


Abstract
Cabotegravir, a novel integrase inhibitor under development for treatment and prevention of HIV, is primarily metabolized by UGT1A1 and UGT1A9 to a direct ether glucuronide metabolite. The aim of these studies was to elucidate the mechanistic basis of cabotegravir–glucuronide disposition in humans. Cabotegravir glucuronidation was predominantly hepatic (>95%) with minimal intestinal and renal contribution. Rat liver perfusions demonstrated that cabotegravir–glucuronide formed in the liver undergoes comparable biliary and sinusoidal excretion, consistent with high concentrations of the glucuronide in human bile and urine. Cabotegravir–glucuronide biliary excretion was mediated by MRP2 (not transported by BCRP or P-gp), whereas hepatic basolateral excretion into sinusoidal blood was via both MRP3 (Ft = 0.81) and MRP4 (Ft = 0.19). Surprisingly, despite high urinary recovery of hepatically-formed cabotegravir–glucuronide, metabolite levels in circulation were negligible, a phenomenon consistent with rapid metabolite clearance. Cabotegravir–glucuronide was transported by hepatic uptake transporters OATP1B1 and OATP1B3; however, metabolite clearance by hepatic uptake from circulation was low (2.7% of hepatic blood flow) and unable to explain the minimal systemic exposure. Instead, circulating cabotegravir–glucuronide undergoes efficient renal clearance, where uptake into the proximal tubule would be mediated by OAT3 (not transported by OAT1), and subsequent secretion into urine by MRP2 (Ft = 0.66) and MRP4 (Ft = 0.34). These studies provide mechanistic insight into the disposition of cabotegravir–glucuronide, a hepatically-formed metabolite with appreciable urinary recovery and minimal systemic exposure, including fractional contribution of redundant transporters to any given process based on quantitative proteomics.
SIGNIFICANCE STATEMENT The role of membrane transporters in metabolite disposition, especially glucuronides, and as sites of unexpected drug–drug interactions, which alter drug efficacy and safety, has been established. Cabotegravir–glucuronide, formed predominantly by direct glucuronidation of parent drug in liver, was the major metabolite recovered in human urine (27% of oral dose) but was surprisingly not detected in systemic circulation. To our knowledge, this is the first mechanistic description of this phenomenon for a major hepatically-formed metabolite to be excreted in the urine to a large extent, but not circulate at detectable levels. The present study elucidates the mechanistic basis of cabotegravir–glucuronide disposition in humans. Specific hepatic and renal transporters involved in the disposition of cabotegravir–glucuronide, with their fractional contribution, have been provided.
Introduction
People living with human immunodeficiency virus (HIV) have a choice in efficacious drug therapies that have led to a life expectancy comparable to that of healthy persons (Samji et al., 2013; Trickey et al., 2017). Furthermore, contemporary HIV drugs have demonstrated robust effectiveness and treatment with improved adherence, toxicity, and dosing profiles. Central to this evolution of HIV therapy has been integrase strand transfer inhibitors (INSTI) and their availability as both oral and, in future, long-acting formulations (Hobson et al., 2018). Improvements in HIV therapy have also led to increased scrutiny of drug–drug interactions due to common polypharmacy treatment of comorbidities in an aging HIV patient population. Identifying drug-metabolizing enzymes and transporters involved in the disposition of a drug and its metabolites is central to understanding the drug–drug interaction profile of a new therapy.
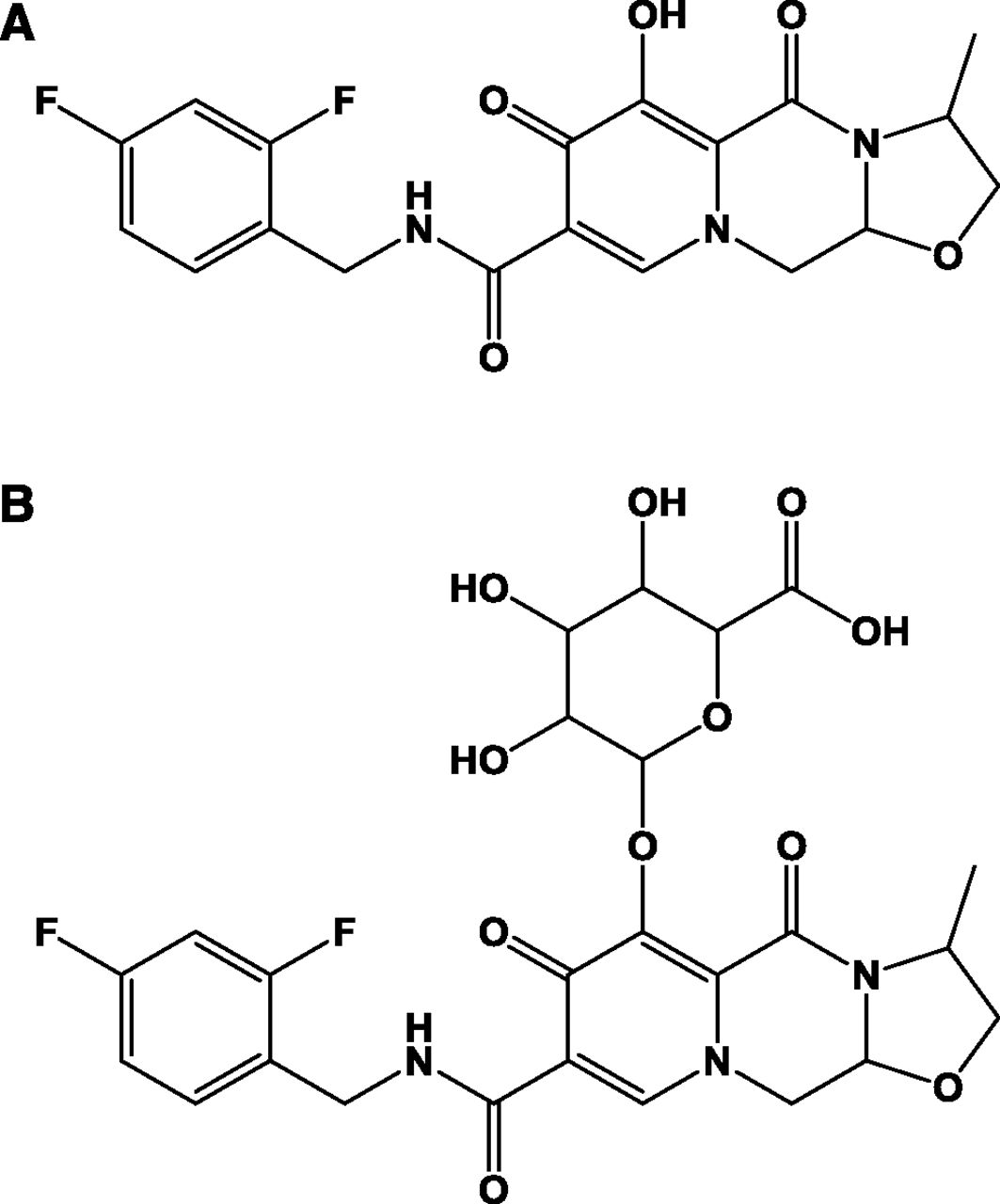
Cabotegravir (Fig. 1) is a novel INSTI in clinical development for the treatment and prevention of HIV infection (Spreen et al., 2013; Margolis et al., 2015; Whitfield et al., 2016). Cabotegravir is principally metabolized by direct conjugation with glucuronic acid, with a fractional contribution of 0.67 and 0.33 from UDP-glucuronosyltransferases 1A1 (UGT1A1) and UGT1A9 to overall glucuronidation (Bowers et al., 2016). Because UGT1A1 and UGT1A9 are expressed in human intestine, liver, and kidneys, cabotegravir–glucuronide could be produced in these metabolic and excretory tissues (Nakamura et al., 2008; Ohno and Nakajin, 2009; Court et al., 2012).
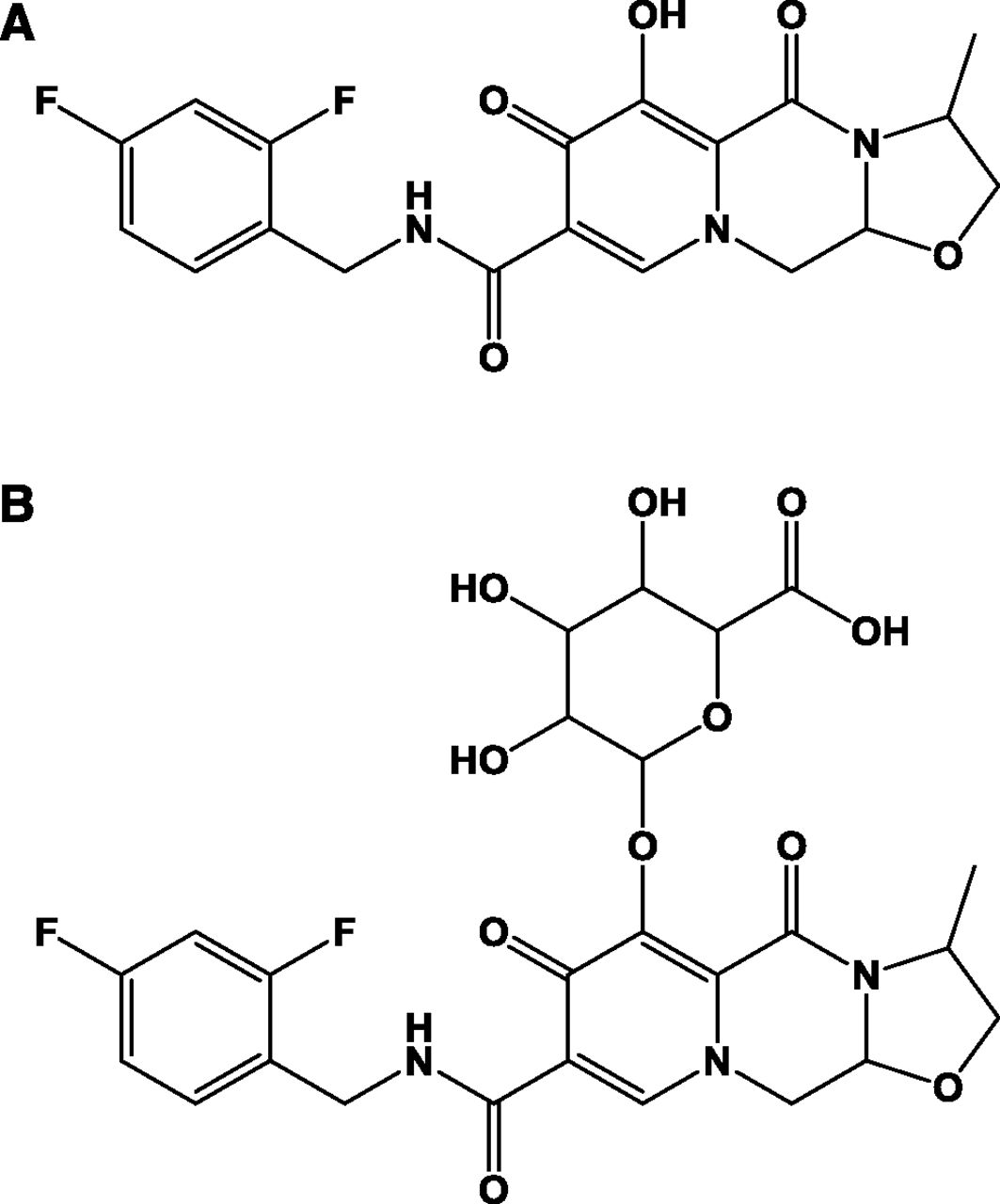
Structure of (A) cabotegravir and (B) cabotegravir–glucuronide.
Cabotegravir human metabolism and excretion were studied following oral administration of 14C-labeled dose (Bowers et al., 2016). Cabotegravir–glucuronide was the predominant metabolite in human bile (assessed via Entero-Test bile string), as expected for a major hepatically-formed metabolite. In this human mass balance study, it was of note that parent cabotegravir was the principal circulating drug-related component, whereas cabotegravir–glucuronide was not detected in systemic circulation, consistent with observations in preclinical species (Bowers et al., 2016). Despite minimal systemic exposure and negligible renal formation, cabotegravir–glucuronide recovery in human urine was surprisingly high (27% of the oral dose). Because cabotegravir–glucuronide was not detected in plasma (below detection limits), in a study that included quantitative urine collection, it is currently not possible to quantify its renal clearance. However, as renal clearance is calculated as the ratio of amount excreted in urine to systemic exposure, the high cabotegravir–glucuronide urinary recovery divided by low systemic exposure is consistent with high renal clearance involving active tubular secretion.
Conjugation with glucuronic acid, a hydrophilic negatively charged sugar, markedly increases molecular polarity and reduces passive membrane permeability. As such, these conjugates are reliant on membrane transporters for excretion from their sites of formation, and subsequently exhibit transporter-mediated disposition (Patel et al., 2016; Yang et al., 2017). For example, glucuronide conjugates formed in the intestinal wall can be excreted into mesenteric circulation via multidrug resistance-associated protein 3 (MRP3) and/or into gastrointestinal lumen via MRP2 and/or breast cancer resistance protein (BCRP) (Donovan et al., 2001; Teng et al., 2012; Yang et al., 2017). Liver, the primary site of most drug metabolism, excretes glucuronide metabolites into bile by MRP2 and/or BCRP, as well as across the basolateral membrane into sinusoidal blood via MRP3 and/or MRP4 (Giacomini et al., 2010; Patel et al., 2016). The liver can also take up glucuronide metabolites from sinusoidal blood via organic anion-transporting polypeptides (OATPs), including reuptake of sinusoidally-excreted conjugates by downstream hepatocytes, a phenomenon known as “hepatocyte hopping,” as demonstrated for bilirubin glucuronides (Giacomini et al., 2010; Yang et al., 2017).
Elimination of a direct glucuronide metabolite by biliary excretion can be physiologically inefficient, as these conjugates can be readily deconjugated in the intestine and reabsorbed as parent drug. As such, hepatically-formed glucuronide conjugates can exhibit appreciable urinary recovery, which at a gross level involves hepatic basolateral excretion of the metabolite from liver into blood for ultimate clearance by the kidneys. Renal clearance consists of passive glomerular filtration of unbound glucuronide metabolite in circulation, and can additionally involve active tubular secretion, a two-step process of uptake from blood (for glucuronides primarily by OAT1 and/or OAT3), followed by secretion from the proximal tubule into urine (for glucuronides mainly by OAT4, MRP2, and MRP4). Tubular reabsorption of glucuronide metabolites is uncommon due to their low passive permeability, which limits reabsorption back to blood from urine.
The objective of the current studies was to establish the mechanistic basis for cabotegravir–glucuronide disposition in humans. Specific transporters mediating the disposition of cabotegravir–glucuronide were elucidated, and where redundant transporters are involved in a single process (e.g., MRP3 and MRP4 in hepatic basolateral excretion), their fractional contribution was determined using quantitative proteomics.
Materials and Methods
Materials
The human embryonic kidney (HEK-MSRII) cells and BacMam baculovirus transduction reagents for human OATP1B1, OATP1B3, OAT1, and OAT3 transporters were obtained from the Biologic Sciences group, GlaxoSmithKline (Collegeville, PA). TransportoCells human OAT4-overexpressing HEK293 cells were commercially obtained from Corning. Human MRP2, MRP3, MRP4, and control vesicles with transport assay kits were obtained from Genomembrane (Yokohama, Japan). Cyropreserved plateable human hepatocytes (pooled) and inVitrogen cell plating media were obtained from Celsis (Baltimore, MD). Human liver, intestinal, and kidney microsomes were purchased from Xenotech (Lenexa, KS). Human liver and kidney tissues for quantitative proteomics were procured from Asterand Bioscience (Royston, UK).
[3H]-Estradiol 17β-D-glucuronide (specific activity 41.4 Ci/mmol) and [3H]-estrone sulfate (specific activity 54 Ci/mmol) were obtained from PerkinElmer (Boston, MA). [14C]-Cabotegravir (specific activity 126 μCi/mg), unlabeled cabotegravir, and stable isotopically labeled cabotegravir-d4 (stable label) were supplied by Chemical Development, GlaxoSmithKline. The purity of these radioactive compounds was ≥97%. Cabotegravir–glucuronide sodium salt was procured from Syngene (Bangalore, India). Rifamycin SV, MK-571 sodium salt, sodium butyrate, unlabeled estradiol 17β-D-glucuronide, bromosulfophthalein, benzbromarone, 6-carboxy fluorescein, imipramine, Krebs–Henseleit buffer, sodium taurocholate, calcium chloride, sodium bicarbonate, and glucose were purchased from Sigma-Aldrich (St. Louis, MO). Collagen and poly-D-lysine–coated 24-well plates were obtained from Corning (Tewksbury, MA). All other chemical compounds and reagents purchased were of analytical grade or higher and used without any further purification.
For dot–blot analysis, MRP2 (M2 III-6) mouse monoclonal antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-MRP3 (M3II-9) and anti-MRP4 (ab56675), both mouse monoclonal antibodies, were procured from Abcam (Cambridge, UK). IRDyeR 800CW anti-mouse antibody, goat polyclonal, was obtained from LI-COR Biosciences (Lincoln, NE).
Methods
Microsomal Incubation.
Metabolic conversion of [14C]-cabotegravir was studied in human liver (0.5 mg/ml), kidney (0.5 mg/ml), and intestinal (1 mg/ml) microsomes in 100 mM Tris-HCl buffer, pH 7.40, supplied with 2 mM magnesium chloride and 1 mM EDTA. Microsomes were activated with alamethicin (50 μg/mg microsomal protein) and placed on ice for 15 minutes. Microsomes were preincubated with [14C]-cabotegravir (5 µM) for 5 minutes, and the reaction was initiated with the addition of 5 mM uridine 5′-diphosphoglucuronic acid. Incubations in liver and kidney microsomes were carried out for 120 minutes and in intestinal microsomes for 60 minutes at 37°C with continuous shaking. At the end of the incubation, an aliquot of microsomes was collected, and the reaction was terminated with the addition of acetonitrile. Simultaneously, the metabolism of [14C]-cabotegravir was studied in the absence of UDPGA and microsomes as controls. Samples were centrifuged at 1600g for 5 minutes, and the supernatant was collected for radio-chromatography.
Microsomal formation rates (picomoles per minute per milligram protein) were scaled to liters per hour, using 40 mg microsomal protein per gram liver and 1800 g total liver weight (70 kg), 12.8 mg microsomal protein per gram kidney and 310 g total kidney weight (70 kg), and 2978 mg microsomal protein per intestine (Davies and Morris, 1993; Barter et al., 2007; Scotcher et al., 2016; Hatley et al., 2017).
In Situ Rat Liver Perfusion.
In situ rat liver perfusion was carried out in accordance with the GlaxoSmithKline Policy on the Care, Welfare, and Treatment of Laboratory Animals, and the protocol was reviewed and approved by the Institutional Animal Care and Use Committee at GlaxoSmithKline. Male Sprague–Dawley rats (350–400 g) were purchased from Charles River (Raleigh, NC) and allowed to acclimatize for at least 5 days prior to the initiation of the study. Rats were kept in a controlled environment and supplied with appropriate diet and water.
On the day of the experiment, rats were anesthetized (ketamine/xylazine 87/13 mg/kg i.p.), and the peritoneal cavity was opened to access the liver, hepatic portal vein, lower inferior vena cava, hepatic artery, and bile duct. The hepatic portal vein and superior vena cava were cannulated with an intravenous catheter. The lower inferior vena cava was ligated, and the bile duct was cannulated with a polyethylene tubing. Rat livers were perfused with oxygenated Krebs–Henseleit buffer at 30 ml/min for 15 minutes (Curtis et al., 1971; Wolkoff et al., 1987). Following acclimation, livers were perfused (single-pass) with oxygenated Krebs–Henseleit buffer containing 10 µM cabotegravir for 60 minutes. Bile and perfusate samples were collected at every 5-minute interval and immediately stored on dry ice. At the end of perfusion (1 hour), livers were flushed and snap frozen in liquid nitrogen. Liver samples were stored at −80°C until homogenization in distilled deionized water and analyzed for cabotegravir and cabotegravir–glucuronide using LC-MS/MS.
Steady-state (S.S.) basolateral and canalicular clearances of cabotegravir–glucuronide from in situ rat liver perfusion study were determined using eqs. 1 and 2.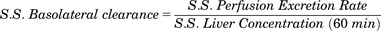 (1)
(1)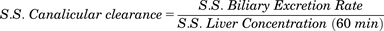 (2)
(2)
Cell Culture
OATP1B1, OATP1B3, OAT1, and OAT3.
HEK-MSRII cells overexpressing OATP1B1, OATP1B3, OAT1, and OAT3 were produced using a protocol previously published in the literature (Patel et al., 2018). Briefly, cells were thawed at 37°C and transferred to Dulbecco’s modified Eagle’s medium and Ham’s F-12 medium supplied with fetal bovine serum (10%). Cells were centrifuged and resuspended in fresh media with geneticin (0.4 mg/ml) and sodium butyrate (2 mM). Cells were plated at a density of 400,000 cells/well in poly-D-lysine–coated 24-well plates and incubated at 37°C, 5% CO2, and 95% humidity for 4 hours, after which they were transduced with OATP- or OAT-BacMam virus in cell media for 48 hours.
OAT4.
HEK-OAT4 cells were rapidly thawed and transferred to Dulbecco’s modified Eagle’s medium supplied with fetal bovine serum (10%) and minimum essential medium nonessential amino acids. Following centrifugation at 100g for 10 minutes, media was removed, and cells were resuspended in fresh medium. Cells (0.4 × 106 cells/well) were plated and maintained at 37°C with 5% CO2 and minimal humidity for 4 hours. Media was replaced, and cells were incubated overnight at 37°C with 5% CO2 and minimal humidity.
Cellular Uptake
The uptake of cabotegravir–glucuronide in singly-overexpressing OATP1B1, OATP1B3, OAT1, OAT3, and OAT4 cells was studied in the presence and absence of a prototypical inhibitor according to a previously published protocol (Patel et al., 2018). Briefly, confluent cell monolayers were washed twice with transport medium and preincubated with a prototypical inhibitor or DMSO at 37°C for 20 minutes. Preincubation solution was removed, and cells were incubated with cabotegravir–glucuronide solution in the presence and absence of a prototypical inhibitor at 37°C. Following incubation, cabotegravir–glucuronide solution was aspirated, and cells were rapidly washed thrice with ice-cold transport media. Cells were lysed with distilled deionized water and stored at −80°C overnight, and cabotegravir–glucuronide was detected by LC-MS/MS.
As a positive control to confirm transporter activity, each cell line was assessed using a prototypical substrate and inhibitor pair. The uptake of [3H]-estradiol 17β-D-glucuronide was conducted in HEK-OATP1B1 and HEK-OATP1B3 cells in the presence and absence of rifamycin, a prototypical inhibitor of these transporters. For OAT4 activity, [3H]-estrone sulfate uptake was studied in HEK-OAT4 cells in the presence and absence of bromosulfophthalein and benzbromarone. Following uptake, HEK-OAT4 cells were lysed with 0.1% (v/v) Triton X-100 in phosphate-buffered saline, and the cell-associated radioactivity was analyzed with liquid scintillation spectroscopy. The uptake of 6-carboxy fluorescein was carried out in HEK-OAT1 and OAT3 cells, in the presence and absence of benzbromarone. HEK-OAT1 and HEK-OAT3 cells were lysed with distilled deionized water at −80°C overnight prior to fluorometric analysis with FLUOstar Omega plate reader.
A set of singly-overexpressing HEK293 cells was collected to quantitate transporter levels using mass spectroscopic analysis.
Vesicular Transport
Commercially available, inside-out vesicles were used to study the substrate potential of cabotegravir–glucuronide toward MRP2, MRP3, and MRP4. Vesicles (0.75 mg/ml) were pretreated with prototypical MRP inhibitors or DMSO at 37°C for 10 minutes. The transport was initiated by incubating cabotegravir–glucuronide with vesicles in 50 mM 4-morpholinepropanesulfonic acid–Tris buffer (pH 7.00), supplied with ATP or AMP, at 37°C for 10 minutes. Then about 200 µl ice-cold stop solution (400 mM 4-morpholinepropanesulfonic acid–Tris, 700 mM KCl) was added to terminate further transport processes, and the reaction mixture was filtered with the aid of a glass filter plate (MultiScreenHTS FB Filter Plate; Millipore) connected to a vacuum apparatus. Vesicles were rapidly washed thrice with ice-cold stop solution, following which filters were carefully collected and immersed in acetonitrile:distilled deionized water (1:1) containing cabotegravir-d4 (internal standard). Samples were stored at −20°C until analysis by high-performance liquid chromatography (HPLC)-MS/MS.
As a positive control to confirm transporter activity, [3H]-estradiol 17β-D-glucuronide transport was also studied according to the procedure described above. Following final wash, the filter plate was dried at 50°C for 30 minutes. About 100 µl/well microscint fluid was added, and the radioactivity was analyzed by TopCount NXT HTS detector (PerkinElmer, Waltham, MA).
P-glycoprotein (P-gp) and BCRP vesicular transport of probe substrates and cabotegravir–glucuronide was studied according to a previously published protocol (Vermeer et al., 2016).
A set of MRP2, MRP3, and MRP4 vesicles was collected to quantitate transporter levels using dot–blot analysis.
Hepatocyte Uptake
Cryopreserved pooled human hepatocytes were thawed and immediately transferred to prewarmed plating media. Cells were plated at a density of 0.375 × 106 cells/well in collagen-coated 24-well plates and maintained at 37°C with 5% CO2 for 5 hours. Following incubation, hepatocytes were washed twice with transport medium and incubated with cabotegravir–glucuronide at 37°C from 0.5 to 15 minutes (six time points). At predetermined time points, solution was aspirated, and hepatocytes were rapidly washed thrice with ice-cold buffer. Cells were lysed with distilled deionized water and stored at −80°C overnight, and cabotegravir–glucuronide in the lysates was analyzed by LC-MS/MS.
As a positive control to confirm transporter activity, [3H]-estradiol 17β-D-glucuronide uptake was studied in the presence and absence of an OATP and OCT inhibitor cocktail (rifamycin and imipramine, 100 µM each).
In vitro uptake clearance from hepatocyte uptake study was scaled from milliliter per minute per million cells to milliliter per minute per kilogram using 120 × 106 hepatocytes per gram liver weight and 25.7 g liver per kilogram body weight (Davies and Morris, 1993; Bayliss et al., 1999). In vivo hepatic clearance was then predicted using well-stirred model corrected for plasma protein binding (eq. 3).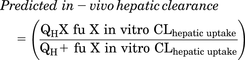 (3)
(3)
where fu represents fraction unbound of cabotegravir–glucuronide in plasma, which is 0.84 (data not published), and QH represents hepatic blood flow as 20.7 ml/min per kilogram body weight (Davies and Morris, 1993).
Sample Preparation and Analysis
Radio HPLC.
[14C]-Cabotegravir and [14C]-cabotegravir–glucuronide–associated radioactivity was analyzed with a HPLC system (1100; Agilent) connected to a Radiomatic Flow Scintillation Analyzer (Packard TR625, 0.5 ml liquid cell). A gradient of water and acetonitrile, both containing 0.1% formic acid, was pumped through a Waters Symmetry C18 (4.6 × 75 mm, 3.5 μm) column, maintained at 40°C, for 45 minutes. [14C]-Cabotegravir and [14C]-cabotegravir–glucuronide eluted at 29.1 and 15.3 minutes, respectively. Compounds were also structurally identified using LC-MS analysis, according to a previously published protocol (Bowers et al., 2016).
HPLC-MS/MS Analysis.
Waters Acquity UPLC system (Waters, Milford, MA) connected to Applied Biosystems/MDS Sciex API 4000 (Ontario, Canada) was employed to analyze unlabeled cabotegravir and cabotegravir–glucuronide. Liver homogenate, bile, perfusate, and cell lysate samples were vigorously mixed with an equal volume of acetonitrile supplied with internal standard (50 ng/ml) and centrifuged at 7000 rpm for 5 minutes, following which the supernatant was collected for HPLC-MS/MS analysis. Acquity BEH C18 column (2.1 × 50 mm 1.7 µm; Milford, MA), maintained at 65°C, was pumped with a gradient of 0.1% formic acid in water and 0.1% formic acid in acetonitrile at 0.75 ml/min for 2 minutes. Cabotegravir and cabotegravir–glucuronide eluted at approximately 1.08 and 0.96 minutes, respectively.
Turbo ionspray ionization in a positive mode was employed to detect cabotegravir and cabotegravir–glucuronide with multiple-reaction monitoring. Transition (m/z) for cabotegravir and cabotegravir–glucuronide was 406/263 and 582.3/406, respectively. Raw data obtained were integrated with Applied Biosystems/MDS Sciex software Analyst v 1.6.1.
Quantitative Transporter Analysis.
Transporter expression in singly-overexpressing HEK293 cells (OAT1, OAT3, and OAT4), vesicles (MRP2, MRP3, and MRP4), and cryopreserved human kidney and liver tissues was quantified to generate a protein-based relative expression factor (REF). Relative transporter quantification of vesicles compared with tissue was carried out by dot–blot analysis, and that for HEK cells compared with tissue was performed by mass spectrometric–based proteomic analysis. Cell pellet, vesicles, and tissue samples were diluted with three volumes of Dulbecco’s PBS and homogenized at 4°C for 15 seconds. Lysis buffer (4% SDS, 50 mM Tris, pH 7.40) and benzonase (2 U/µl) were added, and samples were incubated at 4°C for 60 minutes. Samples were ultracentrifuged, and the supernatant was collected to determine protein concentrations with bicinchoninic acid assay. An aliquot of supernatant was mixed with 4× buffer (200 mM Tris HCl, 250 mM Trizma base, 20% glycerol, 4% SDS, 0.01% bromphenol blue) for dot–blot analysis, whereas another aliquot was treated with NuPAGE Lithium dodecyl sulfate sample-loading buffer for mass spectrometric analysis. Each aliquot was supplemented with 50 mM dithiothreitol and heated at 95°C for 5 minutes.
Mass Spectrometric Analysis.
To quantify transporter (OAT1, OAT3, and OAT4) levels in HEK cells and tissues by mass spectrometric analysis, about 25 µg total protein per sample was in gel digested and labeled with isobaric mass tags, and samples were separated into 25 fractions by high-pH reverse-phase chromatography (Savitski et al., 2014). Samples were analyzed with an Orbitrap Lumos Mass spectrometer (Thermo Fisher Scientific, San Jose, CA) with Multinotch MS3 enabled (McAlister et al., 2014).
Dot–Blot Analysis.
Dot blots were carried out at 20 ng total protein/spot for MRP vesicles (MRP2, MRP3, and MRP4) and 2 µg total protein/spot for tissue samples. Samples were spotted on nitrocellulose membranes and dried for 60 minutes. Membranes were rehydrated in 20% ethanol and washed twice with Dulbecco’s PBS, pH 7.40. Membranes were blocked with Odyssey blocking buffer, and MRP primary antibodies in blocking buffer, supplemented with 0.1% Tween 20, were added to membranes and incubated overnight at 4°C with gentle agitation. Membranes were rinsed with 0.1% Tween in PBS (pH 7.40) four times for 5 minutes and incubated with fluorescent-labeled secondary antibody IRDyeR 800CW (LI-COR) in blocking buffer supplemented with 0.1% Tween and 0.02% SDS, for 60 minutes in dark with continuous shaking. The excess of secondary antibody was removed, and membranes were scanned on a LI-COR Odyssey scanner (LI-COR Biosciences).
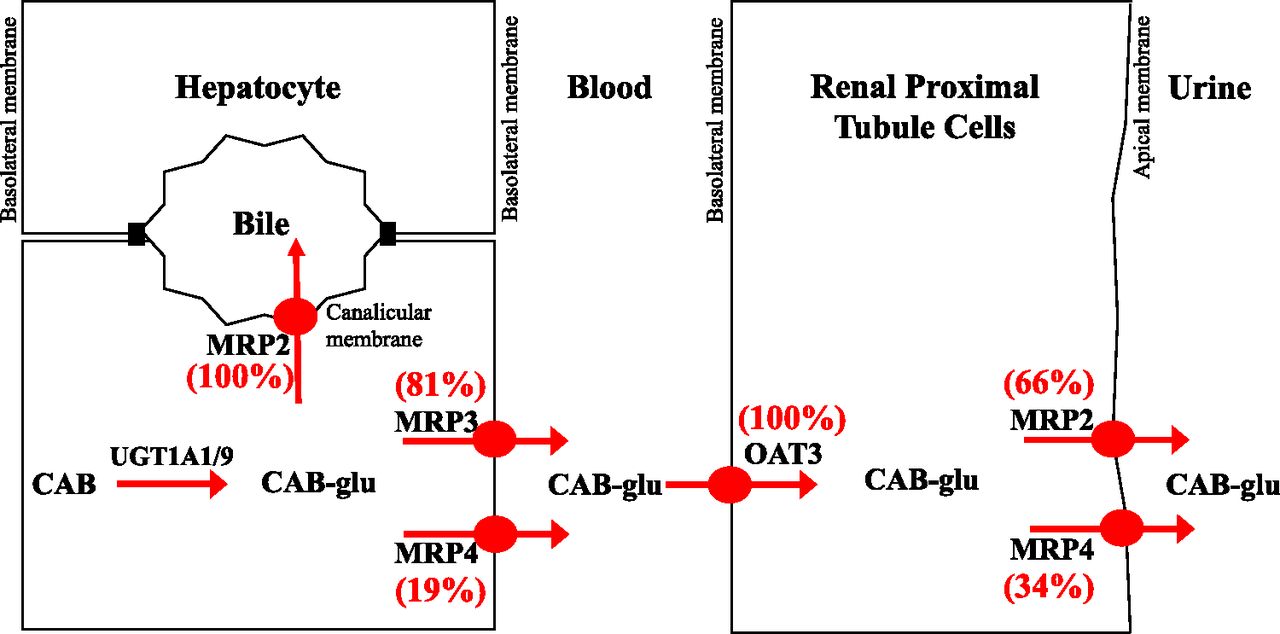
Transport rate (picomoles per minute per milligram protein) of cabotegravir–glucuronide in MRP2, MRP3, and MRP4 vesicles was scaled to microliter per minute per milligram protein and normalized with hepatic or renal REF (Table 2) for these efflux transporters. Scaled clearance was then used to determine fraction transport (Ft) by MRP3 and MRP4 in hepatic basolateral excretion and MRP2 and MRP4 in renal apical excretion of cabotegravir–glucuronide. For example, cabotegravir–glucuronide intrinsic clearance by MRP3 (1.13 µl/min per milligram protein) and MRP4 (0.21 µl/min per milligram protein) was normalized with hepatic REF of 166.0 and 127.9 (Table 2), yielding a scaled intrinsic clearance of 6.81 and 1.64 nl/min per milligram protein, corresponding to a fraction transported of 81% and 19% by MRP3 and MRP4 in hepatic basolateral excretion, respectively. Likewise, normalizing the intrinsic clearance of cabotegravir–glucuronide by MRP2 and MRP4 (1.1 and 0.21 µl/min per milligram protein) with renal REF of 249.8 and 94.0 (Table 2) yielded a scaled intrinsic clearance of 4.40 and 2.23 nl/min per milligram protein, corresponding to a fraction transported of 66% and 34% by MRP2 and MRP4 in the apical proximal tubular excretion, respectively.
Statistical Analysis
One-way ANOVA with correction for multiple comparisons was used to determine statistical significance. In all cases, P < 0.05 was considered statistically significant.
Results
Microsomal Glucuronidation.
Cabotegravir formed a single metabolite in human liver, kidney, and intestinal microsome incubations conducted under glucuronidation conditions. Cabotegravir–glucuronide accounted for 4% to 17% of total drug-related radioactivity at the end of these incubations. The hepatic, renal, and intestinal intrinsic clearance for formation of cabotegravir–glucuronide were estimated to be 12.2, 0.33, and 0.12 l/h, respectively.
In Situ Rat Liver Perfusion.
During single-pass rat liver perfusions with cabotegravir, the biliary and perfusate excretion rates of cabotegravir–glucuronide reached plateaus between 27.5 and 60 minutes, consistent with attainment of steady-state conditions. Cabotegravir–glucuronide biliary and basolateral steady-state excretion rates and clearances, as well as liver concentrations, are summarized in Table 1. Basolateral versus canalicular excretion of cabotegravir–glucuronide in rat livers was approximately equal in magnitude (on average, 55% vs. 45% of total hepatic metabolite excretory clearance, respectively).
Cabotegravir–glucuronide steady-state pharmacokinetic parameters in single-pass perfused livers from male Sprague–Dawley rats
Biliary as well as perfusate excretion rates were calculated between 27.5 and 60 minutes at steady-state (S.S.) conditions, i.e., when these rates reached plateaus. Mean ± S.E.M., n = 3.
Uptake Transport.
Uptake transport of cabotegravir–glucuronide was studied in singly-overexpressing HEK cells in the absence or presence of prototypical inhibitors (Fig. 2). Functional activity of evaluated transporters was confirmed with positive control substrates (≥11-fold increased uptake activity in transporter-overexpressing cells vs. vector control, and which was significantly impaired ≥70% by prototypical inhibitors in transporter-overexpressing cells). Cabotegravir–glucuronide was transported by OATP1B1, OATP1B3, and OAT3. Although transport of cabotegravir–glucuronide by OAT1 was statistically significant, it did not meet the 2-fold threshold for uptake enhancement (OAT1 vs. vector control) to be considered a substrate [FDA guidance, 2012]. Significant cabotegravir–glucuronide uptake by OAT4 was not observed.
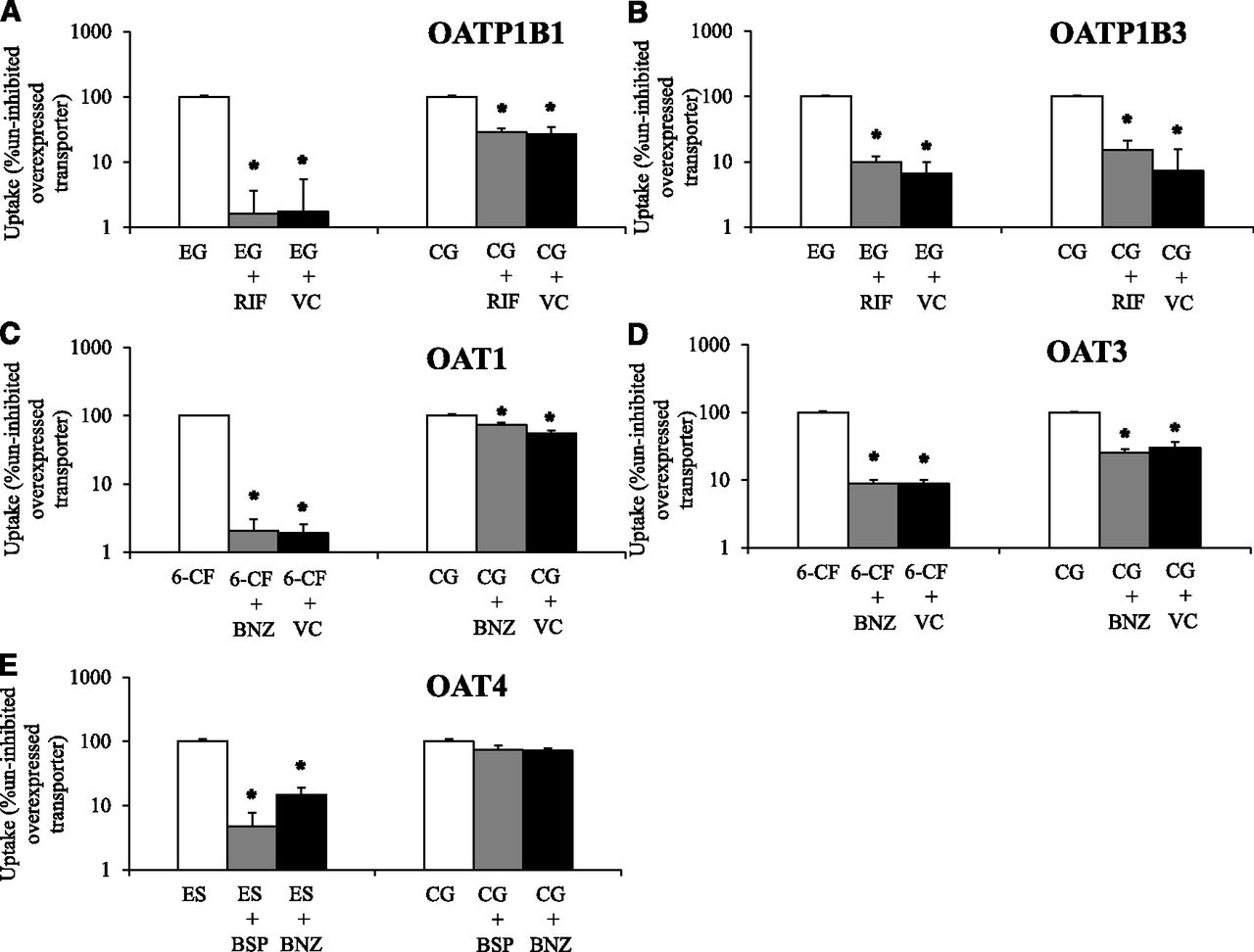
Cabotegravir–glucuronide (CG, 50 µM) uptake was studied in vector control (VC) and transporter singly-overexpressing HEK293 cells in the absence or presence of transporter inhibitors. Uptake of a relevant positive control substrate was simultaneously evaluated with CG by coincubation with (A) [3H]-estradiol 17β-D-glucuronide (EG) in VC and HEK-OATP1B1 cells in the absence or presence of 10 µM rifamycin SV (RIF) for 5 minutes. (B) [3H]-EG in VC and HEK-OATP1B3 cells in the absence or presence of 10 µM RIF for 10 minutes. (C) 6-Carboxy fluorescein (6-CF) in VC and HEK-OAT1 cells in the absence or presence of 30 µM benzbromarone (BNZ) for 10 minutes. (D) 6-CF in VC and HEK-OAT3 cells in the absence or presence of 30 µM BNZ for 10 minutes and (E) [3H]estrone sulfate (ES) in HEK-OAT4 cells in the absence or presence of 10 µM bromosulfophthalein (BSP) and 30 µM BNZ for 3 minutes. (A–D) Open bars represent uptake activity in uninhibited transporter-overexpressing cells; gray bars during coincubation with prototypical inhibitors in transporter-overexpressing cells and black bars in VC cells. CG and ES uptake was not studied in VC cells along with HEK-OAT4 cells. Mean ± S.E.M., n = 4, *P < 0.05.
Efflux Transport.
Efflux transport of cabotegravir–glucuronide was studied as uptake into inside-out plasma membrane vesicles prepared from singly-overexpressing Sf9 cells in the absence or presence of prototypical inhibitors (Fig. 3). Functional activity of evaluated transporters was confirmed with positive control substrates (≥6-fold increased transport activity in the presence of ATP vs. AMP, and which was significantly impaired ≥70% by prototypical inhibitors). Cabotegravir–glucuronide was transported by MRP2, MRP3, and MRP4. In contrast, significant cabotegravir–glucuronide transport by BCRP and P-gp was not observed.

Cabotegravir–glucuronide (CG, 10 µM) efflux transport was studied as uptake into inside-out vesicles prepared from transporter singly-overexpressing Sf9 cells in the presence of AMP, ATP, and ATP plus a prototypical inhibitor. Transport of relevant positive control substrates was simultaneously evaluated with CG by coincubation with (A) [3H]-estradiol 17β-D-glucuronide (EG) in the presence of ATP or AMP and bromosulfophthalein (BSP) with ATP in MRP2 vesicles for 10 minutes. (B) [3H]-EG in the presence of ATP or AMP and MK571 (MK) with ATP in MRP3 vesicles for 10 minutes. (C) [3H]-EG in the presence of ATP or AMP and MK571 (MK) with ATP in MRP4 vesicles for 10 minutes. (D) N-methyl quinidine (NMQ) in the presence of ATP or AMP and verapamil (VER) with ATP in P-gp vesicles for 10 minutes. (E) [3H]-Methotrexate (MTX) in the presence of ATP or AMP and benzbromarone (BNZ) with ATP in BCRP vesicles for 10 minutes. Open bars represent transport activity in AMP-treated vesicles; gray bars in uninhibited ATP-treated transporter-overexpressing vesicles; black bars during coincubation with prototypical inhibitors in ATP-treated transporter-overexpressing vesicles. Mean ± S.E.M., n = 3, *P < 0.05. *Denotes statistically significant increase in the substrate transport in the presence of ATP relative to AMP and decrease with inhibitor coincubation in ATP-treated vesicles, respectively.
Hepatocyte Uptake.
Human hepatocyte uptake of cabotegravir–glucuronide in the time– and concentration–linear range was 10.8 ± 0.9 pmol/min per million cells, which scaled to a human intrinsic clearance of 2.8 ± 0.2 l/h (hepatocyte uptake activity verified with estradiol 17β-D-glucuronide). In vivo hepatic uptake clearance was predicted to be 0.55 ml/min per kilogram (eq. 3), consistent with a low hepatic extraction ratio (∼2.7% of hepatic blood flow).
Transporter REFs.
REFs for transporters involved in cabotegravir–glucuronide disposition were determined by quantification of protein expression levels in the in vitro systems relative to human liver and kidney samples (Table 2).
REF values for transporters involved in hepatic and renal disposition of cabotegravir–glucuronide
REF values for OATs were calculated as expression ratio in HEK293-overexpressing and pooled human kidney samples determined by quantitative proteomics. Protein quantification was based on all identified peptides for the respective protein. REF values for MRPs were calculated as the expression ratio in MRP-overexpressing vesicles and pooled human liver samples (Hepatic REF) or pooled human kidney samples (Renal REF) determined by dot blot. ND, transporter not detected in that tissue. Geometric mean, n = 6; see Supplemental Table 1 for variability on Log2-transformed REF values.
Discussion
Cabotegravir is a novel INSTI in clinical development for the treatment and prevention of HIV infection (Spreen et al., 2013; Margolis et al., 2015; Whitfield et al., 2016). Cabotegravir is principally metabolized by direct conjugation with glucuronic acid (Fig. 1), with a fractional contribution of 0.67 and 0.33 from UGT1A1 and 1A9 to overall glucuronidation (Bowers et al., 2016). Following oral administration in humans, cabotegravir–glucuronide was highly recovered in bile (as expected) and urine (27% of the oral dose) despite minimal systemic exposure to the metabolite (Bowers et al., 2016) and <1% renal formation (data from the present study). Because cabotegravir–glucuronide was not observed in plasma, it is currently not possible to quantify its renal clearance, but it is expected to be high (high urinary recovery divided by low systemic exposure).
The sites of cabotegravir–glucuronide formation were investigated using human intestinal, liver, and kidney microsomes. The scaled clearance values for cabotegravir–glucuronide formation demonstrated >95% hepatic formation. This finding indicates that cabotegravir–glucuronide is formed in the liver and subsequently excreted in the bile (canalicular efflux) as well as into sinusoidal blood (basolateral efflux). To better understand the extent of hepatic basolateral versus biliary excretion of cabotegravir–glucuronide, a single-pass perfusion of parent drug was carried out using rat livers (Pfeifer et al., 2013; Zamek-Gliszczynski et al., 2013). These studies indicated comparable extent of canalicular and sinusoidal excretion of cabotegravir–glucuronide from the liver. Although rats are known to overpredict human clearance of OATP1B substrates (Grime and Paine, 2013), cabotegravir–glucuronide hepatic reuptake was minimized in this single-pass perfusion design that eliminates recirculation of the metabolite through the liver; nonetheless, rat livers may have exaggerated cabotegravir–glucuronide reuptake by downstream hepatocytes, potentially overestimating biliary and underestimating basolateral excretory clearance. Furthermore, the expression of Mrp2/MRP2 is high in normal rat and human liver relative to Mrp3/MRP3 (Hilgendorf et al., 2007; Wang et al., 2015; Fallon et al., 2016), which would be expected to result in greater biliary excretion of cabotegravir–glucuronide. The comparable extent of biliary and sinusoidal excretion of cabotegravir–glucuronide suggests a higher affinity for Mrp3/MRP3 relative to Mrp2/MRP2, as has been proposed for acetaminophen–glucuronide (Xiong et al., 2002; Manautou et al., 2005; Zamek-Gliszczynski et al., 2006b). Notably, cabotegravir–glucuronide is more complex than acetaminophen–glucuronide in that there is additional basolateral excretion via MRP4, but, because it only contributes 19% to the overall process, the present results are overall conceptually consistent with greater affinity for Mrp3/MRP3 than Mrp2/MRP2.
Glucuronide metabolites are polar and poorly permeable molecules that usually necessitate transporter-mediated excretion from the liver into the bile and sinusoidal blood (Zamek-Gliszczynski et al., 2006a). Several efflux transporters are expressed on the basolateral and canalicular membrane of hepatocytes that can mediate basolateral and biliary excretion of cabotegravir–glucuronide (Patel et al., 2016). For instance, cabotegravir–glucuronide can be transported from hepatocytes into the blood by basolateral MRP3 and MRP4 and into the bile by canalicular MRP2, BCRP, and P-gp. The substrate potential of cabotegravir–glucuronide toward these hepatic efflux transporters was investigated using singly-expressing vesicular systems. Cabotegravir–glucuronide was transported by MRP2, MRP3, and MRP4, but not by P-gp and BCRP. These data suggest that cabotegravir–glucuronide is excreted from hepatocytes into bile by MRP2 (Ft = 100%) and into the sinusoidal blood by both MRP3 and MRP4. To determine the extent of MRP3 and MRP4 contribution in the basolateral excretion of cabotegravir–glucuronide, a quantitative proteomics REF was employed. Using this scaling factor, the transport of cabotegravir–glucuronide from hepatocytes into sinusoidal blood was found to be mediated 81% by MRP3 and 19% by MRP4.
Low systemic exposure of cabotegravir–glucuronide could be explained by efficient reuptake by hepatic OATPs (hepatocyte hopping) and/or high renal clearance involving active tubular secretion with uptake via OATs (Giacomini et al., 2010; Bowers et al., 2016). Hence, the substrate potential of cabotegravir–glucuronide toward hepatic uptake (OATP1B1 and OATP1B3) as well as renal (OAT1 and OAT3) basolateral uptake transporters was studied. Cabotegravir–glucuronide was found to be transported by OATP1B1, OATP1B3, and OAT3. These data suggest that OATP1B1 and OATP1B3 could play an important role in the hepatic reuptake of the circulating cabotegravir–glucuronide. However, the hepatic extraction of cabotegravir–glucuronide scaled from human hepatocytes was very low (2.7% of hepatic blood flow), indicating that the systemic removal of the metabolite by hepatic reuptake is very limited; in vitro-to-in vivo correlation of hepatic uptake clearance is established in simple suspended/plated hepatocytes (Soars et al., 2007), whereas more complex two- or three-dimensional cultures are not as well validated in this regard. These data further support that circulating cabotegravir–glucuronide is efficiently cleared by the kidneys. The basolateral uptake of cabotegravir–glucuronide from blood into the renal proximal tubule cells is mediated by OAT3 (Ft = 100%, not a substrate of OAT1) and is subsequently transported from the proximal tubule to urine by MRP2 (Ft = 66%) and MRP4 (Ft = 34%), but not by OAT4.
As regulatory scrutiny of metabolites in unexpected drug–drug interactions with safety and efficacy consequences has intensified especially for direct glucuronides (Zamek-Gliszczynski et al., 2014), the purpose of this work was to provide a mechanistic basis for cabotegravir–glucuronide disposition in humans. Generally, it is highly unusual for a major hepatically-formed metabolite to undergo extensive urinary excretion with negligible levels in circulation. The present work is the first mechanistic description of such a phenomenon, and it was carried out for prospective understanding and derisking. Available evidence, including hepatic and renal impairment studies (Parasrampuria et al., 2019; Shaik et al., 2019), does not support any specific clinical liabilities associated with the glucuronide metabolite of cabotegravir.
In summary, the present study has elucidated various transporter processes that are involved in the disposition of cabotegravir–glucuronide in humans (Fig. 4). Glucuronidation occurred predominantly in the liver, and cabotegravir–glucuronide was excreted from the liver into both bile and sinusoidal blood. Hepatic reuptake clearance of cabotegravir–glucuronide was low, and instead low systemic exposure is attributed to high renal clearance. Lastly, the fractional contribution (Ft) of redundant transporters such as MRP3 and MRP4 in hepatic sinusoidal excretion and MRP2 and MRP4 in urinary excretion was determined using quantitative proteomics.

Illustration of transporter mechanisms mediating the disposition of cabotegravir–glucuronide (CAB-glu) in humans.
Acknowledgments
We thank Vishal Shah and David Wagner for contribution on microsomal incubations and Aarti Patel for constructive critique on various aspects of this article. We also thank Markus Boesche, Tatjana Rudi, Manuela Kloes-Hudak, and Kerstin Kammerer for assistance with mass spectrometry.
Authorship Contributions
Participated in research design: Patel, Polli, Zamek-Gliszczynski.
Conducted experiments: Patel, Wolf, Pierre.
Contributed new reagents or analytic tools: Patel, Eberl.
Performed data analysis: Patel, Zamek-Gliszczynski.
Wrote or contributed to the writing of the manuscript: Patel, Polli, Zamek-Gliszczynski.
Footnotes
- Received March 26, 2019.
- Accepted June 6, 2019.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- BCRP
- breast cancer resistance protein
- Ft
- fraction transport
- HIV
- human immunodeficiency virus
- HPLC
- high-performance liquid chromatography
- INSTI
- integrase strand transfer inhibitor
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MRP
- multidrug resistance-associated protein
- OAT
- organic anion transporter
- OATP
- Organic Anion Transporting Polypeptide
- P-gp
- P-glycoprotein
- REF
- relative expression factor
- UDPGA
- uridine 5′-diphosphoglucuronic acid
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}