifene was approved by the European Medicines Agency in 2009 and indicated for the treatment of osteoporosis in postmenopausal women. Acolbifene is undergoing late-stage clinical trials (Fabian et al., 2015). Currently, it is not known whether the indole and benzopyran classes of SERMs (Supplemental Fig. 1), which differ from the triphenylethylene, benzothiophene, and tetrahydronaphthalene classes of SERMs in their structural scaffold and substituents (Bansal and Lau, 2019), inhibit the catalytic activity of AOX1.
In the present study, the primary objectives were to: 1) compare and contrast the effect of acolbifene (a benzopyran), bazedoxifene (an indole), and lasofoxifene (a tetrahydronaphthalene) on the catalytic activity of AOX1, as assessed by carbazeran 4-oxidation (Xie et al., 2019) catalyzed by human tissue cytosol and recombinant AOX1 enzyme; 2) determine whether the structural analogs (Supplemental Fig. 1) of bazedoxifene and lasofoxifene are inhibitors of AOX1; 3) explore how these SERMs bind to AOX1 active site, as evaluated by molecular-docking analyses; and 4) investigate the effect of estrone sulfate and estrone on the extent of AOX1 inhibition by bazedoxifene, given that bazedoxifene is administered clinically in combination with conjugated estrogens. Our in vitro and in silico data provide new molecular insights into the interaction between specific SERMs and human AOX1.
Materials and Methods
Chemicals, Reagents, and Enzymes.
Acolbifene and arzoxifene hydrochloride were purchased from AdooQ Bioscience (Irvine, CA). Carbazeran, 4-oxo-carbazeran (also known as 4-hydroxycarbazeran), bazedoxifene N-oxide, des(1-azepanyl)ethylbazedoxifene, lasofoxifene, racemic 7-methoxylasofoxifene, cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol, and nafoxidine were purchased from Toronto Research Chemicals (North York, ON, Canada). Bazedoxifene acetate, tamoxifen, raloxifene, toremifene citrate, ospemifene, clomifene citrate, hydralazine, sodium valproate, tolbutamide, and DMSO were purchased from Sigma-Aldrich (St. Louis, MO). Droloxifene citrate was purchased from Abcam (Cambridge, UK). All other commercially available chemicals were of analytical or high performance liquid chromatographic grade. Rabbit anti-AOX1 primary antibody (catalogue TA321294) and recombinant human AOX1 (catalogue TP319221) for protein quantification were purchased from OriGene Technologies (Rockville, MD). Horseradish peroxidase–conjugated anti-rabbit secondary antibody (catalogue 043-426) and Rabbit (12–180 kDa) Size Separation Master Kit (catalogue CBS-01-01) were bought from ProteinSimple (San Jose, CA).
Human liver cytosol (mixed gender; pool of 150 donors; 20 mg/ml; catalogue 452115, lot 38290, Gentest brand; 75 males and 75 females) was purchased from Corning (Corning, NY). Human kidney cytosol (mixed gender; pool of four donors; 5 mg/ml; catalog H0610.RC, lot 1310121) and human lung cytosol [nonsmokers; mixed gender; pool of four donors; 5 mg/ml; catalogue H0610.PC(NS), lot 1310100] were purchased from Sekisui XenoTech (Kansas City, KS). Human recombinant AOX1 enzyme (catalogue CYP150, lot 150011B) and control cytosol (isolated from Escherichia coli host cells; catalogue CYP099, lot INT016E18C) were purchased from Cypex (Dundee, Scotland, UK).
Carbazeran 4-Oxidation Assay.
Incubation mixture (200 μl for assays containing human liver cytosol or 100 μl for assays containing human kidney cytosol, lung cytosol, or recombinant AOX1) consisted of potassium phosphate buffer (100 mM, pH 7.4), carbazeran, and an enzyme. The final concentration of DMSO in all samples was 1% v/v, which was shown not to affect the AOX1 activity (Behera et al., 2014). Each incubation mixture was prewarmed for 3 minutes at 37°C in a shaking water bath. Enzymatic reaction was initiated by adding liver cytosol (20 μg, 0.1 mg/ml final concentration), kidney cytosol (200 μg, 2 mg/ml final concentration), lung cytosol (150 μg, 1.5 mg/ml final concentration), or recombinant AOX1 (30 μg, 0.3 mg/ml final concentration). The mixture was incubated for 5 (liver cytosol), 75 (kidney and lung cytosol), or 15 minutes (recombinant AOX1). The reaction was terminated by adding an equal volume (200 or 100 μl) of ice-cold acetonitrile containing tolbutamide (25 nM final concentration; internal standard). Each sample was mixed and placed immediately in an ice bath. After centrifugation at 16,000g for 15 minutes at 4°C, the supernatant was transferred to a 96-well microplate for analysis of 4-oxo-carbazeran and tolbutamide by ultra-high performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS). To construct a calibration curve for each experiment, 4-oxo-carbazeran stock solutions (1–1000 μM in DMSO) were freshly added to the incubation mixture to give final concentrations of 1–1000 nM (0.2–200 pmol; in 0.1% v/v DMSO) and subjected to the same procedures as described above.
O6-Benzylguanine 8-Oxidation Assay.
The assay was conducted according to our previous study (Xie et al., 2019), except that the substrate concentration was 5–600 μM, the incubation time was 75 minutes, and the enzymatic reaction was initiated by adding kidney cytosol (200 μg, 2 mg/ml final concentration).
Quantification of 4-Oxo-Carbazeran by UPLC-MS/MS.
The UPLC-MS/MS system and the chromatographic and mass spectrometric conditions for analyzing 4-oxo-carbazeran and tolbutamide were reported in detail in our previous study (Xie et al., 2019). A calibration curve was constructed using weighted (1/x2) linear least-squares regression analysis of the peak area ratio (4-oxo-carbazeran to tolbutamide) versus amount of the metabolite standard added into the incubation mixture.
Enzyme Kinetics Analysis of Carbazeran 4-Oxidation.
Enzyme kinetic experiment was performed by conducting the carbazeran 4-oxidation assay at substrate concentrations ranging from 0.125 to 32 μM for human liver cytosol, 0.5 to 40 μM for human kidney cytosol, 1 to 32 μM for human lung cytosol, and 1 to 80 μM for recombinant AOX1 enzyme. The velocity (V)-versus-substrate concentration (S) data were analyzed by nonlinear least-squares regression analysis and fitted to several models (Michaelis–Menten, Hill, substrate inhibition, and substrate activation) using SigmaPlot version 12.5 (Systat Software, San Jose, CA). Based on visual inspection and various measures of goodness of fit, including Akaike information criterion, coefficient of determination (R2), and S.D. of residuals (Sy.x), the values of Vmax and the substrate concentration at half the maximum velocity (apparent Km) were calculated using the Michaelis–Menten model:  or the substrate inhibition model:
or the substrate inhibition model: 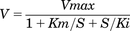 , where Ki represents the equilibrium dissociation constant between the substrate and the binding site of the enzyme. The turnover number, unbound fraction, corrected Km, and unbound intrinsic clearance (Clint,u) were calculated as described in our previous study (Xie et al., 2019).
, where Ki represents the equilibrium dissociation constant between the substrate and the binding site of the enzyme. The turnover number, unbound fraction, corrected Km, and unbound intrinsic clearance (Clint,u) were calculated as described in our previous study (Xie et al., 2019).
Enzyme Inhibition Experiments.
Enzyme inhibition was determined by conducting the carbazeran 4-oxidation assay in the presence of a SERM, a positive control, a negative control, or the vehicle (DMSO) at concentrations specified in each figure legend. Raloxifene, a known potent AOX1 inhibitor (Obach, 2004), was used as a positive control, whereas valproic acid was included as a negative control (Obach et al., 2004). In the concentration–response experiment, the incubation was conducted in the presence of varying concentrations of each chemical in human liver cytosol as described in each figure legend. The IC50 value was determined by nonlinear regression analysis using Sigmaplot 12.5 with the equation: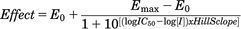 where I is the inhibitor concentration, E0 is the minimum effect, and Emax is the maximum effect.
where I is the inhibitor concentration, E0 is the minimum effect, and Emax is the maximum effect.
To determine the enzyme kinetics of the inhibition of the enzyme by SERMs, the carbazeran 4-oxidation assay was conducted in the presence of multiple concentrations (0.5, 1, 2, or 4 μM) of carbazeran and multiple concentrations of a SERM (bazedoxifene, lasofoxifene, tamoxifen, or raloxifene), as specified in the figure legend. The apparent Ki (apparent equilibrium dissociation constant for the enzyme–inhibitor complex) value and mode of inhibition were determined by nonlinear least-squares regression analysis of the metabolite formation data at various concentrations of the inhibitor and substrate, using equations for full and partial competitive, noncompetitive, uncompetitive, and mixed-mode inhibition (Sigmaplot 12.5). The best-fit model was determined by Akaike information criterion, R2, and visual inspection of the data in the Lineweaver–Burk plot. The equations for the full competitive inhibition model (eq. 1) and the full noncompetitive inhibition model (eq. 2) are as follows: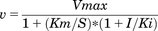 (1)
(1)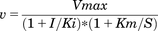 (2)where S represents the substrate concentration, I represents the inhibitor concentration, Vmax represents the apparent maximum reaction velocity, Km represents the substrate concentration at which the reaction rate is half of Vmax, and Ki represents the apparent equilibrium dissociation constant for the enzyme–inhibitor complex.
(2)where S represents the substrate concentration, I represents the inhibitor concentration, Vmax represents the apparent maximum reaction velocity, Km represents the substrate concentration at which the reaction rate is half of Vmax, and Ki represents the apparent equilibrium dissociation constant for the enzyme–inhibitor complex.
Time-Dependent Inhibition Experiment.
Primary incubation mixture (200 μl) contained potassium phosphate buffer (100 mM, pH 7.4), human liver cytosol (100 μg, 0.5 mg/ml), and a SERM (acolbifene, bazedoxifene, lasofoxifene, or tamoxifen, each at 10 μM), a positive control (hydralazine, 10 μM) (Strelevitz et al., 2012), a negative control (raloxifene, 0.02 μM) (Obach, 2004), or vehicle (DMSO, 0.5% v/v). The mixture was prewarmed for 3 minutes at 37°C in a shaking water bath, and the reaction was initiated by adding the enzyme. At 0 and 30 minutes after preincubation, an aliquot (10 μl) from the primary incubation mixture was transferred to 190 μl prewarmed (for 3 minutes at 37°C) secondary incubation mixture (total volume of 200 μl) containing potassium phosphate buffer and carbazeran (16 μM). The enzymatic reaction in the secondary incubation mixture was incubated for 5 minutes at 37°C and terminated by adding 200 μl ice-cold acetonitrile containing tolbutamide (25 nM final concentration; internal standard). The samples were processed in the same manner as that described under Carbazeran 4-Oxidation Assay.
Molecular Docking.
The structure of the AOX1 protein was obtained from the crystal structure of human AOX1 [Protein Data Bank (PDB): 4UHW chain A] (Coelho et al., 2015) at 98.9% identity to the protein sequence (UniProt AC Q06278). The molecular-docking methodology involved contending with the presence of a molybdenum cofactor and several open side channels, as well as a relative scarcity of cocrystallized ligands. To simulate electrostatic interactions in this binding site, a mix of partial charge methods was adopted, applying a charge transfer method, QTPIE (Chen and Martinez, 2007), to the chemically-unusual dioxothiomolybdenum ion , and Antechamber (Wang et al., 2006) for assignment of the other protein charges. The sulfur atom in the dioxothiomolybdenum ion residue was manually protonated. Ligand charges were assigned using an approximation of the Amber AM1-BCC method, called EEM-Cheminf-HF-MPA (Geidl et al., 2015). For the five compounds—bazedoxifene, lasofoxifene, tamoxifen, acolbifene, and raloxifene—an extensive (∼10-hour) docking simulation was applied with constraints, allowing ligands to be flexible but with fixed amino acid side chains, optimizing a customized CHEMPLP objective function using the PLANTS program (Korb et al., 2009). The constraints were placed in the side subpockets and acted to guide the ligand toward the binding subpocket as observed in the 4-[5-(2,6-dioxo-1,2,3,6-tetrahydropyridin-4-yl)-1H-1,2,4-triazol-3-yl]-6-oxo-1,6-dihydropyridine-2-carbonitrile interactions with the crystal structure for bovine xanthine oxidase (PDB:3 AM9). Subsequently, for the five compounds, along with their chemically-related derivatives—the bazedoxifene derivatives des(1-azepanyl)ethylbazedoxifene and bazedoxifene N-oxide, and the lasofoxifene derivatives 7-methoxylasofoxifene, cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol, and nafoxidine—an energy minimization was performed in which the initial pose was based on superposition with the result of the first docking run (Kawabata, 2011). For this energy minimization, the ligand was fixed and receptor side chains were allowed to be fully flexible.
AOX1 Protein Quantification by a Capillary Nano-Proteomic Immunoassay.
The amount of AOX1 in cytosols was quantified in a capillary nano-proteomic immunoassay (SimpleWestern System; ProteinSimple), as described in our previous study (Xie et al., 2019).
Statistical Analysis.
Data were analyzed by one-way or two-way ANOVA and, where appropriate, were followed by the Student–Newman–Keuls multiple comparison test (SigmaPlot 12.5). The level of statistical significance was set a priori at P < 0.05.
Results
Enzyme Kinetics of Carbazeran 4-Oxidation Catalyzed by Human Tissue Cytosols and Recombinant AOX1 Enzyme.
Experiments were performed to determine the linear range of the carbazeran 4-oxidation assay with respect to the amount of cytosolic protein (Supplemental Fig. 2) and incubation time (Supplemental Fig. 3) in different types of human tissue cytosol and recombinant AOX1. Shown in Supplemental Table 1 are the assay conditions used in the carbazeran 4-oxidation assay. The catalysis of carbazeran 4-oxidation by liver cytosol, lung cytosol, and recombinant AOX1 followed the Michaelis–Menten model, whereas that by kidney cytosol followed the substrate inhibition model (Supplemental Fig. 4, A–D). As shown in Supplemental Table 2, the turnover number was considerably greater in liver cytosol than in kidney and lung cytosol. The same rank order also occurred in the abundance of AOX1 protein in these cytosol samples. Liver cytosol, kidney cytosol, lung cytosol, and recombinant AOX1 enzyme catalyzed carbazeran 4-oxidation with corrected Km at low micromolar concentrations. Liver cytosol was also considerably more efficient than kidney cytosol and lung cytosol in catalyzing carbazeran 4-oxidation, as assessed by Clint,u. The relative difference in the Clint,u in liver cytosolic and kidney cytosolic carbazeran 4-oxidation was similar to that obtained in liver cytosolic and kidney cytosolic O6-benzylguanine 8-oxidation (Supplemental Table 2), which is another catalytic marker of human AOX1 (Xie et al., 2019). As expected, the E. coli cytosol, which was the control for the recombinant AOX1 enzyme, did not yield any metabolite (data not shown).
Effects of SERMs on Carbazeran 4-Oxidation Catalyzed by Human Tissue Cytosol and Recombinant AOX1.
To investigate whether acolbifene, bazedoxifene, and lasofoxifene inhibit AOX1 activity and to compare their effects with those of other SERMS (tamoxifen, toremifene, ospemifene, droloxifene, clomifene, raloxifene, and arzoxifene), liver cytosol was incubated with a SERM (25 μM) at a substrate concentration (3 μM) that was near the apparent Km value. As shown in Fig. 1, acolbifene, bazedoxifene, lasofoxifene, tamoxifen, toremifene, ospemifene, droloxifene, clomifene, raloxifene, and arzoxifene decreased liver cytosolic carbazeran 4-oxidation by 26%, 98%, 98%, 88%, 91%, 89%, 94%, 60%, 100%, and 67%, respectively. In contrast, acolbifene showed only little or no inhibition of liver cytosolic carbazeran 4-oxidation.

Comparative effect of acolbifene, bazedoxifene, lasofoxifene, tamoxifen, toremifene, ospemifene, droloxifene, clomifene, raloxifene, and arzoxifene on carbazeran 4-oxidation catalyzed by human liver cytosol. A SERM (25 μM), valproic acid (50 μM; negative control), or DMSO (1% v/v; vehicle) was coincubated with carbazeran (3 μM) and pooled liver cytosol (20 μg protein) at 37°C for 5 minutes. Data are expressed as percentage of activity in the vehicle-treated control group and expressed as mean ± S.E.M. of three independent experiments conducted in duplicate. *Significantly different from the vehicle-treated control group (P < 0.05). The rate of reaction in the vehicle-treated control group was 567 ± 18 pmol/min per milligram protein.
Additional experiments were performed to determine the inhibitory effect of acolbifene, bazedoxifene, and lasofoxifene on carbazeran 4-oxidation catalyzed by human kidney cytosol and recombinant AOX1. Each of these drugs inhibited carbazeran 4-oxidation catalyzed by human kidney cytosol (Supplemental Fig. 5A) and recombinant AOX1 (Supplemental Fig. 5B). The magnitude of the inhibition was similar to that occurred in enzymatic incubations containing liver cytosol (Fig. 1). By comparison, tamoxifen, raloxifene, but not valproic acid (negative control), inhibited carbazeran 4-oxidation by kidney cytosol (Supplemental Fig. 5A) or recombinant AOX1 enzyme (Supplemental Fig. 5B). Given that the acolbifene, bazedoxifene, and lasofoxifene inhibited carbazeran 4-oxidation catalyzed by various tissue cytosols and recombinant AOX1 in a similar pattern (Fig. 1; Supplemental Fig. 5, A and B), subsequent inhibition experiments were conducted with liver cytosol.
Concentration–Response Relationship in the Inhibition of Human Liver Cytosolic AOX1-Mediated Carbazeran 4-Oxidation by Acolbifene, Bazedoxifene, and Lasofoxifene: Comparison with Tamoxifen and Raloxifene.
To determine the inhibitory potency (IC50) and the minimum inhibitory concentration of acolbifene, bazedoxifene, lasofoxifene, tamoxifen, or raloxifene in the inhibition of AOX1 catalytic activity, a concentration–response experiment was conducted with varying concentrations of each SERM. As shown in Fig. 2, these SERMs decreased carbazeran 4-oxidation in a concentration-dependent manner, and with a sigmoidal-shaped concentration–response curve. Among the SERMs investigated, raloxifene was the most potent (IC50 = 0.028 μM), whereas bazedoxifene, lasofoxifene, and tamoxifen inhibited the activity with IC50 values of 0.19, 0.30, and 7.30 μM, respectively (Table 1). In contrast, acolbifene did not completely inhibit AOX1 activity at the highest concentration tested and was the least potent inhibitor of AOX1 (IC50 value of 29.5 μM). Raloxifene, bazedoxifene, and lasofoxifene had comparable minimum AOX1 inhibitory concentrations (0.001–0.03 μM), but they were 0.1%–0.3% of those for tamoxifen and acolbifene (Table 1).

Concentration–response relationship in the inhibition of human liver cytosolic AOX1-mediated carbazeran 4-oxidation by acolbifene, bazedoxifene, lasofoxifene, tamoxifen, and raloxifene. Pooled liver cytosol (20 μg protein) was incubated with carbazeran (3 μM) and varying concentrations of (A) acolbifene (0.3–300 μM), (B) bazedoxifene (0.0003–25 μM), (C) lasofoxifene (0.0003–30 μM), (D) tamoxifen (0.003–100 μM), (E) raloxifene (0.00001–1 μM), or DMSO (1% v/v; vehicle) at 37°C for 5 minutes. Data are expressed as percentage of activity in the vehicle-treated control group and expressed as mean ± S.E.M. of three or four independent experiments conducted in duplicate or triplicate. *Significantly different from the vehicle-treated control group (P < 0.05).
IC50 values and minimum inhibitory concentration in the inhibition of human liver cytosolic AOX1-mediated carbazeran 4-oxidation by SERMs and the structural analogs of bazedoxifene and lasofoxifene
Data are expressed as mean ± S.E.M. of three to seven independent experiments conducted in duplicate or triplicate.
Mode of Inhibition of Human Liver Cytosolic AOX1-Mediated Carbazeran 4-Oxidation by Bazedoxifene, Lasofoxifene, Tamoxifen, and Raloxifene.
To determine the apparent Ki and mode of inhibition of carbazeran 4-oxidation, liver cytosol was incubated with various concentrations of substrate and a SERM. Based on the nonlinear regression analysis and Lineweaver–Burk plots (Fig. 3), bazedoxifene, lasofoxifene, and tamoxifen inhibited carbazeran 4-oxidation by a competitive mode, whereas raloxifene inhibited it by a noncompetitive mode. As shown in Table 2, both bazedoxifene and lasofoxifene yielded submicromolar Ki value of 0.14 ± 0.03 µM, whereas tamoxifen was approximately 20 times less potent and raloxifene was five times more potent than bazedoxifene and lasofoxifene.

Lineweaver–Burk plots for inhibition of human liver cytosolic AOX1-mediated carbazeran 4-oxidation by bazedoxifene, lasofoxifene, tamoxifen, and raloxifene. Pooled liver cytosol (20 μg protein) was incubated with carbazeran (0.5, 1, 2, or 4 μM) and varying concentrations of (A) bazedoxifene (0, 0.1, 0.3, or 1 μM), (B) lasofoxifene (0, 0.1, 0.3, or 1 μM), (C) tamoxifen (0, 1, 3, or 10 μM), or (D) raloxifene (0, 0.03, 0.06, or 0.1 μM) at 37°C for 5 minutes. Data are expressed as percentage of activity in the vehicle-treated control group and expressed as mean ± S.E.M. of four to five independent experiments conducted in duplicate.
Apparent Ki values and mode of inhibition of human liver cytosolic AOX1-mediated carbazeran 4-oxidation by bazedoxifene, lasofoxifene, tamoxifen, and raloxifene
Data are expressed as mean ± S.E.M. for four or five independent experiments conducted in duplicate.
Comparative Effects of a Metabolite and Structural Analog of Bazedoxifene on the Inhibition of Human Liver Cytosolic AOX1-Mediated Carbazeran 4-Oxidation.
The effect of bazedoxifene and its metabolite and structural analog (Supplemental Fig. 1) on the inhibition of carbazeran 4-oxidation was compared (Fig. 4). Bazedoxifene, bazedoxifene N-oxide, and des(1-azepanyl)ethylbazedoxifene, each at 25 μM, decreased carbazeran 4-oxidation by 98%, 95%, and 99%, respectively (Fig. 4A). The IC50 value for bazedoxifene (0.19 ± 0.04 μM) was less than that of bazedoxifene N-oxide (0.29 ± 0.07 μM), but greater than that of des(1-azepanyl)ethylbazedoxifene (0.10 ± 0.02 μM) (Table 1).

Inhibition of human liver cytosolic AOX1-mediated carbazeran 4-oxidation by metabolite/structural analogs of bazedoxifene. (A) Pooled liver cytosol (20 μg protein) was incubated with carbazeran (3 μM) and bazedoxifene, bazedoxifene N-oxide, des(1-azepanyl)ethylbazedoxifene (each at 25 μM), valproic acid (50 μM; negative control), or DMSO (1% v/v; vehicle) at 37°C for 5 minutes. (B and C) Pooled liver cytosol (20 μg protein) was incubated with carbazeran (3 μM) and varying concentrations of (B) bazedoxifene N-oxide (0.0003–25 μM), (C) des(1-azepanyl)ethylbazedoxifene (0.0003–25 μM), or DMSO (1% v/v; vehicle) at 37°C for 5 minutes. Data are expressed as percentage of activity in the vehicle-treated control group and expressed as mean ± S.E.M. of three to five independent experiments conducted in duplicate. *Significantly different from the vehicle-treated control group (P < 0.05).
Comparative Effects of the Structural Analogs of Lasofoxifene on the Inhibition of Human Liver Cytosolic AOX1-Mediated Carbazeran 4-Oxidation.
To elucidate the structural features of lasofoxifene contributes to the inhibition of AOX1, the effect of three structural analogs [7-methoxylasofoxifene, cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol, and nafoxidine; Supplemental Fig. 1] of lasofoxifene on carbazeran 4-oxidation was compared. At 25 µM, lasofoxifene, 7-methoxylasofoxifene, cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol, and nafoxidine decreased carbazeran 4-oxidation by 97%, 92%, 46%, and 77%, respectively (Fig. 5A). Concentration–response experiments (Fig. 5, B–D) indicated that the IC50 value for 7-methoxylasofoxifene was approximately 5-fold greater than that for lasofoxifene (0.30 ± 0.03 μM), whereas it was 8-fold greater than the IC50 value for cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol and nafoxidine (Table 1).

Inhibition of human liver cytosolic AOX1-mediated carbazeran 4-oxidation by the tetrahydronaphthalene class of SERMs. (A) Pooled liver cytosol (20 μg protein) was incubated with carbazeran (3 μM) and lasofoxifene, 7-methoxylasofoxifene, cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol, nafoxidine (each at 25 μM), valproic acid (50 μM; negative control), or DMSO (1% v/v; vehicle) at 37°C for 5 minutes. (B–D) Pooled liver cytosol (20 μg protein) was incubated with carbazeran (3 μM) and varying concentrations of (B) 7-methoxylasofoxifene (0.003–100 μM), (C) cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol (0.03–30 μM), (D) nafoxidine (0.003–100 μM), or DMSO (1% v/v; vehicle) at 37°C for 5 minutes. Data are expressed as percentage of activity in the vehicle-treated control group and expressed as mean ± S.E.M. of three to seven independent experiments conducted in duplicate. *Significantly different from the vehicle-treated control group (P < 0.05). #Significantly different from the vehicle-treated control group and the lasofoxifene-treated group.
Molecular Docking of SERMs and Their Structural Analogs to the Active Site of Human AOX1.
The investigation of the binding of SERMs and structural analogs to human AOX1 by molecular docking revealed that the strong competitive binders demonstrated a combination of specific key interactions that the weaker competitively inhibiting compounds did not (Fig. 6; Table 3). For bazedoxifene (pink, Fig. 6) and lasofoxifene (salmon, Fig. 6), hydrogen bonds were predicted between the ligands and the molybdenum cofactor and Asn-1084. The binding of bazedoxifine appeared to be particularly strong because of its unique hydrogen bond to Glu-888 and its strong van der Waals overlapped with the Phe-885 ring. Phe-885 has previously been predicted to be involved in interactions with substrates of AOX1 (Lepri et al., 2017). The weaker competitive binders, tamoxifen (blue, Fig. 6) and acolbifene (purple, Fig. 6), lacked these predicted interactions. Raloxifene (dark blue, Fig. 6) inhibited strongly, but noncompetitively, and it may bind allosterically to the surface of AOX1 at a location similar to the binding site for 10-{2-[(2R)-1-methylpiperidin-2-yl]ethyl}-2-(methylsulfanyl)-10H-phenothiazine identified in the crystal structures (Coelho et al., 2015). In this study, it was used to some extent as a negative control for docking to this site. Alternate hydrogen bonds were predicted for raloxifene to Lys-893 and Glu-1270, consistent with the observed strong inhibition.

Molecular docking of SERMs and their structural analogs to the active site of human AOX1. The predicted binding of the 10 compounds is shown with the molybdenum cofactor [MOS, dioxothiomolybdenum (VI) ion] visible toward the center of each frame and the distance between it and the ligand central oxygen atom shown in pink. The key residues are shown as labeled. Computed hydrogen bonds are shown in light blue.
Analysis of SERM ligand docking and key interactions with human AOX1
Another key structural predictor that was found to correlate with the experimental IC50 values, at least for the competitive inhibitors, was the distance between the ligand central oxygen atom and the molybdenum cofactor (O-Mo distance), as summarized in Table 3 and shown in Fig. 6. For the tightest bindings, associated with the lowest IC50 values, the O-Mo distance was reduced, suggesting more intimate interaction with the catalytic site of the enzyme. Conversely, SERMs with less favorable fitting within this pocket were forced to adopt docked poses that oriented their central oxygen atom away from the molybdenum cofactor. Raloxifene was again the exception to the trend, further supporting the different binding mechanism indicated by inhibition studies.
For the study of the binding of the analogs, the binding scores generated by the docking algorithm can only sensibly be used as an approximate indication of the relative binding energy of closely structurally related compounds. It can be difficult to globally correlate the predicted binding scores of a docking program with experimental IC50 values or to compare binding scores from different ligands in substantially different orientations. However, in the case of the closely related bazedoxifene and lasofoxifene and their analogs, there was a positive correlation (r2 = 0.78) between log IC50 values and binding efficiency (where the binding efficiency = binding score/mass of ligand) (Fig. 7). Table 4 details the simulated binding efficiency data for bazedoxifene and lasofoxifene and their analogs. Compared with bazedoxifene, the analog compound des(1-azepanyl)ethyl bazedoxifene yielded a relative log (IC50) of 104.2% and a relative binding efficiency of 104.5%. The slightly weaker binder, bazedoxifene N-oxide, yielded a relative log (IC50) of 97.3% and a relative binding efficiency of 94.6%. For the lasofoxifene analogs, an important structural difference was observed in the predicted binding: in lasofoxifene (pink), there was a hydrogen bond to Asn-1084, whereas in the analogs, for example, nafoxidene (purple), this hydrogen bond was not present. The binding efficiencies, although perhaps less predictive than in bazedoxifene, still ranked lasofoxifene as the best of the derivatives, followed by 7-methoxylasofoxifene. Overall, a strong relationship was shown between the in silico determination of binding efficiency and the in vitro biochemical data for the inhibition of human AOX1 by bazedoxifene, lasofoxifene, and their analogs.

Docking analysis in the molecular interaction between human AOX1 and bazedoxifene, lasofoxifene, and their structural analogs
Effect of Conjugated Estrone and Estrone on the Inhibition of Human Liver Cytosolic AOX1-Mediated Carbazeran 4-Oxidation by Bazedoxifene.
Bazedoxifene in combination with conjugated estrogens (Duavee), such as estrone sulfate, is approved by the United States Food and Drug Administration for the treatment of moderate to severe vasomotor symptoms associated with menopause and prevention of postmenopausal osteoporosis (Cada and Baker, 2014). Circulating estrone sulfate can be metabolized in vivo to the active form, estrone, by sulfatases. Previously, estrone was shown to inhibit aldehyde oxidase (Obach, 2004; Obach et al., 2004). Therefore, we compared the effect of estrone sulfate and estrone on the inhibition of AOX1 catalytic activity by bazedoxifene. Concentration–response data indicated that estrone was more potent than estrone sulfate in decreasing human liver cytosolic 4-oxidation, with experimentally derived IC50 values of 0.18 ± 0.01 and 258 ± 51 μM, respectively (Fig. 8A). Estrone at 0.3, 1, 3, and 10 μM and estrone sulfate at 300 and 1000 μM enhanced the inhibitory effect of bazedoxifene on human liver cytosol-catalyzed carbazeran 4-oxidation (Fig. 8B).

Effect of estrone and estrone sulfate on inhibition of human liver cytosolic AOX1-mediated carbazeran 4-oxidation by bazedoxifene. (A) Pooled liver cytosol (20 μg protein) was incubated with carbazeran (3 μM) and varying concentrations of estrone (0.0003–30 μM), estrone sulfate (1–3000 μM), or DMSO (1% v/v; vehicle) at 37°C for 5 minutes. (B) Pooled liver cytosol (20 μg protein) was incubated with carbazeran (3 μM) and the combination of bazedoxifene (0.1 μM) with varying concentrations of estrone sulfate (30–1000 μM), estrone (0.03–10 μM), or DMSO (1% v/v; vehicle) at 37°C for 5 minutes. Data are expressed as percentage of activity in the vehicle-treated control group and expressed as mean ± S.E.M. of three or four independent experiments conducted in duplicate. *Significantly different from the vehicle-treated control group (P < 0.05). #Significantly different from the bazedoxifene-treated group (P < 0.05). The bazedoxifene (0.1 μM) group alone inhibited carbazeran 4-oxidation by 41% ± 3% in the estrone experiment and 38% ± 4% in the estrone sulfate experiment.
Effect of Preincubation of Human Liver Cytosol with a SERM on AOX1-Mediated Carbazeran 4-Oxidation.
To investigate whether acolbifene, bazedoxifene, and lasofoxifene exhibit time-dependent inhibition of AOX1, each of these SERMs was preincubated with liver cytosol for 30 minutes before transferring an aliquot of the primary incubation mixture to a secondary incubation mixture containing the substrate (carbazeran). Preincubation of acolbifene, bazedoxifene, lasofoxifene, or tamoxifen with liver cytosol did not increase the extent of AOX1 inhibition (Supplemental Fig. 6), consistent with the lack of an effect of preincubation on the extent of AOX1 inhibition by raloxifene (Obach, 2004). In contrast, hydralazine, a known time-dependent inhibitor of AOX1 (Strelevitz et al., 2012), yielded the expected result (Supplemental Fig. 6).
Discussion
A novel aspect of the present study is the differential inhibitory effect of SERMs on the catalytic activity of human liver cytosolic AOX1. The rank order in the potency (based on IC50 values) of SERM inhibition of AOX1 was raloxifene > bazedoxifene ∼ lasofoxifene > tamoxifen > acolbifene. Bazedoxifene and lasofoxifene inhibited AOX1 with apparent Ki values at submicromolar concentrations. These two SERMs were 20 times more potent than tamoxifen and five times less potent than raloxifene. Lasofoxifene, bazedoxifene, and tamoxifen inhibited AOX1 activity by a competitive mode, whereas raloxifene inhibited it by a noncompetitive mode. Raloxifene inhibits human liver cytosol-catalyzed oxidation of vanillin, phthalazine, and nicotine-Δ1′(5′)-iminium ion in an uncompetitive manner (Obach, 2004), whereas it inhibits the oxidation of N-[(2-dimethylamino)ethyl]acridine-4-carboxamide by a competitive mode (Barr and Jones, 2013). Collectively, these results indicate a degree of selectivity in human AOX1 inhibition by SERMs, and their mode of AOX1 inhibition appears to be substrate-dependent.
The potency of AOX inhibition by raloxifene has been linked to the bisphenol structure and the hydrophobic alkylamino side chain of this SERM (Obach, 2004). These essential features are conserved in the structure of bazedoxifene, whereas lasofoxifene possesses a monophenol and an alkylamino side chain, which were shown to retain potent inhibitory activity. In contrast, acolbifene has the essential features of a bisphenol structure and alkylamino side chain. However, it was identified as a relatively weak inhibitor and the least potent among the SERMs investigated in the present study. Despite these shared structural elements, acolbifene possesses a six-membered pyran ring, whereas raloxifene and bazedoxifene have a five-membered thiophene and pyrrole ring, respectively. These suggest that the type of backbone structures plays a role in AOX1 inhibition by this class of drugs, although with differing degree of potency and efficacy. Alternatively, the orientation of the alkylamino side chain or the type of side chain in acolbifene may influence the binding to the enzyme and decreases the extent of AOX1 inhibition by acolbifene. The findings of our molecular-docking simulations were in broad agreement with our inhibition data. Correlative trends exist between the in vitro and in silico data, particularly between the measured IC50 values for the competitive inhibitors and the involvement of the molybdenum cofactor and key residues in ligand binding, and also in the indication of a notably different binding mechanism for the noncompetitive inhibitor raloxifene.
Another important finding of the present study is that N-oxidation and removal of the 1-(azepanyl)ethyl moiety affect the inhibitory potency of bazedoxifene. The IC50 in the inhibition of AOX1 by bazedoxifene N-oxide was greater than that of bazedoxifene, indicating that the N-oxide group decreases the potency of bazedoxifene, whereas the IC50 value of des(1-azepanyl)ethylbazedoxifene was less than that of bazedoxifene, indicating that the loss of the 1-(azepanyl)ethyl moiety increases the potency of bazedoxifene. A large positive correlation exists between the measured IC50 values and the calculated binding efficiency values obtained for these compounds from the docking analyses. In another study (Barr et al., 2015), the investigators developed a quantitative structure–activity relationship based on a homology model of human AOX1 derived from the crystal structure of mouse AOX3 (PDB ID 3ZYV) (Coelho et al., 2012). They concluded that dipole, hydrophilic solvent-accessible surface area, and hydrogen bonding-accepting capacity were descriptors that explained the inhibitory potency of naturally-occurring chemicals, such as flavanoids, catechin, stillbenoid, and coumarin (Barr et al., 2015). The importance of hydrogen bonding is reiterated in our study.
The present study identified for the first time the structural features of lasofoxifene that contribute to its potent inhibition of AOX1 activity. The C7 hydroxyl group is the most important for the inhibitory potency of lasofoxifene based on the following experimental evidence: 1) a sole switch of C7 hydroxyl group to methoxy group in 7-methoxylasofoxifene resulted in attenuation of AOX1 inhibition, and the IC50 value of 7-methoxylasofoxifene was ∼5 times greater than that of lasofoxifene; and 2) both cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol and nafoxidine, which possess a C7 methoxy group instead of the hydroxyl group, inhibited carbazeran 4-oxidation to a lesser extent compared with lasofoxifene, and their IC50 values were ∼8 times greater than that of lasofoxifene. The molecular-docking results support the importance of the C7 hydroxyl group for the binding of lasofoxifene, from where a hydrogen bond to Asn-1084 is identified. This hydrogen bond is lost in all of the lasofoxifene analogs where the C7 hydroxyl group is modified. The substituted pyrrolidine ring is also important for the inhibition of AOX1 by lasofoxifene because further attenuation of the inhibitory activity was observed when the substituted pyrrolidine ring was removed in cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol, as compared with 7-methoxylasofoxifene. However, the pyrrolidine ring appeared to be less essential than the C7 hydroxyl group because of the comparable IC50 values of 7-methoxylasofoxifene and cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol. In contrast, the addition of a double bond in nafoxidine did not change the extent of inhibition or the IC50 value, as compared with 7-methoxylasofoxifene. The structural modeling again provides support, with the absence of a computed hydrogen bond between the central oxygen atom [which itself is not present in cis-4-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthalenyl)phenol] and the molybdenum cofactor, whereas this bond is maintained for both 7-methoxylasofoxifene and nafoxidine, where the central oxygen atom is present as it is in lasofoxifene.
Bazedoxifene in combination with conjugated estrogens, termed as tissue-selective estrogen complex, is a new approach to treating/preventing menopausal osteoporosis (Pickar et al., 2018). Estrone sulfate is the major conjugated estrogen in the combination product containing bazedoxifene (Berrodin et al., 2009). It is also the major circulating form in postmenopausal women (Marchand et al., 2018). Estrone was shown as a potent inhibitor of AOX1 with an IC50 value of 0.43 μM (using phthalazine oxidation) in a previous study (Obach, 2004) and 0.18 μM (using carbazeran 4-oxidation) in the present study, whereas the present study shows that the sulfated form of estrone was only a weak AOX1 inhibitor with IC50 value at high micromolar range. Estrone at 0.3–10 μM enhanced the inhibitory effect of bazedoxifene, whereas estrone sulfate enhanced the effect of bazedoxifene only at high micromolar concentrations. The in vivo concentration of unconjugated estrone in humans is in the nanomolar range (∼10 nM or 2.6 ng/ml) (Package insert. Therefore, in vivo, estrone and its sulfate form are not expected to enhance the extent of AOX1 inhibition by bazedoxifene.
Pharmacokinetic studies of SERM conducted on human volunteers have indicated maximal plasma concentrations of 6.2–7.2 ng/ml (∼0.015 μM) for bazedoxifene (McKeand, 2017), 6.43 ng/ml (0.0155 μM) for lasofoxifene (Gardner et al., 2006), 164–494 ng/ml (0.44–1.33 μM) for tamoxifen (Morello et al., 2003), and 1.36 ng/ml (0.003 μM) for raloxifene (EVISTA (Raloxifene Hydrochloride), 2007). SERMs are well-distributed and concentrated in the liver (Morello et al., 2003). Although the in vivo concentrations of SERMs in human liver are not known, the concentrations of bazedoxifene, lasofoxifene, tamoxifen, and raloxifene have been reported to be 43- to 55-fold (Chandrasekaran et al., 2010), 14- to 25-fold (Prakash et al., 2008), ∼40- to 60-fold (Lien et al., 1991), and 8-fold (Lindstrom et al., 1984) greater in rat liver than in plasma, respectively. If these SERMs are concentrated to a similar extent in human liver, the hepatic concentration of bazedoxifene, lasofoxifene, tamoxifen, and raloxifene is estimated to be 0.73, 0.39, 80, and 0.023 μM, respectively. Overall, our reported apparent Ki values for bazedoxifene, lasofoxifene, tamoxifen, and raloxifene (Table 2) suggest potential hepatic drug interactions with AOX1 substrates.
In summary, SERMs differentially inhibited the catalytic activity of human AOX1. Bazedoxifene and lasofoxifene inhibited AOX1-catalyzed carbazeran 4-oxidation by a competitive mode, whereas acolbifene, which has a different orientation of the alkylamino side chain, only weakly inhibited it. The inhibitory potency of bazedoxifene was decreased by N-oxidation, whereas it was increased by the loss of the 1-(azepanyl)ethyl moiety. The 7-hydroxy group and the substituted pyrrolidine ring of lasofoxifene contributed to the potent inhibition of AOX1 by lasofoxifene. Estrone and its sulfated form are not expected to increase any potential in vivo inhibitory effect of bazedoxifene. Our findings suggest that bazedoxifene and lasofoxifene may interact with other drugs (e.g., methotrexate, idelalisib) or endogenous chemicals (e.g., retinaldehyde) (Garattini et al., 2009) known to be human AOX1 substrates. Future clinical studies would be needed to determine whether these interactions occur in vivo. To date, studies have been reported on AOX1 protein structure–drug metabolism relationships to predict human AOX1 substrates (Lepri et al., 2017; Cruciani et al., 2018), but limited information on human AOX1 structure–enzyme inhibitor relationships determined based on the crystal structure of human AOX1 (Takaoka et al., 2018; Deris-Abdolahpour et al., 2019). The molecular-docking approach developed in this study, by virtue of its consistent performance and the ability it allows for meaningful comparative analyses of chemical inhibitors, provides a robust in silico framework for future investigation of the binding of chemical inhibitors with AOX1. Therefore, our novel biochemical findings, together with molecular-docking analyses, provide new insights into the differential inhibition of AOX1 by SERMs and how SERMs bind to AOX1, and increase our understanding of the AOX1 pharmacophore for the inhibition of AOX1 by drugs and other chemicals.
Authorship Contributions
Participated in research design: Chen, Lau.
Conducted experiments: Chen, Austin-Muttitt, Zhang.
Performed data analysis: Chen, Austin-Muttitt, Zhang, Mullins, Lau.
Wrote or contributed to the writing of the manuscript: Chen, Austin-Muttitt, Mullins, Lau.
Footnotes
- Received April 22, 2019.
- Accepted July 5, 2019.
This work was supported by the Singapore Ministry of Education Academic Research Fund Tier 1 [Grant R-148-000-218-112 to A.J.L.], the National University of Singapore [Start-Up Grant R-148-000-185-133 to A.J.L.], and the Singapore Ministry of Health’s National Medical Research Council under its Cooperative Basic Research Grant scheme [Grant R-148-000-225-511 to A.J.L.]. S.C. was supported by a National University of Singapore Research Scholarship (President’s Graduate Fellowship).
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AOX1
- aldehyde oxidase-1
- Clint,u
- unbound intrinsic clearance
- PDB
- Protein Data Bank
- SERM
- selective estrogen receptor modulator
- UPLC-MS/MS
- ultra-high performance liquid chromatography–tandem mass spectrometry
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}